

Abstract
AUF1 is an AU-rich element (ARE)-binding protein that recruits translation initiation factors, molecular chaperones, and mRNA degradation enzymes to the ARE for mRNA destruction. We recently found chaperone Hsp27 to be an AUF1-associated ARE-binding protein required for tumor necrosis factor alpha (TNF-α) mRNA degradation in monocytes. Hsp27 is a multifunctional protein that participates in ubiquitination of proteins for their degradation by proteasomes. A variety of extracellular stimuli promote Hsp27 phosphorylation on three serine residues—Ser15, Ser78, and Ser82—by a number of kinases, including the mitogen-activated protein (MAP) pathway kinases p38 and MK2. Activating either kinase stabilizes ARE mRNAs. Likewise, ectopic expression of phosphomimetic mutant forms of Hsp27 stabilizes reporter ARE mRNAs. Here, we continued to examine the contributions of Hsp27 to mRNA degradation. As AUF1 is ubiquitinated and degraded by proteasomes, we addressed the hypothesis that Hsp27 phosphorylation controls AUF1 levels to modulate ARE mRNA degradation. Indeed, selected phosphomimetic mutants of Hsp27 promote proteolysis of AUF1 in a proteasome-dependent fashion and render ARE mRNAs more stable. Our results suggest that the p38 MAP kinase (MAPK)-MK2–Hsp27 signaling axis may target AUF1 destruction by proteasomes, thereby promoting ARE mRNA stabilization.
INTRODUCTION
AU-rich elements (AREs) target degradation or translational suppression of many mRNAs encoding oncoproteins, cytokines, cell cycle regulators, and signaling proteins. This is achieved via the association of AREs with one or more ARE-binding proteins and, in some cases, microRNAs (7, 33). The AUF1/hnRNP D family of ARE-binding proteins promotes degradation of mRNAs encoding cytokines, such as tumor necrosis factor alpha (TNF-α) and interleukin 1β (IL-1β), and cell cycle regulators, such as p16INK4a, p21WAF1/CIP1, and cyclin D1. AUF1 consists of four isoforms of 37, 40, 42, and 45 kDa generated by alternative pre-mRNA splicing of a single pre-mRNA. They associate with heat shock proteins Hsc70-Hsp70 and Hsp27, translation initiation factor eIF4G, poly(A)-binding protein (PABP), and other unidentified proteins to form a multisubunit complex we refer to as AUF1- and signal transduction-regulated complex (ASTRC) (15, 27). This complex recruits messenger ribonucleases to degrade ARE mRNAs. RNA interference (RNAi)-mediated knockdown of either AUF1 or Hsp27 stabilizes TNF-α ARE mRNA, indicating that both proteins are essential for ARE-mediated mRNA degradation (AMD) (27).
Hsp27 is a ubiquitously expressed, multifunctional protein involved in molecular chaperoning, actin polymerization, and protection from oxidative stress (9). It also participates in ubiquitination of proteins to promote their degradation by proteasomes (21, 22). A variety of extracellular stimuli promote phosphorylation on three serine residues—Ser15, Ser78, and Ser82—by a number of kinases, including p38 mitogen-activated protein (MAP) kinase (MAPK), MAP kinase-activated protein kinase 2 (MK2), Akt, and protein kinase C (2, 6, 14, 17, 23, 38). p38 MAP kinase and MK2 are particularly relevant for mRNA degradation, as activating either kinase stabilizes ARE mRNAs (5, 34). For example, ectopic expression of phosphomimetic mutant forms of Hsp27 stabilizes a reporter mRNA containing a 123-nucleotide (nt) fragment of the cyclooxygenase 2 (COX2) ARE mRNA; this ARE fragment also binds AUF1 (19).
In this work, we continued our examination of the contributions of Hsp27 to mRNA degradation. As AUF1 is ubiquitinated and degraded by proteasomes (15, 16), we addressed the hypothesis that Hsp27 phosphorylation may control AUF1 abundance, thereby controlling ARE mRNA degradation. We created Hsp27 phosphomimetic mutants by replacing Ser15, Ser78, and Ser82 with aspartic acid. This triple substitution does indeed promote proteolysis of AUF1 in a proteasome-dependent fashion and renders ARE mRNAs more stable. Upon further screening of individual phosphomimetic mutants, we found that Ser15 phosphorylation of Hsp27 is sufficient to destabilize the AUF1 protein and promote mRNA stabilization. Thus, the p38 MAP kinase-MK2-Hsp27 signaling axis may target AUF1 destruction by proteasomes to reduce its abundance, thereby promoting ARE mRNA stabilization.
MATERIALS AND METHODS
Materials.
The human promonocytic leukemia cell line THP-1 was provided by Charles McCall (Wake Forest University School of Medicine). Anti-FLAG M2 and anti-α-tubulin monoclonal antibodies were from Sigma. Hsp27 and Hsp70 antibodies were from Stressgen. Anti-AUF1 antibody was prepared by immunization of rabbits with purified His6-p37AUF1 protein by Pocono Rabbit Farms, PA. Horseradish peroxidase-conjugated secondary antibodies were from Promega Corporation. All primers were synthesized by IDT Technologies (Coralville, IA). Restriction enzymes were purchased from New England BioLabs or Promega Corporation.
Cloning of FLAG-Hsp27.
The FLAG-tagged coding region of Hsp27 was amplified by PCR from plasmid pCMVtag2B-FLAG-Hsp27 (19) with the following primers: 5′-GCACGTTTAAACGCCACCATGGATTACAAGGAT-3′ (forward) and 5′-CCCGGGTTACTTGGCGGCAGTCTCATC-3′ (reverse). The forward primer contains a PmeI site, and the reverse primer contains a SmaI site (underlined). The amplicon was subsequently inserted into plasmid pGEM-T Easy (Promega) for further modifications. The wild-type and modified FLAG-Hsp27 coding regions were removed from the pGEM-T Easy plasmids with PmeI and EcoRI (the 5′ and 3′ ends, respectively). They were ligated into plasmid pEF3, which contains a truncated human EF1A promoter (25). The pEF3 vector was prepared for ligation of FLAG-Hsp27 cDNAs by KpnI digestion, blunting with Klenow enzyme (Promega), and subsequent digestion with EcoRI.
Mutation of Hsp27.
Known phosphorylation sites of Hsp27, Ser15, Ser78, and Ser82, were mutated to either alanine (the Ser codon AGC was replaced with the Ala codon GCC [underlined]) or aspartate (the Ser codon AGC was replaced with the Asp codon GAC [underlined]) with the QuikChange kit (Stratagene) according to the manufacturer's directions. Deoxyoligonucleotides with the following sequences were designed with the QuikChange Primer Design tool (Stratagene): S15A (codon AGC to codon GCC), 5′-TGCGGGGCCCCGCCTGGGACCCCT-3′; S78A (codon AGC to codon GCC), 5′-AGCCGCGCGCTCGCCCGGCAACTCAG-3′; S82A (codon AGC to codon GCC), 5′-CAGCCGGCAACTCGCCAGCGGGGTCTCG-3′; S15D, 5′-CTGCGGGGCCCCGACTGGGACCCCTT-3′; S78D, 5′-GACCCGCGCGCTCGACCGGCAACTCAGC-3′; S82D, 5′-CAGCCGGCAACTCGACAGCGGGGTCTCG-3′. All mutations were verified by DNA sequencing.
Cell culture and stable transfections.
THP-1 cells were cultured in RPMI 1640 medium (Cellgro Mediatech, Herndon, VA) supplemented with 10% defined, endotoxin-free fetal bovine serum (HyClone, Logan, UT) and 1× penicillin-streptomycin-glutamine (Gibco) at 37°C in 5% (vol/vol) CO2. Stable transfections were performed as follows: 2 × 107 THP-1 cells were harvested and resuspended in 500 μl RPMI 1640 supplemented with fetal bovine serum without antibiotics. The cells were transferred to 0.4-cm electroporation cuvettes (Bio-Rad Laboratories, Hercules, CA), and 5 μg linearized plasmid containing a FLAG-Hsp27 mutant was added. After incubation on ice for 10 min, electroporation was performed at 360 V and 500 μF with a GenePulser (Bio-Rad). The electroporated cells were then resuspended in 10 ml warm, serum-supplemented RPMI 1640 medium without antibiotics. After 72 h, the transfected cells were transferred to RPMI medium with penicillin/streptomycin. Selection of stably transfected cells was achieved by culturing them with 500 μg/ml Geneticin (Gibco) for 3 weeks.
Immunoprecipitations.
All immunoprecipitations were performed by utilizing the Catch and Release kit v2.0 (Millipore) according to the manufacturer's recommendations. Briefly, the cells were either left untreated, treated with vehicle (dimethyl sulfoxide [DMSO]; J. T. Baker), or treated with 10 nM tetradecanoyl phorbol acetate (TPA) (Sigma) for 1 h. Twenty to 40 million cells were lysed in modified RIPA buffer (150 mM NaCl, 1% NP-40, 0.5% deoxycholic acid, 50 mM Tris-HCl, pH 7.5) and protease inhibitors (10 μg/ml leupeptin, 10 μg/ml pepstatin A, 1 mM phenylmethylsulfonyl fluoride) at 4°C for 20 min. The lysates were centrifuged at 13,000 × g for 10 min at 4°C, and the supernatants (cytoplasms) were collected. The cytoplasmic lysates were treated with 4 μg/μl (final concentration) RNase A for 15 min at 30°C. Complexes were immunopurified with anti-AUF1 or preimmune rabbit serum (final dilution, 1:500) and resolved by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were transferred to nitrocellulose membranes and analyzed by Western blotting with the SuperSignal West Chemiluminescent Substrate kit (Pierce) according to the manufacturer's protocol. The antibodies used and their dilutions were as follows: AUF1, 1:10,000; Hsp27, 1:3,000; Hsp70, 1:30,000; α-tubulin, 1:8,000.
Plasmid construction, transfections, and spectrum deconvolutions for live-cell fluorescence resonance energy transfer (FRET) analyses.
Previously, enhanced cyan fluorescent protein (ECFP) and enhanced yellow fluorescent protein (EYFP) cDNAs were fused to the 5′ end of wild-type Hsp27 and to the 3′ end of p37AUF1 cDNA (27). Each fluorescent protein carries an A206K mutation, which limits their autodimerization (36). For the experiments described in this report, two additional sets of plasmid pairs were created. First, the ECFP and EYFP cDNAs were fused to the 3′ end of p40AUF1 cDNA exactly as described for p37AUF1 (27). Second, the ECFP and EYFP cDNAs were fused to the 5′ ends of Hsp27-S15,78,82A, Hsp27-S15,78,82D, and Hsp27-S15D cDNAs exactly as described for wild-type Hsp27 (27). All fusion constructs were cloned into plasmid pEF3.
To test the expression of fusion proteins, 10 million K562 cells were electroporated with 10 μg of each ECFP or EYFP plasmid in RPMI 1640 medium supplemented with 10% fetal calf serum in the absence of antibiotics. After 48 to 72 h, whole-cell lysates of cells were prepared in 1× SDS loading buffer for Western blot analyses of fusion protein expression with anti-Hsp27 or anti-AUF1 antibody.
For live-cell FRET assays, 1 million THP-1 cells were transiently cotransfected with the desired ECFP and EYFP plasmid pairs with Effectene reagent (Qiagen) according to the manufacturer's protocol. Twenty-four hours after transfection, cells were harvested, washed with phosphate-buffered saline (PBS), and water mounted onto coverslips. Protein-protein interactions were determined by confocal microscopy coupled with fluorescence spectroscopy, as described previously (11, 12). FRET experiments were performed, and the data were deconvolved with a previously described algorithm (27), slightly modified here (see below) to ensure more accurate calculation of FRET efficiency. The efficiency of energy transfer, EFRET, was calculated from the amounts of donor fluorescence (FDA) and acceptor florescence (FAD) present in the deconvolved spectrum by the following equation: EFRET = FAD/(FAD + FDAΦA), in which ΦA is the quantum yield of acceptor fluorescence emission; ΦA is 0.61 for EYFP. The distance between the two proteins (R) was estimated by the following equation: R = Ro[(1 − EFRET)/EFRET)]1/6, in which Ro is the Förster distance, defined as the radius between freely rotating donor and acceptor molecules, that yields an EFRET value of 0.5; Ro = 49.2 Å for the ECFP-EYFP pair.
After deconvolving emission spectra into ECFP fluorescence, EYFP fluorescence, and endogenous cellular fluorescence, a plot of acceptor fluorescence upon donor excitation versus acceptor fluorescence upon acceptor excitation revealed that a diagonal minimum within the data scatter exists, increasing from lower to higher acceptor fluorescence. Since the acceptor is excited by the 442-nm laser (albeit inefficiently), direct excitation effects in the deconvolutions were corrected by subtracting a fraction of the acceptor fluorescence seen upon excitation with the 488-nm laser. This fraction was chosen so that the few data points closest to the diagonal contained effectively no remaining EYFP fluorescence. As expected, when we applied this new correction to our previously published results (27), average EFRET values for p37AUF1-p37AUF1 and p37AUF1-Hsp27 interactions decreased slightly but remained significant for each pair. Photobleaching controls were also performed as described previously (27). The data were analyzed statistically for comparison among plasmid pairs with Student's t test. P values of <0.05 were considered significantly different.
Determination of mRNA half-life.
For each cell culture, Actinomycin D (Calbiochem) was added to a final concentration of 5 μg/ml to inhibit transcription. Cells were harvested at each time point and processed with the QIAshredder kit (Qiagen). Total RNA was purified with the RNeasy kit (Qiagen). A population of cDNA from total RNA was prepared with the High-Capacity cDNA reverse transcription kit (Applied Biosystems). TaqMan MGB probes to TNF-α (Hs00174128_m1; 5′-6-carboxyfluorescein [FAM] reporter dye and 3′ nonfluorescent quencher), IL-1β (Hs00174097_m1; 5′ FAM and 3′ nonfluorescent quencher), and primer-limited GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (4326317E; 5′ VIC reporter dye and 3′ nonfluorescent quencher) mRNA were obtained from Applied Biosystems. Real-time quantitative reverse transcription (RT)-PCR (qPCR) was performed with the Stratagene MX3005P qPCR system and TaqMan Gene Expression Assay Master Mix (Applied Biosystems). In some experiments, the SYBR green procedure was employed for quantitative RT-PCR. mRNA levels were normalized to GAPDH mRNA and plotted as a percentage of the value at time zero (set at 100%). The data were analyzed by nonlinear regression, and mRNA half-lives (t1/2) were determined from first-order decay constants (k) utilizing PRISM software as previously described (27, 35). When appropriate, comparisons were performed between mRNA decay constants using the unpaired two-tailed t test, with differences yielding P values of <0.05 considered significant.
Determination of protein half-life.
Cells were cultured with 100 μg/ml cycloheximide (Sigma-Aldrich) to inhibit translation, 10 μM Lactacystin (Sigma-Aldrich) to block proteasome activity, or both drugs simultaneously. Cells were harvested at each time point and lysed in SDS sample buffer (0.05 M Tris-HCl [pH 6.8], 1% SDS, 5% glycerol, 0.005% bromophenol blue, 10 mM dithiothreitol [DTT]), and 3 × 105 cell equivalents of proteins were resolved by 10% SDS-PAGE. Proteins were detected by Western blot analyses as described above. Image quantification was performed with ImageJ software (http://rsb.info.nih.gov/ij/). The data were analyzed by nonlinear regression to determine first-order decay constants, k, and to calculate half-lives as described above for the mRNA half-life. The standard error about the regression solution is linear about k and therefore hyperbolic about the protein half-life: t1/2 = ln2/k. Assignment of cellular half-lives to very stable proteins is difficult, since the hyperbolic nature of t1/2 relative to the first-order decay constant inflates errors in t1/2 when k approaches zero. Thus, there is no meaningful difference between “no decay” and a long half-life, such as 20 h, in these analyses.
RNA-binding assays.
The Hsp27 coding region devoid of the FLAG tag was amplified from plasmid pCMVtag2B-FLAG-Hsp27 and inserted into plasmid pBAD/HisB (Invitrogen, Carlsbad, CA). Recombinant His6-Hsp27 and His6–Hsp27-TriD (where TriD refers to Ser15Asp, Ser78Asp, and Ser82Asp replacement of serine with aspartate) were prepared from Escherichia coli TOP10 cells transformed with either pBAD/HisB-Hsp27 or pBAD/HisB–Hsp27-TriD as previously described (32). RNA oligonucleotides containing a 38-nt core ARE from TNF-α mRNA or a similar-size fragment from rabbit β-globin (Rβ) mRNA were synthesized by Dharmacon (Lafayette, CO) as previously described (31). For electrophoretic mobility shift assays (EMSA), TNF-α or Rβ RNA was 5′ labeled with [γ-32P]ATP and T4 polynucleotide kinase. Experiments were performed with His6-Hsp27 and His6–Hsp27-TriD as described previously (31). Gels were analyzed with a Typhoon 9410 PhosphorImager (Molecular Dynamics, Amersham Biosciences).
RESULTS
Phosphomimetic mutant forms of Hsp27 alter AUF1 expression.
Our previous studies showed that posttranslational modifications of AUF1 and Hsp27 within ASTRC occur in response to activation of protein kinase C in THP-1 cells (27, 30). Likewise, activation of the p38 signaling pathway in HeLa cells promotes phosphorylation of Hsp27 and stabilization of ARE reporter mRNA (19). To examine the effects of Hsp27 phosphorylation on AUF1 and AMD, several phosphomimetic mutants and phosphorylation-resistant mutants were created by individually replacing the critical serines with aspartates (S15D, S78D, or S82D) or alanines (S15A, S78A, or S82A), respectively. Since phosphorylation at one site may not be sufficient to evoke a change in protein function, double mutations were constructed in which Ser78 and Ser82 were replaced with either aspartate (referred to as Hsp27-DblD) or alanine (referred to as Hsp27-DblA). Triple mutations were constructed, as well, with all three serines changed to aspartate (referred to as Hsp27-TriD) or alanine (referred to as Hsp27-TriA). These expression plasmids were stably transfected into THP-1 cells; the FLAG epitope shifts protein mobility to distinguish the mutants from endogenous Hsp27 on Western blots. Each cell line stably expressed the respective mutant protein (data not shown). FLAG-tagged wild-type Hsp27 served as a control in subsequent experiments.
Since triple-phosphomimetic Hsp27 confers a significant stabilizing effect upon an ARE mRNA (19), initial experiments focused on THP-1 cells expressing the Hsp27-TriA or -TriD mutant compared to wild-type FLAG-Hsp27. THP-1 cells express all four AUF1 isoforms. Isoform p37AUF1 is the least abundant; p40AUF1/p42AUF1 comigrate on gels but are easily separated from p45AUF1 (30). Isoforms p40AUF1/p42AUF1 and p45AUF1 were readily observed in whole-cell lysates of cells expressing wild-type Hsp27 (Fig. 1, top, lane 1); their levels were unaffected by expression of Hsp27-TriA (data not shown). By contrast, expression of Hsp27-TriD reduced total AUF1 abundance, though a form with higher mobility was apparent (Fig. 1, top, compare lane 2 with lane 1). Since AUF1 isoforms are subject to posttranslational modifications that alter their electrophoretic mobilities, including phosphorylation (4, 30, 37) and ubiquitination (15, 16), it is not clear if this faster-migrating band represents p37AUF1 or another isoform without modifications (see Discussion). Nonetheless, these results suggest that expression of the triple phosphomimetic Hsp27 mutant reduces total AUF1 abundance in THP-1 cells. Hsp27-TriD appeared to reduce the abundance of cellular Hsp27, as well (Fig. 1, middle, compare lane 2 with lane 1). Since AUF1 is normally subject to ubiquitination (15, 16) and Hsp27 directs ubiquitinated proteins to the proteasome for degradation (21, 22), we next examined the effects of Hsp27 mutants on AUF1 protein decay rates.
Fig. 1.

Stable expression of Hsp27 phosphomimetic in THP-1 cells alters AUF1 protein levels. Shown are representative Western blots of whole-cell lysates from THP-1 cells overexpressing the indicated FLAG-Hsp27 phosphomimetic mutants and probed with antibody to AUF1 (top), Hsp27 (middle), or Hsp70 (bottom, as a loading control). Each lane contains 105 cell equivalents of protein. Endo, endogenous Hsp27; FLAG, FLAG-tagged Hsp27. Arrowheads denote the FLAG-tagged and endogenous Hsp27 proteins.
Hsp27-TriD promotes proteasome-dependent degradation of the AUF1 family of proteins.
To determine whether the reduced abundance of AUF1 proteins in THP-1/Hsp27-TriD cells is caused in part by their accelerated degradation, we analyzed protein stability. Cells were cultured with cycloheximide to halt protein synthesis. Cells were harvested and lysed at each time point, and the proteins were resolved by SDS-PAGE and analyzed by Western blotting. Graphs of the percentage of protein remaining versus time were analyzed by nonlinear regression to determine the first-order decay constant, k, and protein half-lives. While p37AUF1 is not visible in these exposures, THP-1/Hsp27 and THP-1/Hsp27-TriA cells presented little or no discernible degradation of AUF1 proteins (Fig. 2 A, B, and E) during the 7-h time course. We note that this result contrasts with the measurements of Laroia and Schneider, who demonstrated AUF1 isoform half-lives ranging from 60 min for p40/42AUF1 to ∼8 h for p45AUF1 (16). However, their studies were performed with HeLa cells, while we utilized THP-1 monocytes; thus, there may be cell-type-specific differences in decay rates. Nonetheless, total AUF1 proteins decayed with a half-life of 3.7 h in THP-1/Hsp27-TriD cells (Fig. 2C and E). Likewise, FLAG-tagged Hsp27-TriD decayed with a half-life of 2.2 h (Fig. 2C and F), while FLAG-tagged wild-type and Hsp27-TriA both exhibited little or no decay (Fig. 2A, B, and F).
Fig. 2.

Hsp27-TriD expression destabilizes the AUF1 protein. Cells were cultured with cycloheximide for the indicated times. AUF1 and FLAG-Hsp27 protein levels were assessed by Western blot analyses of whole-cell lysates (3 × 105 cell equivalents/lane) with antibodies to AUF1 and FLAG. α-Tubulin was used as an internal control. (A) THP-1/Hsp27 cells. (B) THP-1/Hsp27-TriA cells. (C) THP-1/Hsp27-TriD cells. (D) THP-1/Hsp27-TriD cells were cocultured with 10 μM lactacystin to inhibit proteasome activity. (E and F) AUF1 (E) and Hsp27 (F) levels were quantified and normalized to α-tubulin in the indicated cell lines and further analyzed with nonlinear regression to determine the protein half-life. − Lact, cells cultured without lactacystin; + Lact, cells cultured with lactacystin; WT, wild type.
To determine whether the Hsp27-TriD mutant enhanced degradation of AUF1 proteins via the proteasome pathway, both protein synthesis and proteasome-mediated protein degradation were inhibited by concurrent treatment of cells with cycloheximide and the proteasome inhibitor lactacystin. Lactacystin blocked degradation of AUF1 proteins and Hsp27-TriD (compare Fig. 2D to C; see quantitations in Fig. 2E and F). These results suggest that proteasomes accelerate degradation of the AUF1 protein family, as well as Hsp27-TriD, in THP-1/Hsp27-TriD cells (see Discussion).
Posttranslational modification of Hsp27 affects AUF1-Hsp27 protein-protein interactions.
We previously observed AUF1-Hsp27 association by coimmunoprecipitation of Hsp27 in cell lysates with AUF1 antibody and by live-cell FRET (27). The importance of AUF1-Hsp27 protein-protein interactions is underscored by two additional observations: both proteins are required for AMD, as knockdown of either stabilizes TNF-α mRNA, and both proteins bind the TNF-α ARE with high affinity (Kd [dissociation constant] = 0.8 for AUF1 and 1.6 nM for Hsp27, measured by fluorescence anisotropy).
The aforementioned immunoprecipitation experiments were performed with a 130,000 × g pellet fraction (P130) of cytoplasmic extracts. This P130 fraction contains ribosome/polyribosome-associated mRNAs, mRNA degradation enzymes, Hsp27, and AUF1. Notably, AUF1-Hsp27 interactions are also observed in the 130,000 × g supernatant fraction (S130) (unpublished observations). Thus, AUF1-Hsp27 interactions can be readily detected in cytoplasmic extracts without the 130,000 × g fractionation step, and consequently, they were utilized for immunoprecipitations. Since activation of protein kinase C with phorbol ester 12-O-tetradecanoylphorbol-13-acetate (TPA) can affect the association of Hsp27 with the AUF1 complex of proteins (27), AUF1 immunoprecipitations were compared using lysates from untreated and TPA-treated cells. While FLAG-tagged wild-type Hsp27 and Hsp27-TriA both coimmunoprecipitated with AUF1 from lysates regardless of TPA treatment, Hsp27-TriD did not coimmunoprecipitate with AUF1, either with or without TPA treatment. Even highly prolonged exposures of immunoblots did not reveal any coimmunoprecipitation (Fig. 3A, compare lanes 7 and 8 with lanes 3 to 6). As expected, Hsp27 was undetectable in immunoprecipitations with preimmune serum (Fig. 3A, lanes 1 and 2).
Fig. 3.

Coimmunoprecipitation analyses of Hsp27-TriA and Hsp27-TriD with AUF1. (A) Antibody to AUF1 or preimmune (Pre im) serum was utilized for immunoprecipitations (IP) with equal cell equivalents of cytoplasmic lysates from nonactived (−TPA) or activated (+TPA; 10 nM for 1 h) cells expressing the FLAG-tagged Hsp27 proteins indicated at the top. The immunoprecipitates were then analyzed by Western blotting (WB) with antibody to the FLAG epitope. A darker exposure of the blot is also shown to detect low levels of Hsp27-TriD in immunoprecipitates. (B) Western blots of the indicated immunoprecipitates were probed with anti-AUF1 antibody. (C) The indicated cytoplasmic lysates were immunoprecipitated with nonimmune antibody or antibody to the FLAG epitope. The immunoprecipitates were then analyzed by Western blotting with antibody to the FLAG epitope. Cyt, cytoplasmic lysate; Nuc, nuclear lysate (to compare Hsp27 levels).
Two controls were performed to ensure fidelity of the immunoprecipitations. First, immunoprecipitations were done with antibody to AUF1, followed by Western blot detection of AUF1 in pellets. AUF1 was detected in immunoprecipitates of lysates from cells expressing the wild type, Hsp27-TriA, and Hsp27-TriD (Fig. 3B, lanes 3 in the respective panels). Thus, the inability to detect Hsp27-TriD in immunoprecipitates was not due simply to failure of AUF1 to immunoprecipitate. For the second control, all three lysates were immunoprecipitated with antibody to the FLAG epitope and examined for the presence of each FLAG-tagged Hsp27 protein by Western blotting. FLAG-tagged wild-type, -TriA, and -TriD proteins were readily observed (Fig. 3C, lanes 4 in the respective panels), demonstrating that they are soluble and themselves readily immunoprecipitable. We thus conclude that the Hsp27-TriD mutant specifically fails to stably coimmunoprecipitate with AUF1. However, as coimmunoprecipitation infers only protein-protein interactions that survive cell rupture and extensive protein dilution, we wished to examine AUF1-Hsp27 interactions with an independent live-cell method.
Analyses of AUF1-Hsp27 interactions by live-cell FRET.
We previously utilized a strategy that combined confocal microscopy and fluorescence spectroscopy with FRET as a biosensor to demonstrate p37AUF1-p37AUF1 and p37AUF1-Hsp27 interactions in live cells (27). We thus examined protein-protein interactions in live THP-1 cells expressing p37AUF1 fused at its C terminus with ECFP or EYFP, together with wild-type Hsp27, Hsp27-TriA, or Hsp27-TriD fused at its N terminus with ECFP or EYFP. These fusion protein configurations were chosen based upon previous observations (e.g., ECFP and EYFP fused to p37AUF1 at its N terminus were exclusively nuclear, unlike endogenous p37AUF1). For each tested protein pair, ECFP is the fluorescence donor and EYFP is the fluorescence acceptor. Analyses of Western blots of lysates from transfected cells confirmed that all fusion proteins have the expected apparent molecular weights (data not shown).
As in our previous work, we employed a 2-fold strategy. First, spectral scans of a random section of the cytoplasm (where mRNA decay takes place) were performed across the 450- to 650-nm spectral range for a number of cells. ECFP and EYFP fluorescence was corrected by deconvolving spectra to remove contributions of intrinsic fluorescent components (e.g., nicotinamide and riboflavin derivatives). This permitted calculation of more accurate FRET efficiencies (EFRET). Second, FRET efficiencies were calculated for each cell to permit rational refinement of the obtained data, averaging, and statistical comparisons among different protein pairs.
Figure 4 A shows a single transfected THP-1 cell exposed to the different lasers. As observed by confocal microscopy coupled with fluorescence spectroscopy, excitation of p37AUF1-ECFP at 442 nm led to emission peaking at 475 nm (Fig. 4A, left); it also led to emission of EYFP–Hsp27-TriA peaking at 527 nm (Fig. 4A, right). The latter emission is indicative of a FRET signature and suggested p37AUF1-ECFP interaction with EYFP–Hsp27-TriA. Direct excitation of EYFP–Hsp27-TriA at 488 nm led to emission at 527 nm, as expected (Fig. 4A, middle). Figure 4B shows the spectral deconvolution of one selected region of the cytoplasm in the THP-1 cell shown in Fig. 4A. As observed in the deconvolved spectrum, excitation of ECFP at 442 nm produced a peak at 475 nm indicative of p37AUF1-ECFP expression (Fig. 4B, left); excitation of EYFP at 488 nm produced a peak at 527 nm, indicative of EYFP–Hsp27-TriA expression (Fig. 4B, right). These data indicate that p37AUF1-ECFP and EYFP-TriA were indeed coexpressed in the cell. Excitation of p37AUF1-ECFP at 442 nm in this cell also produced a peak of yellow emission at 527 nm, consistent with a FRET signature (Fig. 4B, left, arrow). The calculated EFRET from the deconvolved spectrum was 0.24 for this particular cell. Analyses of several cells resulted in an average EFRET of 0.34 ± 0.03 for the transfected population (Fig. 4C) to yield a mean distance of ∼55 Å between the proteins. This EFRET value is not statistically different from that for the p37AUF1-ECFP and EYFP-Hsp27 (wild-type) protein pair, 0.42 ± 0.03 (R = 51 Å) (Fig. 4C; P = 0.0526). These results are consistent with the immunoprecipitation data, demonstrating comparable interactions between AUF1 and both wild-type Hsp27 and Hsp27-TriA.
Fig. 4.

Analyses of p37AUF1–Hsp27-TriA interactions in live cells by FRET. (A) Confocal fluorescence imaging of a THP-1 cell coexpressing p37AUF1-ECFP and EYFP–Hsp27-TriA. Shown are emission of p37AUF1-ECFP upon excitation at 442 nm (left), emission of EYFP–Hsp27-TriA upon excitation at 488 nm (middle), and emission of EYFP–Hsp27-TriA upon excitation of p37AUF1-ECFP at 442 nm (right), indicative of a FRET signature. (B) Deconvolutions of fluorescence emission spectra of the cell in panel A. (Left) The cell was irradiated with 442-nm light, and the spectrum was deconvolved into its major components. After the raw spectrum (black line) was transformed so that it possessed even wavelength intervals (red line), it was separated into the following components: ECFP (i.e., p37AUF1-ECFP expression, green line), EYFP (FRET signature from EYFP–Hsp27-TriA, yellow line and arrow), nicotinamide background at 500 nm (navy blue line), and fiboflavin background at 550 nm (pink line). Addition of the four components (light-blue line) closely approximated the observed spectrum. From this spectrum, the EFRET was 0.24, with a calculated distance of 59 Å between the two fluorescence-tagged proteins. (Right) Deconvolved spectrum of the same cell upon direct EYFP excitation at 488 nm. The yellow line represents EYFP–Hsp27-TriA expression. For both spectra and all deconvolutions shown in subsequent figures, fluorescence units are listed for ECFP, EYFP, and the cell at 550 nm (cell550); these were determined by integration of the curves at the respective emission wavelength maxima ± 10 nm (the band pass width of the filters). (C) Statistical analyses of the p37AUF1-ECFP-EYFP-Hsp27 (wild-type) interaction and the p37AUF1-ECFP-EYFP-Hsp27-TriA interaction. Scatter plots of deconvolved spectra of the indicated cells were derived from FRET analyses. Each point represents EFRET calculated from one deconvolved spectrum. P was equal to 0.0526 for this comparison.
By contrast, association of Hsp27-TriD was not observed by AUF1 immunoprecipitation (Fig. 3A). However, there are precedents for loss of protein-protein interactions during disruption of cells. For example, in the case of the gamma interferon receptor complex, preassembly of receptor subunits was not detected by immunoprecipitation in the absence of ligands but was observed with live-cell FRET experiments (1, 8, 10, 12). Accordingly, FRET experiments were performed to investigate potential interaction between p37AUF1 and Hsp27-TriD in live cells. Cells were cotransfected with plasmids encoding p37AUF1-ECFP and EYFP–Hsp27-TriD. Coexpression of p37AUF1-ECFP (Fig. 5 A, left) and EYFP–Hsp27-TriD (Fig. 5A, middle) resulted in a FRET signal (Fig. 5A, right), suggesting an interaction between the proteins in the cell. A spectral scan from 450 to 650 nm at a selected point within the cytoplasm of the cell and data deconvolution produced the expected 475-nm peak indicative of p37AUF1-ECFP (Fig. 5B, left); excitation of EYFP at 488 nm resulted in a peak at 527 nm, corresponding to EYFP–Hsp27-TriD expression (Fig. 5B, right). A peak at 527 nm was also observed upon excitation at 442 nm, indicative of a FRET signature (Fig. 5B left, arrow). The calculated EFRET for this specific cell was 0.30. Examination of multiple cells resulted in a mean EFRET of 0.21 ± 0.03 for the transfected population (Fig. 5C) to yield an average distance of ∼61 Å between the proteins (compared to 51 Å for the p37AUF1–Hsp27 wild-type pair; P < 0.0001).
Fig. 5.

Analyses of p37AUF1–Hsp27-TriD interactions in live cells by FRET. (A) Confocal fluorescence imaging of a THP-1 cell coexpressing p37AUF1-ECFP and EYFP–Hsp27-TriD. Shown are emission of p37AUF1-ECFP upon excitation at 442 nm (left), emission of EYFP–Hsp27-TriD upon excitation at 488 nm (middle), and emission of EYFP–Hsp27-TriD upon excitation of p37AUF1-ECFP at 442 nm (right), indicative of a FRET signature. (B) Deconvolutions of fluorescence emission spectra from the cell in panel A. (Left) The cell was irradiated with 442-nm light, and the spectrum was deconvolved into its major components as described in the legend to Fig. 4B (FRET signature from EYFP–Hsp27-TriD, yellow line and arrow). For this cell, EFRET was 0.30, with a distance of 56 Å between the two fluorescence-tagged proteins. (Right) Deconvolved spectrum of the same cell upon direct EYFP excitation at 488 nm. The yellow line represents EYFP–Hsp27-TriD expression. (C) Statistical analyses of the p37AUF1-ECFP-EYFP–Hsp27-TriA interaction (from Fig. 4C, for comparison) and the p37AUF1-ECFP-EYFP–Hsp27-TriD interaction, as described in the legend to Fig. 4C. P was <0.0001 for this comparison.
To ensure the mathematical treatment of spectra accurately estimated FRET between the donor and acceptor protein, selective photobleaching of EYFP–Hsp27-TriD was performed in cells coexpressing p37AUF1-ECFP. As expected, the photobleached regions exhibited more ECFP fluorescence than they did prior to photobleaching (Fig. 6A) (3,888 versus 3,270 fluorescence units, respectively; compare Fig. 6A right and left, green lines). This demonstrates that EYFP emission upon 442-nm (ECFP) excitation was due to FRET (i.e., true protein-protein interactions) and not direct excitation of EYFP. Three independent cells yielded similar outcomes (data not shown). Similar photobleaching results were obtained with the p37AUF1–Hsp27-TriA pair (Fig. 6B) (4,869 versus 3,706 fluorescence units, respectively; compare Fig. 6B right and left, green lines). Taken together, the results in Fig. 4 to 6 demonstrate that serine-to-aspartate phosphomimetic substitutions of Hsp27 alter, but do not abolish, the association of Hsp27 with p37AUF1 in live cells, in contrast to the immunoprecipitation results (Fig. 3A).
Fig. 6.

Control photobleaching experiments for interactions of p37AUF1 with Hsp27-TriD and Hsp27-TriA. Shown are deconvolution analyses of a THP-1 cell coexpressing p37AUF1-ECFP and EYFP–Hsp27-TriD (A) and p37AUF1-ECFP and EYFP–Hsp27-TriA (B). The spectra on the left are before photobleaching of EYFP, and those on the right are after photobleaching of EYFP. An increase in ECFP fluorescence after photobleaching in both cases is indicative of a true FRET signature. Three cells were analyzed for each protein pair with comparable results.
We next expanded our live-cell FRET experiments to include p40AUF1 for two reasons: first, p40AUF1 and p37AUF1 are the predominant cytoplasmic isoforms in THP-1 monocytes (29, 30). Second, it appears that all AUF1 proteins are subject to degradation in cells expressing Hsp27-TriD (Fig. 2). This suggests that Hsp27 may associate with p40AUF1, just as it does with p37AUF1, since all three of these proteins are cytoplasmic. Indeed, live-cell FRET experiments revealed comparable EFRET values between p40AUF1–Hsp27 wild-type pairs and p40AUF1–Hsp27-TriA pairs (Fig. 7 A) (P = 0.0540). However, the interaction of p40AUF1 with Hsp27-TriD was reduced somewhat compared to its interaction with wild-type Hsp27 (Fig. 7A) (P = 0.0328). Nonetheless, this still represents an appreciable association of the proteins, in contrast to the immunoprecipitation results (Fig. 3). Like p37AUF1, the p40AUF1-p40AUF1 homodimer-like association was also readily observed in cells by live-cell FRET, as were p37AUF1-p40AUF1 heterodimer-like interactions (Fig. 7B). Experiments by Sarkar and colleagues suggest that p37AUF1 and p40AUF1 can associate with p42AUF1 and p45AUF1, as well, permitting the two smaller isoforms to shuttle the two larger isoforms from the cytoplasm to the nucleus (24). These heterocomplex arrangements may ensure degradation of all AUF1 isoforms regardless of their intracellular distribution.
Fig. 7.
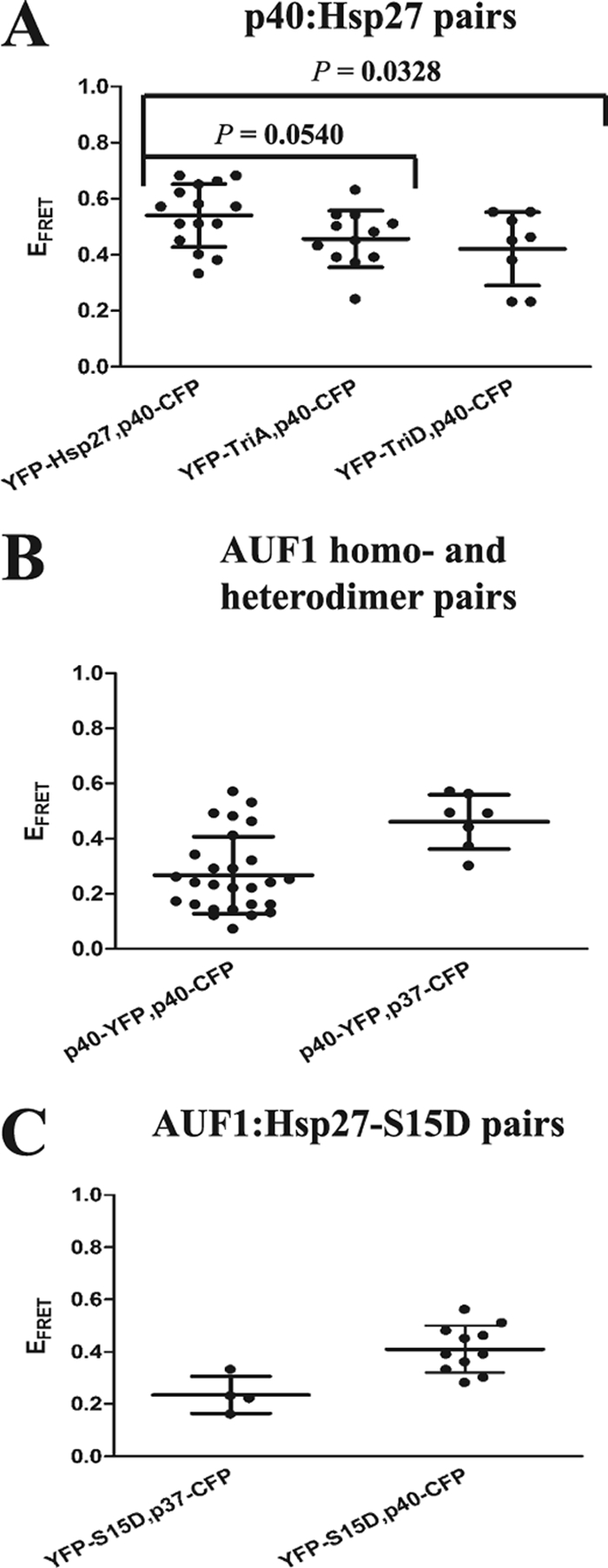
Analyses of additional AUF1-AUF1 and AUF1-Hsp27 interactions in live cells by FRET. (A, B, and C) Cells expressing the indicated fusion protein pairs were analyzed for EFRET and plotted as described in the legend to Fig. 4C. Where relevant, statistical analyses between protein pairs are shown.
Differential phosphomimetic substitution of Hsp27 influences its association with ASTRC.
We next examined the contributions of individual Hsp27 phosphorylation sites to association with AUF1 complexes. To this end, cytoplasmic extracts from THP-1 cell lines expressing a panel of FLAG-tagged Hsp27 mutants were screened to find mutants that coimmunoprecipitate with AUF1. Hsp27 was detected by Western blotting with anti-FLAG antibody. Only Hsp27-TriD and Hsp27-S15D mutants did not stably associate with AUF1 (Fig. 8). This observation suggests that phosphorylation of Ser15 is sufficient to modify Hsp27 association with AUF1 complexes, since both Hsp27-S15D and Hsp27-TriD proteins have this mutation in common. Even so, live-cell FRET experiments with the p37AUF1-ECFP-YFP-Hsp27-S15D pair and the p40AUF1-ECFP-YFP-Hsp27-S15D pair revealed their association in cells (Fig. 7C). Similar to Hsp27-TriD, we speculate that the S15D modification may reduce the stability of the Hsp27-AUF1 association, at least in immunoprecipitation reactions.
Fig. 8.

Different phosphomimetic substitutions in Hsp27 affects its coimmunoprecipitation with AUF1. AUF1-associated Hsp27 mutants were immunoprecipitated from cytoplasmic lysates. THP-1 cells were stably transfected with plasmids expressing the indicated FLAG-Hsp27 mutants. AUF1 protein complexes were immunopurified from equal cell equivalents of cytoplasmic cell lysates utilizing antibody to AUF1 and resolved by 10% SDS-PAGE. The blots were probed with an antibody against FLAG epitope.
Since Hsp27-TriD affects the abundance of the AUF1 family of proteins, as well as their degradation (Fig. 1 and 2C, respectively), we examined the effects of Hsp27-S15D on AUF1 expression in whole-cell lysates. Like Hsp27-TriD, expression of Hsp27-S15D altered the abundance or migration of AUF1 isoforms. The p45AUF1 isoform was no longer apparent (Fig. 1, lane 3), while the fastest-migrating band increased relative to cells expressing wild-type Hsp27 (Fig. 1, compare lane 3 to lane 1). As we noted above, since AUF1 isoforms are subject to posttranslational modifications that alter their electrophoretic mobilities, we cannot be sure if the fastest-migrating band is p37AUF1 or larger, unmodified isoforms (see Discussion). Hsp27-S15D appeared to reduce the abundance of cellular Hsp27, as well (Fig. 1, middle, compare lane 3 with lane 1,).
Since Hsp27-S15D seems to exert effects on AUF1 and Hsp27 expression similar to those of Hsp27-TriD, we examined its effects on protein turnover with cycloheximide chase assays. While AUF1 did not detectably decay in cells ectopically expressing wild-type Hsp27, expression of Hsp27-S15D reduced the AUF1 half-life to 4.9 h (Fig. 9A and C). The proteasome contributes to degradation, as its inhibition extended the AUF1 half-life to 10 h (Fig. 9B and C). Like the Hsp27-TriD mutant, Hsp27-S15D was unstable (half-life, 3.1 h) (Fig. 9A and D), and proteasome inhibition completely stabilized it (Fig. 9B and D). Together, these experiments indicate that Hsp27 phosphorylation on Ser15 may be sufficient to promote degradation of AUF1 and Hsp27 proteins.
Fig. 9.

Degradation of AUF1 protein is significantly accelerated in THP-1/Hsp27-S15D cells. Cells were cultured with cycloheximide for the indicated times. AUF1 and FLAG-Hsp27 protein levels were assessed by Western blotting of whole-cell lysates of 3 × 105 cells/lane with αAUF1 and αFLAG antibodies, respectively. Tubulin blots were performed to internally control for the total protein content. (A) AUF1 and Hsp27-S15D protein levels without proteasome inhibition. (B) AUF1 and Hsp27-S15D protein levels in cells with concurrent proteasome inhibition (10 μM lactacystin for the indicated times). (C and D) Protein levels were quantified and normalized to those of tubulin and then analyzed by nonlinear regression to determine the protein half-life for AUF1 (C) and Hsp27-S15D (D). − Lact, sample without proteasome inhibition; + Lact, sample with proteasome inhibitor.
Phosphomimetic mutants of Hsp27 affect ARE mRNA decay kinetics.
As noted above, Hsp27 binds the TNF-α ARE with high affinity, and knockdown of Hsp27 in THP-1 cells stabilizes the mRNA (27). Phosphomimetic mutants of Hsp27 alter AUF1 expression and reduce the abundance of Hsp27 (Fig. 1). To determine whether these mutants affect ARE mRNA stability, we examined the decay kinetics of TNF-α and IL-1β mRNAs in cells expressing wild-type Hsp27, Hsp27-TriA, Hsp27-TriD, or Hsp27-S15D. Cells were treated with actinomycin D to block transcription; RNA was isolated from the cells at various times and analyzed by qRT-PCR, with GAPDH mRNA serving as an internal control. Nonlinear regression analyses of the percent mRNA remaining versus time yields the first-order decay constant, k, from which the mRNA half-life is calculated. TNF-α mRNA half-lives were 20 min and 14 min in cells expressing wild-type or Hsp27-TriA, respectively (Fig. 10A); these differences were not statistically significant (P > 0.05). These values are also comparable to that observed with untransfected cells (21 min) (reference 27 and data not shown). By contrast, expression of either Hsp27-TriD or Hsp27-S15D extended the TNF-α mRNA half-life to 70 min and 100 min, respectively. These Hsp27 phosphomimetic mutants also extended the half-life of the IL-1β ARE mRNA to over 5 h, compared to a 2-h half-life in cells expressing either wild-type Hsp27 or Hsp27-TriA (Fig. 10B). Thus, posttranslational modification of Hsp27 slows TNF-α and IL-1β mRNA decay kinetics, just as it does for cyclooxygenase 2 (COX2) mRNA (19).
Fig. 10.

Expression of Hsp27-TriD or Hsp27-S15D stabilizes TNF-α and IL-1β mRNAs. THP-1 cells stably expressing wild-type Hsp27, Hsp27-TriA, Hsp27-TriD, or Hsp27-S15D were cultured with actinomycin D (ActD) to block transcription. Total RNA was purified at the indicated time points. mRNAs were quantified by real-time RT-PCR and analyzed with nonlinear regression to determine mRNA half-lives for TNF-α (A) and IL-1β (B) mRNAs. Due to the higher decay rate of TNF-α mRNA, its half-lives are shown in minutes, while those of IL-1β are shown in hours.
Phosphomimetic substitution of Hsp27 does not reduce ARE-binding affinity.
To determine if the increased stability of TNF-α mRNA in THP-1 cells expressing phosphomimetic mutants of Hsp27 might be due in part to their impaired ARE-binding activity, EMSA were performed. Recombinant His6-Hsp27 proteins and a 38-nt RNA oligonucleotide with the core TNF-α ARE were utilized for the assays. Increasing amounts of His6-Hsp27 supported the formation of a ribonucleoprotein complex in vitro with the TNF-α ARE (Fig. 11, left), and comparable results were obtained with His6-Hsp27-TriD (Fig. 11, right). The apparent dissociation constants, Kd, defined as 50% of input RNA being bound (3), were 400 nM versus 300 nM for wild-type Hsp27 and Hsp27-TriD, respectively. This result indicates that aspartate substitutions in Hsp27 do not impair its ARE-binding affinity in this assay. Thus, putative differences in ARE-binding affinities likely cannot account for the stabilization of ARE mRNA in cells expressing Hsp27-TriD (see Discussion).
Fig. 11.

Wild-type and Hsp27-TriD bind the TNF-α ARE with comparable affinities. Electrophoretic mobility shift assays were performed with 32P-labeled TNF-α ARE and the indicated concentrations of His6-Hsp27 (left) and His6-Hsp27-TriD (right). The reaction mixtures were fractionated by native gel electrophoresis. Free RNA is indicated, and the positions of the protein-RNA complexes are indicated by the arrowheads.
DISCUSSION
Our earlier work identified Hsp27 as a subunit of the AUF1 complex of proteins. Hsp27 is an ARE-binding protein that is essential for AMD, as RNAi-mediated reduction of Hsp27 abundance stabilized TNF-α ARE mRNA in the human monocyte cell line THP-1. Additional work has also established a role for Hsp27 in AMD. (i) Shchors and colleagues found that AUF1 and Hsp27 associate with cell death-inhibiting RNA (CDIR), a U-rich transcript derived from the 3′ untranslated region (UTR) of a gene with unknown function (26). Binding of the AUF1-Hsp27 complex to CDIR was associated with reduced AMD and an antiapoptotic phenotype. (ii) Sommer and colleagues showed that overexpression of Hsp27 reduced the levels of ARE mRNA encoding the c-Yes oncoprotein (28). (iii) Lasa and colleagues showed that activation of the p38 MAPK pathway stabilized an ARE reporter mRNA (19). This was due to subsequent p38-dependent activation of MK2, which in turn phosphorylates Hsp27. Indeed, ectopic expression of an Hsp27 phosphomimetic (glutamic acid substitutions at Ser15, Ser78, and Ser82), but not the wild-type protein, was sufficient to stabilize a COX2-ARE reporter transcript in the absence of p38 MAPK activation.
Based upon this and other work, we sought a molecular basis for links between phosphomimetic mutants of Hsp27 and stabilization of at least some ARE mRNAs. We initially found that expression of Hsp27 with replacement of Ser15, Ser78, and Ser82 by aspartates, but not alanines, led to increased turnover of AUF1 and Hsp27 proteins by proteasomes, as a proteasome inhibitor fully stabilized the proteins (Fig. 2). Expression of this phosphomimetic Hsp27 mutant also led to stabilization of TNF-α and IL-1β mRNAs (Fig. 10). We subsequently found that replacement of Ser15 alone with aspartate was sufficient to accelerate the turnover of the AUF1 and Hsp27 proteins. However, in this case, the proteasome inhibitor only partially stabilized AUF1, suggesting that additional, unknown proteases may contribute to AUF1 turnover, as well (Fig. 9). Nonetheless, since Hsp27 phosphomimetics conferred ARE mRNA stabilization, signaling events may act in part to alter AUF1 and/or Hsp27 expression, thereby stabilizing selected cytokine mRNAs.
As noted above, it is not clear exactly which AUF1 isoforms are present in cells expressing the Hsp27 phosphomimetic mutants compared to normal THP-1 cells. Western blots of whole-cell lysates of THP-1 cells expressing wild-type Hsp27 revealed bands with mobilities consistent with p40AUF1/p42AUF1 (which are difficult to resolve) and p45AUF1 (Fig. 1); p37AUF1 is least abundant in THP-1 cells and can be observed by Western blotting only under conditions that overexpose the other isoforms. Expression of either Hsp27-TriD or Hsp27-S15D alters the distribution of bands observed on Western blots (Fig. 1) and leads to a fast-migrating polypeptide that may or may not be p37AUF1. As AUF1 isoforms undergo posttranslational modifications that alter their electrophoretic mobilities (16, 37), this band potentially represents an unmodified, larger AUF1 isoform. Also, various isoforms may undergo proteolytic-processing events that produce smaller polypeptides. For example, the single-strand DNA-binding protein UP2 is approximately 6 kDa smaller than p37AUF1; cDNA cloning and sequence comparisons revealed that it contains sequence derived from p40AUF1 (reference 13 and our unpublished observations). Regardless of the ultimate identities of the AUF1 isoforms, or putative processing products thereof, it is important to note that the Hsp27 phosphomimetics clearly accelerate turnover of all expressed AUF1 proteins (Fig. 2 and 9). Further work will be required to establish how Hsp27 phosphorylation controls the expression of individual AUF1 isoforms and/or their posttranslational modification(s).
We showed previously by coimmunoprecipitation experiments that Hsp27 is a subunit within the AUF1 complex of proteins (27). Also, live-cell FRET experiments revealed an average intermolecular distance of ∼49 Å between Hsp27 and p37AUF1 in the cytoplasm. Based upon these observations and the role of Hsp27 in ubiquitination/degradation, we predicted that the Ser15- and Ser15-Ser78-Ser82-Hsp27 phosphomimetics, which accelerate AUF1 and Hsp27 turnover, would likely present increased association with the AUF1 complex of proteins. However, we found these two mutants did not detectably coimmunoprecipitate with AUF1 (Fig. 3 and 8), even in cells in which proteasomes were inhibited with lactacystin (data not shown). However, FRET experiments indicated close association of these proteins in live cells. We interpret these results to mean that the phosphomimetic substitutions may reduce the affinity of Hsp27-AUF1 association. Indeed, we have observed other low-affinity, protein-protein interactions by live-cell FRET (10, 12) that were refractory to coimmunoprecipitation, e.g., gamma interferon receptor chains in the absence of ligands (1, 8). Perhaps proteasome subunits or an E3 ubiquitin ligase directed by Hsp27 to AUF1 may alter protein-protein affinities within the AUF1 complex of proteins. Clearly, more work will be required to address the molecular basis for subunit remodeling promoted by posttranslational modification of Hsp27.
The finding of a physical interaction between AUF1 and phosphomimetic Hsp27 in cells by FRET presents a simple model by which these proteins undergo codegradation by proteasomes and perhaps other proteases. One rationale for their codegradation is that, since both AUF1 and Hsp27 are subject to signaling-induced changes in the phosphorylation state, their codegradation may act to limit the duration of the signaling stimulus on mRNA decay. Codegradation of proteins is certainly not without precedent. For example, degradation of a protein associated with a ubiquitin-dependent target protein is exemplified by degradation of the c-Fos oncoprotein during targeted degradation of c-Jun by proteasomes (20). Interestingly, phosphorylation of c-Jun impedes c-Fos degradation. This raises the possibility that differential phosphorylation of Hsp27 or AUF1 within Hsp27-AUF1 complexes might affect the degradation of one or both proteins. Further work will be required to fully address this, however.
While phosphomimetic substitutions in Hsp27 accelerate Hsp27 and AUF1 turnover, their expression also stabilizes TNF-α and IL-1β mRNAs (Fig. 10). What effect does this have on mRNA abundance? We find that long-term expression of these mutants in stably transfected THP-1 cells reduces steady-state TNF-α and IL-1β mRNA abundance by 80 to 90%; we observed a similar effect upon long-term knockdown of either AUF1 or Hsp27 by stable expression of short hairpin RNAs (shRNAs) (unpublished observations). These results indicate that the Hsp27 phosphomimetics recapitulate the effects of Hsp27 or AUF1 knockdown. Thus, what is the biological relevance of mRNA stabilization upon expression of phosphorylated Hsp27? Normally, phosphorylation of Hsp27 is an acute response to activation of the p38 MAPK pathway, for example, by exposure of cells to IL-1β, as was shown previously (18). As such, our results allow the prediction that under conditions of acute activation of p38, Hsp27 phosphorylation serves to transiently stabilize cytokine ARE mRNAs to increase their abundance. In cell lines stably expressing phosphomimetic Hsp27 mutants, we observe the effects of long-term Hsp27 phosphorylation. Future work will expand upon the biological ramifications of signaling-induced Hsp27 phosphorylation, AUF1 isoform expression/stability, and cytokine mRNA stabilization and abundance.
What is the mechanism by which phosphomimetic Hsp27 stabilizes TNF-α and IL-1β mRNAs? The binding affinity of recombinant Hsp27-TriD to TNF-α ARE RNA is similar to that of wild-type Hsp27 (Fig. 11). We attempted to perform mRNA RNP (mRNP) immunoprecipitations for FLAG-Hsp27 or AUF1 to examine mRNA-protein affinities in the native cellular context. However, these experiments were unsuccessful. We believe the problem is due in part to the vanishingly small amounts of TNF-α and IL-1β mRNAs in nonactivated THP-1 cells (30). On top of this, expression of the Hsp27 phosphomimetics reduces TNF-α and IL-1β mRNA levels 80 to 90%, as noted above, and they reduce total Hsp27 abundance. Thus, interpretable results from these experiments have proven elusive so far. Nonetheless, at least we know that the recombinant proteins bind the TNF-α ARE with similar affinities in vitro. This observation appears to refute the hypothesis that differential binding affinities are responsible for the differences in mRNA decay kinetics conferred by these two proteins. Rather, the simplest explanation for the mRNA stabilization effects conferred by the Hsp27 phosphomimetics is their ability to reduce total Hsp27 abundance compared to ectopically expressed, wild-type Hsp27; this in itself is sufficient to stabilize ARE mRNA (27). Clearly, further work will be required to elucidate the exact mechanisms (RNA binding or something else) through which Hsp27 controls mRNA decay.
In conclusion, we have demonstrated that specific phosphomimetic substitutions in Hsp27 can promote turnover of AUF1 by proteasomes to reduce or alter its expression and thereby stabilize ARE mRNAs. This may provide a molecular basis for earlier observations by Lasa et al. (19), who showed that activation of the p38 MAPK-MK2-Hsp27 signaling axis stabilizes an ARE reporter mRNA. Future work will both address molecular mechanisms by which Hsp27 promotes AUF1 turnover and search for additional signaling pathways that may converge upon Hsp27 to affect degradation of AUF1 and ARE mRNAs.
ACKNOWLEDGMENTS
This work was supported by NIH grants R01 CA052443 to G.B., P01 AI057596 to S.P, and R01 AI059465 to S.P. A.M.K. and F.M.G. were supported by Integrative Graduate Education and Research Traineeship (IGERT) DGE0333196 from the NSF to Prabhas Moghe (Rutgers University Department of Biomedical Engineering). F.M.G. was also supported by Initiative for Minority Students R25 GM058389 from the NIH to Michael Leibowitz (UMDNJ).
Footnotes
Published ahead of print on 18 January 2011.
REFERENCES
- 1. Bach E. A., et al. 1996. Ligand-induced assembly and activation of the gamma interferon receptor in intact cells. Mol. Cell. Biol. 16:3214–3221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dai S., et al. 2008. Comprehensive characterization of heat shock protein 27 phosphorylation in human endothelial cells stimulated by the microbial dithiole thiolutin. J. Proteome Res. 7:4384–4395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. DeMaria C. T., Brewer G. 1996. AUF1 binding affinity to A+U-rich elements correlates with rapid mRNA degradation. J. Biol. Chem. 271:12179–12184 [DOI] [PubMed] [Google Scholar]
- 4. Fawal M., et al. 2006. A “liaison dangereuse” between AUF1/hnRNPD and the oncogenic tyrosine kinase NPM-ALK. Blood 108:2780–2788 [DOI] [PubMed] [Google Scholar]
- 5. Frevel M. A., et al. 2003. p38 Mitogen-activated protein kinase-dependent and -independent signaling of mRNA stability of AU-rich element-containing transcripts. Mol. Cell. Biol. 23:425–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gaestel M., et al. 1991. Identification of the phosphorylation sites of the murine small heat shock protein hsp25. J. Biol. Chem. 266:14721–14724 [PubMed] [Google Scholar]
- 7. Guhaniyogi J., Brewer G. 2001. Regulation of mRNA stability in mammalian cells. Gene 265:11–23 [DOI] [PubMed] [Google Scholar]
- 8. Ishiharu C., et al. 1998. Interferon-gamma receptor beta-chain expression and formation of alpha- and beta-chain complexes after receptor conjugation on human peripheral eosinophils. Int. Arch. Allergy Immunol. 117(Suppl. 1):72–76 [DOI] [PubMed] [Google Scholar]
- 9. Konstantinova I. M., Tsimokha A. S., Mittenberg A. G. 2008. Role of proteasomes in cellular regulation. Int. Rev. Cell Mol. Biol. 267:59–124 [DOI] [PubMed] [Google Scholar]
- 10. Krause C. D., et al. 2006. Preassembly and ligand-induced restructuring of the chains of the IFN-gamma receptor complex: the roles of Jak kinases, Stat1 and the receptor chains. Cell Res. 16:55–69 [DOI] [PubMed] [Google Scholar]
- 11. Krause C. D., et al. 2006. Interactions among the components of the interleukin-10 receptor complex. Biochem. Biophys. Res. Commun. 340:377–385 [DOI] [PubMed] [Google Scholar]
- 12. Krause C. D., et al. 2002. Seeing the light: preassembly and ligand-induced changes of the interferon gamma receptor complex in cells. Mol. Cell Proteomics 1:805–815 [DOI] [PubMed] [Google Scholar]
- 13. Lahiri D. K., Thomas J. O. 1986. A cDNA clone of the hnRNP C proteins and its homology with the single-stranded DNA binding protein UP2. Nucleic Acids Res. 14:4077–4094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Landry J., et al. 1992. Human HSP27 is phosphorylated at serines 78 and 82 by heat shock and mitogen-activated kinases that recognize the same amino acid motif as S6 kinase II. J. Biol. Chem. 267:794–803 [PubMed] [Google Scholar]
- 15. Laroia G., Cuesta R., Brewer G., Schneider R. J. 1999. Control of mRNA decay by heat shock-ubiquitin-proteasome pathway. Science 284:499–502 [DOI] [PubMed] [Google Scholar]
- 16. Laroia G., Schneider R. J. 2002. Alternate exon insertion controls selective ubiquitination and degradation of different AUF1 protein isoforms. Nucleic Acids Res. 30:3052–3058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Larsen J. K., Yamboliev I. A., Weber L. A., Gerthoffer W. T. 1997. Phosphorylation of the 27-kDa heat shock protein via p38 MAP kinase and MAPKAP kinase in smooth muscle. Am. J. Physiol. 273:L930–L940 [DOI] [PubMed] [Google Scholar]
- 18. Lasa M., Abraham S. M., Boucheron C., Saklatvala J., Clark A. R. 2002. Dexamethasone causes sustained expression of mitogen-activated protein kinase (MAPK) phosphatase 1 and phosphatase-mediated inhibition of MAPK p38. Mol. Cell. Biol. 22:7802–7811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lasa M., et al. 2000. Regulation of cyclooxygenase 2 mRNA stability by the mitogen-activated protein kinase p38 signaling cascade. Mol. Cell Biol. 20:4265–4274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Papavassiliou A. G., Treier M., Chavrier C., Bohmann D. 1992. Targeted degradation of c-Fos, but not v-Fos, by a phosphorylation-dependent signal on c-Jun. Science 258:1941–1944 [DOI] [PubMed] [Google Scholar]
- 21. Parcellier A., et al. 2006. HSP27 favors ubiquitination and proteasomal degradation of p27Kip1 and helps S-phase re-entry in stressed cells. FASEB J. 20:1179–1181 [DOI] [PubMed] [Google Scholar]
- 22. Parcellier A., et al. 2003. HSP27 is a ubiquitin-binding protein involved in I-kappaBalpha proteasomal degradation. Mol. Cell. Biol. 23:5790–5802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rane M. J., et al. 2003. Heat shock protein 27 controls apoptosis by regulating Akt activation. J. Biol. Chem. 278:27828–27835 [DOI] [PubMed] [Google Scholar]
- 24. Sarkar B., Lu J. Y., Schneider R. J. 2003. Nuclear import and export functions in the different isoforms of the AUF1/heterogeneous nuclear ribonucleoprotein protein family. J. Biol. Chem. 278:20700–20707 [DOI] [PubMed] [Google Scholar]
- 25. Sarkar S., Sinsimer K. S., Foster R. L., Brewer G., Pestka S. 2008. AUF1 isoform-specific regulation of anti-inflammatory IL10 expression in monocytes. J. Interferon Cytokine Res. 28:679–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shchors K., et al. 2002. Cell death inhibiting RNA (CDIR) derived from a 3′-untranslated region binds AUF1 and heat shock protein 27. J. Biol. Chem. 277:47061–47072 [DOI] [PubMed] [Google Scholar]
- 27. Sinsimer K. S., et al. 2008. Chaperone Hsp27, a novel subunit of AUF1 protein complexes, functions in AU-rich element-mediated mRNA decay. Mol. Cell. Biol. 28:5223–5237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sommer S., Cui Y., Brewer G., Fuqua S. A. 2005. The c-Yes 3′-UTR contains adenine/uridine-rich elements that bind AUF1 and HuR involved in mRNA decay in breast cancer cells. J. Steroid Biochem. Mol. Biol. 97:219–229 [DOI] [PubMed] [Google Scholar]
- 29. Wilson G. M., et al. 2003. Phosphorylation of p40AUF1 regulates binding to A + U-rich mRNA-destabilizing elements and protein-induced changes in ribonucleoprotein structure. J. Biol. Chem. 278:33039–33048 [DOI] [PubMed] [Google Scholar]
- 30. Wilson G. M., et al. 2003. Regulation of A + U-rich element-directed mRNA turnover involving reversible phosphorylation of AUF1. J. Biol. Chem. 278:33029–33038 [DOI] [PubMed] [Google Scholar]
- 31. Wilson G. M., Sun Y., Lu H., Brewer G. 1999. Assembly of AUF1 oligomers on U-rich RNA targets by sequential dimer association. J. Biol. Chem. 274:33374–33381 [DOI] [PubMed] [Google Scholar]
- 32. Wilson G. M., Sutphen K., Bolikal S., Chuang K. Y., Brewer G. 2001. Thermodynamics and kinetics of Hsp70 association with A + U-rich mRNA-destabilizing sequences. J. Biol. Chem. 276:44450–44456 [DOI] [PubMed] [Google Scholar]
- 33. Wilusz C. J., Wormington M., Peltz S. W. 2001. The cap-to-tail guide to mRNA turnover. Nat. Rev. Mol. Cell Biol. 2:237–246 [DOI] [PubMed] [Google Scholar]
- 34. Winzen R., et al. 1999. The p38 MAP kinase pathway signals for cytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targeted mechanism. EMBO J. 18:4969–4980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ysla R. M., Wilson G. M., Brewer G. 2008. Assays of adenylate uridylate-rich element-mediated mRNA decay in cells. Methods Enzymol. 449:47–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zacharias D. A., Violin J. D., Newton A. C., Tsien R. Y. 2002. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science 296:913–916 [DOI] [PubMed] [Google Scholar]
- 37. Zhang W., et al. 1993. Purification, characterization, and cDNA cloning of an AU-rich element RNA-binding protein, AUF1. Mol. Cell. Biol. 13:7652–7665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zheng C., et al. 2006. MAPK-activated protein kinase-2 (MK2)-mediated formation and phosphorylation-regulated dissociation of the signal complex consisting of p38, MK2, Akt, and Hsp27. J. Biol. Chem. 281:37215–37226 [DOI] [PubMed] [Google Scholar]