

Abstract
The Human Proteome Organization (HUPO) Proteomics Standard Initiative has been tasked with developing file formats for storing raw data (mzML) and the results of spectral processing (protein identification and quantification) from proteomics experiments (mzIndentML). In order to fully characterize complex experiments, special data types have been designed. Standardized file formats will promote visualization, validation and dissemination of data independent of the vendor-specific binary data storage files. Innovative programmatic solutions for robust and efficient data access to standardized file formats will contribute to more rapid wide-scale acceptance of these file formats by the proteomics community.
In this work, we compare algorithms for accessing spectral data in the mzML file format. As an XML file, mzML files allow efficient parsing of data structures when using XML-specific class types. These classes provide only sequential access to files. However, random access to spectral data is needed in many algorithmic applications for processing proteomics datasets. Here, we demonstrate implementation of memory streams to convert a sequential access into random access. Our application preserves the elegant XML parsing capabilities. Benchmarking file access times in sequential and random access modes show that while for small number of spectra the random access is more time efficient, when retrieving large number of spectra sequential access becomes more efficient. We also provide comparisons to other file accessing methods from academia and industry.
Keywords: mzML, XML, Sequential file access, Random file access, Proteomics datasets
Introduction
Mass spectrometry-based proteomics experiments generate large amounts of data relevant to the protein content and complexity of a sample. An important element of proteomics research is the computational analyses of the data for protein identification [1,2], post-translational modifications [3,4], protein-protein interactions and relative/absolute protein expression [5-7]. For expeditious analyses of a dataset, visualization as well as validation and determining statistical significance, bioinformatics software must have efficient and high-speed access to the raw data generated by the mass spectrometers. Over the years, manufacturers of mass spectrometers have developed their own native binary formats for storing, accessing and analyzing data. Files in such formats usually cannot be viewed, processed or printed without expensive proprietary vendor software. An earlier attempt to unify file formats into a character/text based format, JCAMP-DX [8], was not widely implemented. In turn, bioinformatics software that has been developed to process mass spectral data (database search engines, for example) has been using different file formats for their inputs and outputs. Many popular search engines, including SEQUEST [2], Mascot [9], OMSSA [10] and X!Tandem [11], use different file formats. The practice of using different output types necessitates use of different software for parsing database search results [12,13] and determining false discovery rates [14-17].
Rapid and extensive development of mass spectrometry-based proteomics created a need for unified file formats for storage, dissemination, processing, visualization and validation of datasets generate by such research [18]. Several open, XML [19] -based formats have been developed to allow users to share data more easily by standardizing peak lists in one data format [20]. Two of the most popular open formats, mzData [21] (developed by PSI-MS: Mass Spectrometry Standards Working Group) and mzXML [20] (developed by Pedrioli et al.) have been combined into a joint format called mzML. The mzML format was officially released at the annual meeting of the American Society for Mass Spectrometry in 2008. It was designed to contain all the information about a single mass spectrometry run, including metadata about the spectra [22] and all binary spectra themselves. Since it is a text-based XML document, mzML is easily accessible by software. The mzML format is based on controlled vocabulary (CV) through cvParam elements. The CV defines very thoroughly the terminology of MS proteomics acquisition and spectrum description and each term has an explicit and detailed definition. The controlled vocabulary can be easily extended, in order to include additional parameters without modifying the mzML schema. The main advantage of this data format is that information from tens of thousands of spectra may be saved in one text file of manageable size. The mzML format seems to be quite stable but its acceptance will depend on the availability of applications and software libraries that implement this format. Software for visualizing spectra in mzML format is freely available [23]. Further developments to improve data storage have also been reported [24]. Conversion from vendor specific binary files (storing mass spectra) to mzML file format has been facilitated by freely available software tools from trans-proteomics pipeline (TPP) [25] and ProteoWizard [26]. Thus, msconvert executable converts mass spectral data from proprietary .WIFF (ABI/Sciex), .BAF (Bruker), .RAW (ThermoFisher Scientific), .D (Agilent) and others into an mzML file. ProteoWizard also provides tools for accessing mass spectra in the mzML file format. These tools are intended to be platform independent (LINUX or WINDOWS). However, currently available functions do not allow easily implementable file access to extract mass spectra.
A large number of software have been developed to support numerous proteomics workflows [27]. In this work, we describe a software approach for efficient access to mzML files. Efficient access to mzML files is important because these files are encoded in XML format. XML file access tools provided by WINDOWS .NET methods are sequential file readers and sequential reading of large data files is generally quite slow. Our programmatic solution turns the sequential access into a random access. For this purpose, we used the indexing provided by the mzML file scheme. We benchmark time performance of sequential versus random file access modes. Comparison of our approach to file access with those from industry and academia are also presented.
Materials and Methods
We used two datasets to benchmark file accessing. The first dataset was obtained from extracts of mouse renal cortex [28], with heavy and light peptides mixed in a 1:1 concentration [29]. The second dataset included peptides of four proteins; bovine serum albumin, cytochrome c, alpha- and beta-caseins mixed in five different concentrations. The first dataset was acquired using an LTQ ion trap mass spectrometer, the second using an LTQ-Orbitrap hybrid mass spectrometer. The data acquisition methods have been described elsewhere [28]. To convert our data from RAW format to mzML we used the ProteoWizard’s22 msconvert tool which provides general file format conversion, including zlib [30] compression and indexing.
To compare our method with other approaches, we used two software tools, one from academia and one from industry. The readmzXML tool by Institute for Systems Biology (Seattle, Washington) extracts mass scans from mzML, mzXML and mzData file formats. The functionality to retrieve spectra from mzML files was used in our comparisons. The xrawfile2 tool of Thermo Fisher Scientific (San Jose, CA) is a part of Xcalibut software suit for accessing mass spectra from their RAW files. Specifically we used, the raw_GetMassListFromScanNum functionality of xrawfile2.dll.
Our programs were written in C++ language of Visual Studio 9 and all source codes are distributed freely. The source codes can be obtained by contacting the communicating author. The programs have been developed in the form of dynamic libraries and can easily be incorporated into different applications.
Results and Discussions
The parser that we developed reads and actively interprets several CV parameters, which are necessary for correct decoding binary spectrum data. According to the mzML specifications, all binary spectrum data, in the compressed or uncompressed form, have to be encoded by Base64 scheme, which is commonly used when there is a need to encode binary data that needs to be stored in the text XML format. The mzML file can also be indexed. The main part of the mzML document is contained within the <mzML> </mzML> tags and then it is wrapped within an <indexedmzML> </indexedmzML> construct, which contains the random access index at the bottom of the file.
To read mzML schema, we used a standard .NET XmlTextReader [19] class that provides fast, non-cached, forward-only access to an XML file. For non-interactive applications this implementation is reasonable, but for interactive applications (such as visualization or quantification) the access times are very slow. We also implemented random access to mzML data, which allows fast access to an arbitrary spectrum. Usually, to speed up parsing of an XML file we can read an entire file into memory for fast access to the referenced elements. However, more and more frequently, mzML files exceed several gigabytes in size, which makes it impossible to use existing methods with a standard PC. To resolve this problem, we implement a native mzML indexing scheme.
At the beginning, our parser reads the <indexListOffset> element, which is located at the end of mzML file and contains the byte offset of the <indexList> tag. Once we know the byte offset of the <indexList>, which includes a list of all spectrum numbers followed by byte offsets of corresponding <spectrum> element, we skip all data between <mzML> and </mzML> tags and store the offsets of all <spectrum> elements into an array. Then we use a FileStream∷Seek method to jump to the <spectrum> tag assigned to the given spectrum number, as shown in Figure 1. In the next steps we read all spectrum information, including Base64 encoded spectrum binary data, into a memory stream. Then we define this stream as the input for XmlTextReader and parse selected parts of mzML file in the same way like a regular XML file.
Figure 1.

Code snippet showing the handling the <spectrum> element and usage of memory stream as the input for XmlTextReader.
The XmlTextReader∷ ReadBase64 method decodes the <binary> tag contents into a byte array. Then this array, depending on compression mzML accession code, is decompressed by ManagedZlib∷CompressionStream and/or converted into float numbers by the BitConverter method, as shown in Figure 2. If the <indexedmzML> tag doesn’t exist, all offset information is skipped and XmlTextReader reads the mzML file using a typical sequential access method. This type of random-access of the XML file has been used for several years in the mzXML format, demonstrating that indexing problems are rare and benefits are enormous. In our case, a random access index, which is generally against XML philosophy, is completely safe because we are using mzML files for reading only. There are two typical situations which occur during the reading of mzML files. The first occurs when before accessing the data file, we are able to generate a list of required spectra numbers. In some cases, we know in advance the spectrum numbers that the program will read and we can generate a list and sort in ascending order. Once this is done, we know that the next requested spectrum will be in the file, immediately following the spectrum that has most recently been read. To speed up parsing in this situation we can open the mzML file at the beginning of the process and close it after retrieving all of the needed spectral data. Depending on computer/hard disk performance, this type of sequential reading can typically process 500 spectra out of about 11,000 in 22 to 150 seconds. However, by implementing our parser random access method, the same mass spectra can be retrieved about 10 times faster (2-12s).
Figure 2.

Code snippet illustrating decoding of the <binary> tag by XmlTextReader/ReadBase64 method, decompressing data from memory stream using ManagedZLib∷CompressionStream and conversion into double array.
In the second typical situation, which occurs when we want to access a particular spectrum with a number we can’t predict in advance. In this case, it may be necessary to parse the whole mzML file for each mass spectrum. When the spectrum list is not known in advance, as in the case of interactive applications for graphical viewing of spectra, the sequential access method is problematic. In order to retrieve a particular mass spectrum, we would have to open and then close the mzML file each time. In this situation, sequential access to the file is totally useless because reading 500 spectra would take more than an hour. Our random-access routine can complete the same task in a couple of seconds and access time is independent of application type. In both situations (with and without a prior list of spectra) it is working with the same speed. These observations are illustrated in Figure 3, where we show the spectra retrieval times using different access modes. Times are for retrieval of spectra from 12 mzML files obtained by converting LTQ-Orbitrap .raw files. Each chromatographic run included more than ten thousands spectra. Full survey scans were acquired at the high resolution mode, while tandem mass spectra were recorded in low resolution. Note that in the case of interactive access, the random file access (black line in Figure 3) is by far the most efficient way to retrieve spectra from an mzML. For non-interactive spectral retrievals, sequential access (blue line in Figure 3) becomes faster than random access when most spectra in an mzML file need to be accessed. This could be explained by the overhead associated with random access file, as it is necessary to “re-wind” the file each time a spectrum is accessed. However, in absolute terms, the overhead is negligible (less than few seconds).
Figure 3.
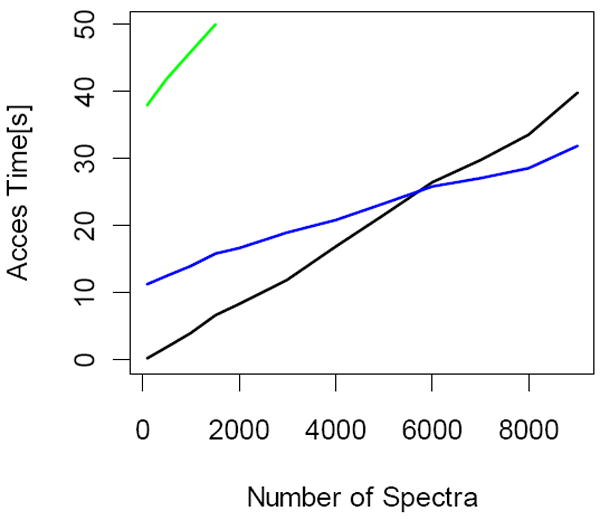
Spectra retrieval times under different access methods for increasing set of requested mass spectra. The data sets for this figure were from high mass resolution instruments, LTQ-Orbitrap. The green line shows results from readmzXML tool of TPP. The program accepts only about 1800 scans as argument. Therefore, we were not able to extend the curve to larger number of spectra. As the number of spectra are increase the sequential access mode (blue line) becomes more efficient than random access mode (black line).
Also shown in Figure 3 are the estimates of retrieval times (green line) of spectra using readmzXML tool of Institute of Systems Biology. The tool is a part of TPP suit of programs. The estimates were obtained using “-n” option of the program. Note that the program accepts only a certain number of spectra as an input (this limitation could be due to the WINDOWS environment). The maximum number of spectra that we could retrieve at a single run was 1800. Therefore, we could not extend the curve to include larger number of spectra. However, the access time curves are normally linearly dependent on the number of spectra, and the slope of the line is indicative of generalization to larger number spectra. As seen from the Figure, readmzXML access times were longer than those from either sequential or random access modes employed in this presentation.
We next tested if there were any overhead associated with compressing mzML files (--zlib option in msconvert). Our experiments showed about 20% overhead associated with the file compression. The access times to uncompressed files were shorter.
We computed access times by using xrawfile2 from XCalibur software suit. The access times for any number of spectra that we tested (from 100 to 10000) were always less than a second. Thus, currently the most efficient way of accessing spectra seems to be possible by the vendor software. It is expected, given the fact that in the mzML format the spectra are encoded in Base64 format. Decoding spectra is an important overhead in terms of efficiency of file access.
In another experiment we compared file access modes for spectra acquired at low mass resolution. These are datasets obtained using LTQ mass spectrometers of Thermo Fisher Scientific. The results are shown in Figure 4. Spectra access times from these datasets were faster than those obtained from high mass resolution instruments (Figure 3), especially for random (black line) and sequential access modes (blue line) employed in this work. This is probably explained by the larger sizes of data in survey scans in high mass resolution datasets. The access times changed only marginally for readmzXML tool.
Figure 4.
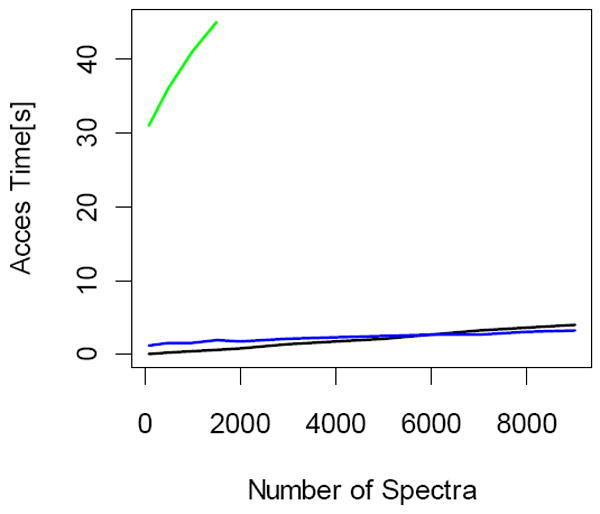
Spectra retrieval times for datasets acquired on low mass resolution instruments (LTQ). The access times are much smaller than those to high mass resolution datasets, especially for random (black line) and sequential (blue line) access modes. For larger number of spectra the sequential access becomes more efficient. However, the improvement in comparison to random access mode is not as dramatic as it is for the high resolution data. The access times for readmzXML tool (green line) change marginally compared to high resolution datasets.
For low mass resolution data, too, for small number of spectra the access times for random mode are shorter that those of sequential mode. However, for larger number of spectra both modes have similar access times, and there is no substantial advantage by the sequetial mode, as was the case for high mass resolution data.
In applications such as quantification, the list of spectra to be retrieved from a given mzML file is known. In this case, most of the processing time is associated with signal processing, peak detection and integration, and generation of isotopic distributions. As long as spectra can be arranged such that mzML files only need to be opened once, file access time is only a small fraction of the processing time. However, even for these applications, random access is a preferable approach, since it generalizes better than sequential access.
Conclusion
We implemented an efficient and easily adaptable approach to accessing mass spectral data in mzML formatted files. Our implementation combines a WINDOWS .NET method for sequentially accessing XML files with memory streaming methods that allow us to convert to random access. We compared times for retrieving spectra from mzML files in sequential and random access modes. We observed that access times are dependent on the specific needs of each application. When a small number of spectra are retrieved, random access is faster than sequential access. However, time differences between random and sequential access decrease as the number of retrieved spectra increases. For large numbers of spectra, sequential access is faster than random access. For interactive file access (such as visualization applications), random access is always the fastest method by far. In conclusion, random access is more efficient and generalizes better to different environments.
Acknowledgments
We are thankful Alisha Goldberg for critically editing the manuscript. This work was supported in part by “Clinical Proteomics Centers in Biodefense and Emerging Infectious Diseases” (NIAID contract HHSN272200800048C).
Footnotes
Publisher's Disclaimer: This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Mann M, Wilm M. Error-tolerant identification of peptides in sequence databases by peptide sequence tags. Anal Chem. 1994;66:4390–4399. doi: 10.1021/ac00096a002. [DOI] [PubMed] [Google Scholar]
- 2.Eng JK, McCormack AL, Yates JR., III An Approach to Correlate Tandem Mass Spectral Data of Peptides with Amino Acid Sequences in a Protein Database. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 3.Tanner S, Shu H, Frank A, Wang LC, Zandi E, et al. InsPecT: identification of posttranslationally modified peptides from tandem mass spectra. Anal Chem. 2005;77:4626–4639. doi: 10.1021/ac050102d. [DOI] [PubMed] [Google Scholar]
- 4.Tabb DL, Saraf A, Yates JR., 3rd GutenTag: high-throughput sequence tagging via an empirically derived fragmentation model. Anal Chem. 2003;75:6415–6421. doi: 10.1021/ac0347462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bakalarski CE, Elias JE, Villén J, Haas W, Gerber SA, et al. The impact of peptide abundance and dynamic range on stable-isotope-based quantitative proteomic analyses. J Proteome Res. 2008;7:4756–4765. doi: 10.1021/pr800333e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 7.Bellew M, Coram M, Fitzgibbon M, Igra M, Randolph T, et al. A suite of algorithms for the comprehensive analysis of complex protein mixtures using high-resolution LC-MS. Bioinformatics. 2006;22:1902–1909. doi: 10.1093/bioinformatics/btl276. [DOI] [PubMed] [Google Scholar]
- 8.Mcdonald RS, Wilks PA. Jcamp-Dx - A Standard Form for Exchange of Infrared-Spectra in Computer Readable Form. Applied Spectroscopy. 1988;42:151–162. [Google Scholar]
- 9.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 10.Geer LY, Markey SP, Kowalak JA, Wagner L, Xu M, et al. Open mass spectrometry search algorithm. J Proteome Res. 2004;3:958–964. doi: 10.1021/pr0499491. [DOI] [PubMed] [Google Scholar]
- 11.Craig R, Cortens JP, Beavis RC. Open source system for analyzing, validating, and storing protein identification data. J Proteome Res. 2004;3:1234–1242. doi: 10.1021/pr049882h. [DOI] [PubMed] [Google Scholar]
- 12.Cociorva D, L Tabb D, Yates JR. Validation of tandem mass spectrometry database search results using DTASelect. Curr Protoc Bioinformatics. 2007;Chapter 13(Unit 13.4) doi: 10.1002/0471250953.bi1304s16. [DOI] [PubMed] [Google Scholar]
- 13.Tabb DL, McDonald WH, Yates JR., 3rd DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J Proteome Res. 2002;1:21–26. doi: 10.1021/pr015504q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moore RE, Young MK, Lee TD. Qscore: an algorithm for evaluating SEQUEST database search results. J Am Soc Mass Spectrom. 2002;13:378–386. doi: 10.1016/S1044-0305(02)00352-5. [DOI] [PubMed] [Google Scholar]
- 15.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 16.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 17.Käll L, Canterbury JD, Weston J, Noble WS, MacCoss MJ. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat Methods. 2007;4:923–925. doi: 10.1038/nmeth1113. [DOI] [PubMed] [Google Scholar]
- 18.McDonald WH, Tabb DL, Sadygov RG, MacCoss MJ, Venable J, et al. MS1, MS2, and SQT-three unified, compact, and easily parsed file formats for the storage of shotgun proteomic spectra and identifications. Rapid Commun Mass Spectrom. 2004;18:2162–2168. doi: 10.1002/rcm.1603. [DOI] [PubMed] [Google Scholar]
- 19.Fraser S, Reamy D. Pro Visual C++/CLI and the .NET 3.5 platform. Apress; Berkeley, California: 2009. [Google Scholar]
- 20.Pedrioli PG, Eng JK, Hubley R, Vogelzang M, Deutsch EW, et al. A common open representation of mass spectrometry data and its application to proteomics research. Nat Biotechnol. 2004;22:1459–1466. doi: 10.1038/nbt1031. [DOI] [PubMed] [Google Scholar]
- 21.Orchard S, Montechi-Palazzi L, Deutsch EW, Binz PA, Jones AR, et al. Proteomics; Five years of progress in the Standardization of Proteomics Data 4th Annual Spring Workshop of the HUPO-Proteomics Standards Initiative; April 23-25, 2007; Ecole Nationale Superieure (ENS), Lyon, France. 2007. pp. 3436–3440. [DOI] [PubMed] [Google Scholar]
- 22.Taylor CF, Paton NW, Lilley KS, Binz PA, Julian RK, Jr, et al. The minimum information about a proteomics experiment (MIAPE) Nat Biotechnol. 2007;25:887–893. doi: 10.1038/nbt1329. [DOI] [PubMed] [Google Scholar]
- 23.Côté RG, Reisinger F, Martens L. jmzML, an open-source Java API for mzML, the PSI standard for MS data. Proteomics. 2010;10:1332–1335. doi: 10.1002/pmic.200900719. [DOI] [PubMed] [Google Scholar]
- 24.Shah AR, Davidson J, Monroe ME, Mayampurath AM, Danielson WF, et al. An Efficient Data Format for Mass Spectrometry-Based Proteomics. J Am Soc Mass Spectrom. 2010;21:1784–1788. doi: 10.1016/j.jasms.2010.06.014. [DOI] [PubMed] [Google Scholar]
- 25.Deutsch EW, Mendoza L, Shteynberg D, Farrah T, Lam H, et al. A guided tour of the Trans-Proteomic Pipeline. Proteomics. 2010;10:1150–1159. doi: 10.1002/pmic.200900375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kessner D, Chambers M, Burke R, Agus D, Mallick P. ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics. 2008;24:2534–2536. doi: 10.1093/bioinformatics/btn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mueller LN, Brusniak MY, Mani DR, Aebersold R. An assessment of software solutions for the analysis of mass spectrometry based quantitative proteomics data. J Proteome Res. 2008;7:51–61. doi: 10.1021/pr700758r. [DOI] [PubMed] [Google Scholar]
- 28.Sadygov RG, Zhao Y, Haidacher SJ, Starkey JM, Tilton RG, et al. Using Power Spectrum Analysis to Evaluate 18O-Water Labelilng Data Acquired from Low Resolution Mass Spectrometers. Journal of Proteome Res. 2010;9:4306–4312. doi: 10.1021/pr100642q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao Y, Denner L, Haidacher SJ, LeJeune WS, Tilton RG. Comprehensive analysis of the mouse renal cortex using two-dimensional HPLC – tandem mass spectrometry. Proteome Science. 2008;6:15. doi: 10.1186/1477-5956-6-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.zlib. 2010 [Google Scholar]