

Abstract
Familial hemiplegic migraine type 1, a monogenic migraine variant with aura, is linked to gain-of-function mutations in the CACNA1A gene encoding CaV2.1 channels. The S218L mutation causes severe channel dysfunction, and paroxysmal migraine attacks can be accompanied by seizures, coma, and hemiplegia; patients expressing the R192Q mutation exhibit hemiplegia only. Familial hemiplegic migraine knock-in mice expressing the S218L or R192Q mutation are highly susceptible to cortical spreading depression, the electrophysiological surrogate for migraine aura, and develop severe and prolonged motor deficits after spreading depression. The S218L mutants also develop coma and seizures and sometimes die. To investigate underlying mechanisms for these symptoms, we used multielectrode electrophysiological recordings, diffusion-weighted magnetic resonance imaging, and c-fos immunohistochemistry to trace spreading depression propagation into subcortical structures. We showed that unlike the wild type, cortical spreading depression readily propagated into subcortical structures in both familial hemiplegic migraine type 1 mutants. Whereas the facilitated subcortical spread appeared limited to the striatum in R192Q, hippocampal and thalamic spread was detected in the S218L mutants with an allele-dosage effect. Both strains exhibited increased susceptibility to subcortical spreading depression and reverberating spreading depression waves. Altogether, these data show that spreading depression propagates between cortex, basal ganglia, diencephalon, and hippocampus in genetically susceptible brains, which could explain the prolonged hemiplegia, coma, and seizure phenotype in this variant of migraine with aura.
Introduction
Familial hemiplegic migraine (FHM) is a monogenic subtype of migraine with aura characterized by transient neurological signs and symptoms including hemiparesis, aphasia, seizures, and coma. Different aura signs and symptoms can coexist, typically transforming into each other (e.g., visual followed by sensory and motor), and resolve in the order they appear (Thomsen et al., 2002). FHM type 1 (FHM1) is caused by missense mutations in the CACNA1A gene (e.g., R192Q, S218L) encoding the pore-forming α1A subunit of neuronal voltage-gated CaV2.1 channels (Ophoff et al., 1996; van den Maagdenberg et al., 2007). In cultured neurons, FHM1 mutations shift channel opening toward more negative membrane potentials and delay channel inactivation. Channels open with smaller depolarization and stay open longer (Tottene et al., 2002, 2005), presumably allowing more Ca2+ to enter presynaptic terminals, resulting in enhanced glutamate release. Consistent with this, excitatory neurotransmission is enhanced in pyramidal cell synapses of R192Q knock-in mice (Tottene et al., 2009).
Spreading depression (SD), widely viewed as the electrophysiological event underlying migraine aura, is an intense wave of neuronal depolarization that propagates (∼3 mm/min) by way of gray matter contiguity, regardless of functional divisions. SD is characterized by massive K+ efflux, Ca2+ influx, and glutamate release, which are believed to depolarize adjacent neurons and glia, thereby facilitating its spread. The transmembrane ionic and water shifts also lead to characteristic apparent diffusion coefficient (ADC) changes on diffusion-weighted magnetic resonance imaging (MRI), and upregulation of the immediate-early gene c-fos, as surrogate measures of intense neuronal depolarization. Mutant mouse models expressing the pathogenic R192Q or S218L FHM1 mutation exhibit increased susceptibility to cortical SD (van den Maagdenberg et al., 2004, 2010; Eikermann-Haerter et al., 2009; Tottene et al., 2009). The S218L mutation shows larger gain-of-function, in vitro, and higher SD susceptibility, in vivo, compared with the R192Q variant (van den Maagdenberg et al., 2004, 2010; Eikermann-Haerter et al., 2009). In contrast to pure hemiplegic migraine associated with the R192Q mutation, attacks in patients carrying the S218L mutation are sometimes accompanied by coma or stupor and generalized seizures (Ophoff et al., 1996; Kors et al., 2001). Accordingly, experimentally induced cortical SD induces pure hemiplegia in R192Q mutant mice, whereas S218L mutants additionally develop coma and often fatal seizures (Eikermann-Haerter et al., 2009; van den Maagdenberg et al., 2010).
Experimental SD produces seizures and coma when evoked in hippocampus or thalamus, respectively (Jakobartl and Huston, 1977). We investigated whether the severe clinical phenotype observed only with the S218L mutation could be caused by subcortical SD involving hippocampus and thalamus. We now provide evidence for enhanced subcortical SD susceptibility in FHM1 mutant mice that facilitates SD propagation bidirectionally between cortex, hippocampus, and thalamus in the S218L mutant strain. Our findings suggest a role for subcortical SD as a potential mechanism to explain hemiplegia, seizures, and coma in FHM1.
Materials and Methods
Experimental groups.
A total of 75 male and female wild-type (WT) or FHM1 knock-in mice [homozygous (HOM) R192Q, or HOM or heterozygous (HET) S218L] (van den Maagdenberg et al., 2004) were used (n = 44 for electrophysiological recordings, n = 22 for c-fos immunostaining, n = 4 for MRI, and n = 5 for laser speckle flowmetry). R192Q mutant mice were compared with their WT littermates or C57BL/6J mice, on which the mutants were backcrossed for >10 generations. Because none of the end points differed between WT littermates and C57BL/6J mice, data from WT strains were pooled. S218L mutant mice were compared with their WT littermates. In female mice, we did not control for the estrus stage as we did not observe high variability in SD susceptibility in randomly tested female mice in our previous study (Eikermann-Haerter et al., 2009). To test whether enhanced glutamatergic activity contributes to subcortical spread in FHM1 mutants, a separate group of female WT and S218L HOM mice was treated with guanosine (7.5 mg/kg, i.p., 30 min before SD induction), an anticonvulsant that enhances astrocytic glutamate uptake and suppresses glutamatergic transmission (Frizzo et al., 2001, 2005; Lara et al., 2001; Soares et al., 2004). All experiments were performed with the investigator blinded to genotype, followed by confirmatory genotyping.
General surgical preparation.
Experimental procedures were approved by the institutional review committee. Mice were housed under diurnal lighting conditions and allowed food and tap water ad libitum. The femoral artery was catheterized for blood pressure (BP) monitoring, and the trachea was intubated for mechanical ventilation (SAR-830; CWE) under isoflurane anesthesia (2.5% induction, 1% maintenance in 70% N2O/30% O2). Arterial blood gases and pH were measured every 20 min and maintained within normal limits by adjusting the ventilation. Systemic physiological parameters did not differ among groups (supplemental Table 1, see Notes). There was no mortality during these experiments.
Electrophysiology.
Mice were placed in a stereotaxic frame, and burr holes were drilled (Fig. 1). Up to four glass microelectrodes were placed to simultaneously record extracellular steady (DC) potential and fast neuronal activity (electrocorticogram) at the following coordinates (posterior, lateral, and ventral from bregma; in mm): cortex: 3.5, 1, 0.3; striatum: −0.5, 2, 3; hippocampus: 1.8, 1.2, 1.2; thalamus: 1.8, 1.2, 3. Table 2 shows the numbers of mice and SD recordings in each experimental group. In a subset of S218L HOM mice, simultaneous bilateral recordings from cortex (n = 9), hippocampus (n = 3), or thalamus (n = 4) were obtained to examine contralateral spread. In a separate group, SD-induced cortical blood flow changes were measured simultaneously in both hemispheres using laser speckle imaging to detect contralateral cortical spread (n = 5), as described previously (Ayata et al., 2004). Because isoflurane and N2O, but not pentobarbital, suppress SD susceptibility (Kudo et al., 2008), after surgical preparation anesthesia was switched to pentobarbital, and the ventilation gas was switched to 70% N2/30% O2, for all experiments. Pentobarbital was chosen over urethane because it allowed us to better maintain a normal systemic physiological state in prolonged experiments performed in this study. After 30 min, one SD was induced through a parietal window (1.5 mm diameter, 2.5 mm posterior, and 3 mm lateral from bregma) by an epidural cotton ball (1.5 mm) soaked with 300 mm KCl. After the onset of the DC shift, KCl was removed by extensive saline wash. Fifteen minutes were allowed between each SD induction whether induced by topical KCl or mechanically during electrode insertion (Koroleva et al., 1991). In a separate group of mice, we induced the cortical SD by a single pin prick and obtained similar data (not shown).
Figure 1.

Electrophysiological recording sites. SD was triggered by brief topical KCl (300 mm) application onto parietal cortex and recorded with up to four glass micropipettes (arrowheads) placed into cortex (c), striatum (s), hippocampus (h), thalamus (t). [Adapted from Allen Mouse Brain Atlas (Internet), 2009. Seattle: Allen Institute for Brain Science. Available at http://mouse.brain-map.org].
Table 2.
Latency between KCl application and the onset of SD
| Strain | Gender | Genotype | Cortex | Striatum | Hippocampus | Thalamus |
|---|---|---|---|---|---|---|
| R192Q | Female | WT | 90 ± 30 | 294 ± 32 | ||
| HOM | 52 ± 3* | 110 ± 19* | ||||
| S218L | Female | WT | 110 ± 26 | 345 ± 110 | ||
| HET | 41 ± 5* | 98 ± 15* | 131 ± 33 | |||
| HOM | 31 ± 4* | 80 ± 17* | 97 ± 12† | 148 ± 56 | ||
| WT + guanosine | 73 ± 8 | 280, 280 | ||||
| HOM + guanosine | 29 ± 4* | 72 ± 7 | 94 ± 21 | 90 | ||
| Male | WT | 99 ± 22 | 267 ± 112 | |||
| HOM | 37 ± 8*# | 77 ± 8* | 86 ± 21 | 165 ± 73 |
Values indicate the latency (seconds) between topical cortical KCl application and SD onset in indicated structures. Only two cortical SDs were recorded in striatum in S218L female WT treated with guanosine; individual values are shown.
*p < 0.01 versus WT;
†p < 0.05 versus S218L HET;
#p = 0.10 versus S218L female HOM. Values are mean ± SD.
Magnetic resonance imaging.
MRI was performed in S218L HOM mice (n = 4) using a 9.4T horizontal bore (Magnex Scientific) scanner with a custom-made surface-radiofrequency coil, under pentobarbital anesthesia. The ADC was measured with a sequence of four diffusion-weighted echoplanar images (weighting along Z direction, b = 0–1287.9 s/mm2; echo time, 20 ms; repetition time, 5000 ms; field of view, 14 mm; slice thickness, 0.50 mm; matrix, 64 × 64). Sixteen contiguous coronal slices were acquired to cover cerebrum with a temporal resolution of 20 s. After surgical preparation and the onset of sequential MRI, cortical SD (CSD) was evoked by topical application of 300 mm KCl on the parietal cortex via a polyethylene tubing mounted above an open cranial window. Ten seconds after topical application of 300 mm KCl, cortex was washed with saline via a separate polyethylene tubing, to minimize cortical exposure to high levels of K+. Imaging was continued for 15 min after CSD induction.
c-fos immunostaining.
Under pentobarbital anesthesia, one cortical SD was induced every 15 min for a total of three cortical SDs and was electrophysiologically confirmed (n = 6, 4, and 12 female WT, R192Q HOM, and S218L HOM mutants, respectively). Three hours after the induction of the first cortical SD, mice were deeply anesthetized and transcardiac perfusion fixed. Sham animals underwent the same procedure, duration, and form of anesthesia, but cotton balls soaked with saline instead of KCl were placed on the cortex. Brains were postfixed in 2.5% paraformaldehyde, and 30 μm cryosections were immunostained for c-fos (Moskowitz et al., 1993) and qualitatively examined at two coronal levels through (1) the striatum and septal nuclei and (2) the hippocampus, thalamus, and amygdala.
Data analysis.
Data were analyzed using SPSS (version 11.0; SPSS). Using pooled data from all mice and a general linear model of variance analysis, we tested for an effect of independent-variable mutation and genotype (fixed factors) on physiological parameters of dependent variables, latency between KCl application and occurrence of SD, and duration and amplitude of SD in the respective structures. The incidence of subcortical SD and mechanical SD induction in respective structures were compared among groups using the χ2 test. Data are presented as mean ± SD. p < 0.05 was considered statistically significant. Strong statistical trends (0.05 ≤ p ≤ 0.10) were also shown.
Results
Electrophysiology
Topical application of 300 mm KCl consistently induced cortical SD (Fig. 2). In WT mice, only 20–30% of cortical SDs propagated into the striatum, and none propagated into the hippocampus or the thalamus (Table 1). In R192Q mutant mice, over 70% of cortical SDs propagated into the striatum but not into other subcortical structures. In contrast, all cortical SDs propagated into the striatum, and a substantial proportion propagated into the hippocampus in S218L mutant mice, with an allele-dosage effect. Moreover, propagation into thalamus was found only in S218L HOM mice. The subcortical propagation rate did not significantly differ between male and female S218L HOM mice. Guanosine (7.5 mg/kg, administered intraperitoneally 30 min before SD recordings) suppressed hippocampal and thalamic spread in female S218L HOM mice. In contrast, striatal spread was not affected in either WT or S218L HOM. Consistent with the faster propagation speed reported previously (Eikermann-Haerter et al., 2009; van den Maagdenberg et al., 2010), the latency between cortical and subcortical SD was significantly shorter in both FHM1 mutants compared with WT (Table 2). Although the overall morphology of SDs differed between cortex and subcortical structures, SD amplitudes and durations were comparable among WT and FHM1 mutants within each structure (Table 3).
Figure 2.

Subcortical propagation of cortical SD into hippocampus and thalamus in S218L mutant mice. Representative microelectrode recordings show SD propagation from cortex into striatum and hippocampus or thalamus in HET or HOM S218L mutants but not in WT mice. Thalamic or hippocampal propagation was not found in R192Q HOM mice (see Table 2). Hippocampal SDs were often prolonged compared with other structures. Thalamic SDs were often multiphasic, sometimes coalescing into a prolonged depolarization. The multiphasic waveform may be attributable to multiple corticothalamic propagation pathways arriving sequentially at the thalamic recording site or caused by circling within thalamic nuclei.
Table 1.
Propagation rate of cortical SDs into subcortical structures
| Strain | Gender | Genotype | Striatum | Hippocampus | Thalamus |
|---|---|---|---|---|---|
| R192Q | Female | WT | 28% (7/25 CSDs in 3/4 mice) | 0% (0/11 CSDs in 0/4 mice) | 0% (0/14 CSDs in 0/4 mice) |
| HOM | 71%* (10/14 CSDs in 4/4 mice) | 0% (0/7 CSDs in 0/4 mice) | 0% (0/7 CSDs in 0/4 mice) | ||
| S218L | Female | WT | 22% (5/22 CSDs in 4/4 mice) | 0% (0/11 CSDs in 0/5 mice) | 0% (0/7 CSDs in 0/4 mice) |
| HET | 100%*† (51/51 CSDs in 5/5 mice) | 27%*† (7/26 CSDs in 5/5 mice) | 0% (0/25 CSDs in 0/5 mice) | ||
| HOM | 100%*† (35/35 CSDs in 7/7 mice) | 72%*†‡ (13/18 CSDs in 7/7 mice) | 39%*†‡ (15/39 CSDs in 9/10 mice) | ||
| WT + guanosine | 29% (2/7 CSDs in 2/2 mice) | NR | NR | ||
| HOM + guanosine | 95%* (42/44 CSDs in 5/5 mice) | 41%*+ (16/39 CSDs in 5/5 mice) | 0%+ (0/10 CSDs in 1/2 mice) | ||
| Male | WT | 20% (4/20 CSDs in 3/4 mice) | 0% (0/10 CSDs in 0/4 mice) | 0% (0/10 CSDs in 0/4 mice) | |
| HOM | 91%*# (39/43 CSDs in 5/5 mice) | 70%* (14/20 CSDs in 5/5 mice) | 21% (4/19 CSDs in 4/5 mice) |
Structures were recorded in random order. Values indicate the proportion of CSDs propagating into indicated subcortical structures while recording from that structure and the proportion of mice that showed subcortical propagation into indicated structures.
*p < 0.05 versus WT;
†p < 0.05 versus R192Q HOM;
‡p < 0.05 versus S218L HET; +p < 0.05 and
#p = 0.06 versus untreated S218L female HOM. NR, Not recorded.
Table 3.
Amplitudes and durations of SD
| Strain | Gender | Genotype | SD amplitude (mV) |
SD duration (sec) |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cortex | Striatum | Hippocampus | Thalamus | Cortex | Striatum | Hippocampus | Thalamus | |||
| R192Q | Female | WT | 10 ± 4 | 30 ± 2 | 38 ± 6 | 41 ± 6 | ||||
| HOM | 13 ± 6 | 31 ± 10 | 41 ± 6 | 45 ± 9 | ||||||
| S218L | Female | WT | 9 ± 4 | 21 ± 7 | 35 ± 2 | 37 ± 2 | ||||
| HET | 11 ± 4 | 35 ± 4 | 34 ± 7 | 34 ± 5 | 51 ± 14 | 67 ± 24 | ||||
| HOM | 15 ± 6 | 28 ± 12 | 29 ± 8 | 24 ± 4 | 29 ± 5* | 35 ± 10 | 53 ± 9 | 29 ± 9 | ||
| WT + guanosine | 13 ± 2 | 13 ± 6 | 35 ± 5 | 27 ± 3 | ||||||
| HOM + guanosine | 15 ± 4 | 17 ± 11 | 24 ± 12 | 25 | 28 ± 6 | 34 ± 6 | 62 ± 12 | 45 | ||
| Male | WT | 15 ± 4 | 16 ± 9 | 32 ± 5 | 33 ± 6 | |||||
| HOM | 15 ± 2 | 18 ± 4# | 28 ± 3 | 12 ± 4† | 33 ± 11 | 29 ± 8 | 72 ± 27 | 32 ± 17 | ||
Values are mean ± SD.
*p < 0.05 versus R192Q HOM;
†p < 0.05 and
#p = 0.10 versus female S218L HOM.
An SD was occasionally evoked in cortex or subcortical structures during electrode insertion. These mechanical SDs occurred with higher incidence in FHM1 mutants compared with WT (Table 4), and when evoked in cortex, they spread into subcortical structures similar to KCl-induced SDs. Moreover, when directly evoked in a subcortical structure (i.e., via electrode insertion), mechanical SDs did reciprocally spread into the cortex and other subcortical structures, suggesting that cortico-subcortical propagation can be bidirectional (Fig. 3; Table 4).
Table 4.
Incidence of mechanical SD and reciprocal spread of subcortical SD into cortex
| Strain | Gender | Genotype | Incidence of mechanical SD (%) (mechanical SD/electrode insertion) |
Incidence of reciprocal spread (%) (reciprocal spread into cortex/mechanical subcortical SD) |
|||||
|---|---|---|---|---|---|---|---|---|---|
| Cortex | Striatum | Hippocampus | Thalamus | Striatum | Hippocampus | Thalamus | |||
| R192Q | Female | WT | 25% (1/4 in 1/4 mice) | 0% (0/4 in 0/4 mice) | 25% (1/4 in 1/4 mice) | 0% (0/4 in 0/4 mice) | 0% (0/1 in 0/1 mouse) | ||
| HOM | 29% (2/7 in 2/4 mice) | 83%* (5/6 in 4/4 mice) | 43% (3/7 in 2/4 mice) | 29% (2/7 in 2/4 mice) | 40% (2/5 in 2/4 mice) | 0% (0/3 in 0/2 mice) | 0% (0/2 in 0/2 mice) | ||
| S218L | Female | WT | 0% (0/8 in 0/4 mice) | 0% (0/8 in 0/4 mice) | 10% (1/10 in 2/4 mice) | 13% (1/8 in 1/4 mice) | 0% (0/1 in 0/2 mice) | 0% (0/1 in 0/1 mouse) | |
| HET | 71%* (5/7 in 3/5 mice) | 67%* (4/6 in 4/5 mice) | 50% (3/6 in 3/5 mice) | 38% (3/8 in 2/5 mice) | 25% (1/4 in 1/4 mice) | 33% (1/3 in 1/3 mice) | 0% (0/3 in 0/2 mice) | ||
| HOM | 58%* (7/12 in 4/6 mice) | 73%* (8/11 in 5/6 mice) | 67%* (12/18 in 6/6 mice) | 60%* (9/15 in 6/6 mice) | 25% (2/8 in 2/5 mice) | 33% (4/12 in 3/6 mice) | 78%† (7/9 in 5/6 mice) | ||
| WT + guanosine | 0% (0/3 in 0/2 mice) | 0% (0/3 in 0/2 mice) | |||||||
| HOM + guanosine | 40%$ (2/5 in 2/5 mice) | 75%* (3/4 in 3/5 mice) | 50%@ (4/8 in 3/5 mice) | 0% (0/2 in 0/2 mice) | 0% (0/3 in 0/5 mice) | 50% (2/4 in 2/5 mice) | |||
| Male | WT | 0% (0/7 in 0/4 mice) | 0% (0/7 in 0/4 mice) | 0% (0/7 in 0/4 mice) | 0% (0/7 in 0/4 mice) | ||||
| HOM | 33%∧ (3/9 in 3/5 mice) | 55%* (6/11 in 4/5 mice) | 8%‡ (1/12 in 1/5 mice) | 28%# (5/18 in 3/5 mice) | 17% (1/6 in 1/5 mice) | 0% (0/1 in 0/5 mice) | 40% (2/5 in 2/5 mice) | ||
Values indicate the proportion of electrode insertions and the proportion of mice that showed a mechanical SD and reciprocal spread.
*p < 0.05,
∧p = 0.09,
$p = 0.05, and
@p = 0.06 versus WT;
†p < 0.05 versus S218L HET and R192Q HOM;
‡p < 0.05 and
#p = 0.06 versus S218L HOM female.
Figure 3.

Reverse propagation of subcortical SD into cortex in S218L HOM mutants. A representative tracing shows a thalamic SD triggered during glass micropipette insertion in S218L HOM mice (dashed line) that preceded slow potential changes in the striatum and cortex, suggesting that SDs can propagate from thalamus to cortex as well.
In all S218L HOM mutants (n = 7), we observed repetitive cortical SD waves after a single KCl application despite a vigorous saline wash (median, three SDs; interquartile range, two to five SDs) (Fig. 4). None of the other groups showed repetitive SDs. Recurrent cortical SDs occurred after 19 and 38% of all SD inductions in male and female S218L HOM mice, respectively, and spread into the striatum (43 and 31%), hippocampus (0 and 11%), and thalamus (33 and 10%) in clusters. Inter-SD intervals were relatively regular, suggesting reverberating SD waves (average inter-SD intervals were 126 ± 20 and 118 ± 22 s in cortex, 202 ± 40 and 229 ± 54 s in striatum, 86 ± 16 and 174 s in hippocampus, and 144 ± 64 and 187 s in thalamus in male and female S218L HOM, respectively). Importantly, striatal but not hippocampal or thalamic propagation was required for recurrent SDs to occur, suggesting that they were reentry waves between cortex and striatum, as described previously (Vinogradova et al., 1991).
Figure 4.

Recurrent cortical and subcortical SDs in S218L HOM mutants. A, B, Representative microelectrode recordings show clusters of SD in cortex, striatum, and hippocampus (A) or thalamus (B) after a single brief epidural KCl application (dashed line) in S218L HOM mice. The arrowhead shows a typical large-amplitude spike burst observed only in the S218L HOM mutant strain. These afterdischarges usually lasted <1 min and started within a few minutes after the recovery of slow potential shift.
Cortical SD propagation into the contralateral cortex was not detected (n = 2 cortical SDs in two female and n = 5 cortical SDs in five male S218L HOM). These results using bilateral electrophysiological recordings were also confirmed using full-field laser speckle imaging to detect SD-evoked cortical blood flow changes (n = 5 cortical SDs in five S218L HOM mice) (supplemental Fig. 1 and supplemental Movie 1, http://www2.massgeneral.org/NCS/Supplemental_Online_Movie.mpeg) (Chang et al., 2010). Hippocampal SDs were similarly limited to the ipsilateral hemisphere (n = 3 hippocampal SDs in three S218L HOM mice). In contrast, all thalamic SDs (n = 4 thalamic SDs in four S218L HOM mice) propagated into the contralateral thalamus with a latency of 13 ± 3 s between bilateral thalamic electrodes (∼2.4 mm apart).
Last, we detected large-amplitude (1–2 mV) and rhythmic (up to 7 Hz) spike activity, reminiscent of epileptiform afterdischarges, within 1–2 min after the recovery of an SD in S218L mutants only (Figs. 2, 4). Spike activity was present in all four structures recorded, displayed an allele-dosage effect, and was more frequent in female mutants compared with males (supplemental Table 2, see Notes). Sustained seizure activity was never observed under our recording conditions.
MRI
We consistently observed a wave of reduced ADCs, slowly propagating into the striatum and the hippocampus after topical KCl application on to the parietal cortex in all mice (n = 4 female S218L HOM) (Fig. 5; supplemental Table 3, see Notes). In two animals only, we detected spread into the thalamus, which propagated into the contralateral thalamus in both cases. SD-related ADC changes consistently propagated in a lateral-to-medial direction within the striatum and in a medial-to-lateral direction within the hippocampus. It was not possible to precisely determine the propagation pathways because of the relatively rapid SD propagation in S218L HOM mice (∼7 mm/min) (Eikermann-Haerter et al., 2009; van den Maagdenberg et al., 2010) compared to the slow MRI acquisition time and repetition rate (once every 20 s).
Figure 5.
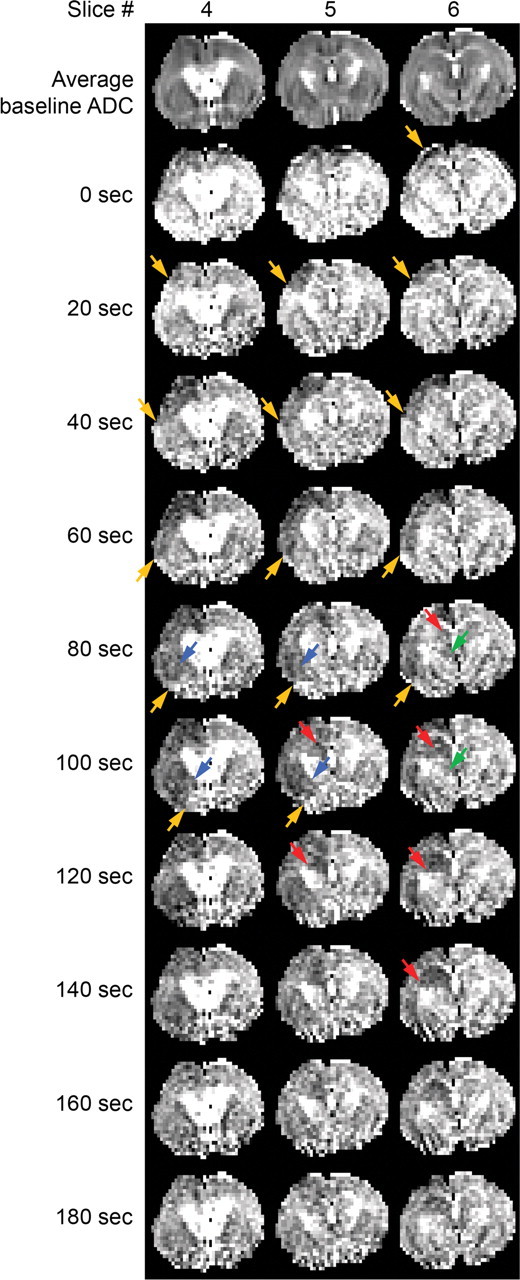
Diffusion-weighted MRI of SD propagation. Serial magnetic resonance images in a representative S218L HOM mouse show ADC changes during cortical SD (orange) spreading into striatum (blue), hippocampus (red), and dorsomedial thalamus (green). Three consecutive coronal slices are shown over time (acquired at 20 s interval; voxel dimensions: 0.5, 0.2, 0.2 mm). The top row shows average baseline ADC maps to serve as an anatomical reference. Arrows indicate the SD wavefront. Striatal ADC changes corresponding to SD propagation typically occurred in a lateral-to-medial direction, whereas changes in hippocampus propagated in a medial-to-lateral direction. Hippocampal ADC changes persisted longer than in all other regions.
c-fos expression
Compared with sham controls and contralateral cortex, ipsilateral cortical c-fos expression was increased in all mice after cortical SD (Fig. 6). c-fos was upregulated in the striatum in all 12 S218L HOM and 3 of 4 R192Q HOM mice but in only 1 of 6 WT mice. c-fos upregulation was found in hippocampus (10 of 12 mice), and in thalamus and lateral hypothalamic area (3 of 12 mice), in S218L HOM mutants only. Consistent with the electrophysiological data, cortical, striatal, and hippocampal c-fos upregulation was strictly unilateral, whereas thalamic c-fos upregulation was bilateral. In addition, all FHM1 mutants showed strong ipsilateral c-fos upregulation in amygdala, basal forebrain nuclei, nucleus accumbens, and the septal nuclei, as well as in regions interposed among these structures, whereas in WT there was only a slight c-fos staining present in three of six mice in the amygdala and in two of six in the septal nuclei only (Fig. 6). In sham controls, staining was very light and did not differ between WT and S218L HOM (n = 3 each; data not shown), thereby ruling out an effect of anesthesia.
Figure 6.
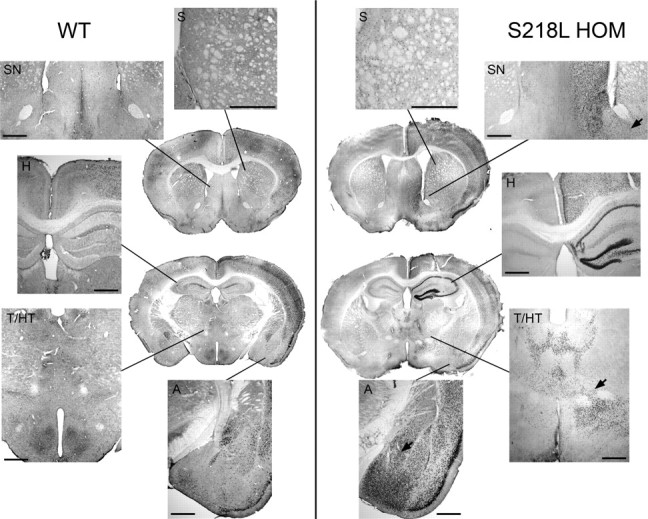
Enhanced c-fos expression in cortex and subcortical structures in S218L mutant mice. c-fos immunohistochemistry in coronal sections taken from representative WT and S218L HOM mice 3 h after three consecutive cortical SDs triggered 15 min apart is shown. Coronal sections were taken from two different levels (striatum and hippocampus) in whole brain. Both the number of labeled cells plus staining intensity were increased in S218L mutant brain compared with wild type. Higher-magnification images show that c-fos was expressed throughout cortex in both strains, although this increase appeared more prominent in the S218L HOM mutant. c-fos expression was upregulated in striatum (S) in all mutants (n = 12) but only one WT mouse (n = 6). c-fos upregulation in hippocampus (H) or thalamus (T) was observed in the mutants only. Mutants also showed c-fos upregulation in septal nuclei (SN; 100%), hypothalamus (HT; 50%), and amygdala (A; 100%). c-fos expression was observed in cells bridging contiguous structures such as striatum and amygdala, striatum and septal nuclei, and thalamus and hypothalamus (arrows). Scale bar, 500 μm.
Cardiovascular physiology
Thalamic SDs were associated with abrupt BP transients 75% of the time in all S218L HOM mice. These BP transients lasted ∼1 min and were hypertensive, increasing BP >25% without accompanying changes in heart rate (Fig. 7). Such BP fluctuations never occurred when a cortical SD failed to propagate into thalamus and thus were present only in S218L HOM mice.
Figure 7.

Thalamic SDs were associated with transient hypertension. Representative electrophysiological tracings are shown with simultaneous arterial BP recordings in an S218L HOM mutant. A, When an SD propagated into thalamus, transient hypertensive episodes were observed in S218L HOM mice. B, In the absence of thalamic SD, BP increases did not occur, even when SD propagated into the striatum. Hence, hypertensive episodes were never observed in R192Q mutant mice.
Discussion
The mechanisms underlying the signs and symptoms of severe aura in FHM patients are poorly understood. We recently reported that after an SD, S218L mutant mice exhibit neurological signs highly reminiscent of clinical attacks in FHM1 patients carrying this mutation, including hemiplegia, coma, and seizures, whereas the R192Q mutant mice developed pure hemiplegia (Kors et al., 2001; Eikermann-Haerter et al., 2009). Here, we present evidence that SD propagation is enhanced in subcortical structures in the S218L mutant mouse, facilitating the propagation of SD throughout the basal ganglia, hippocampus, and diencephalon and predisposing to reverberating SD waves. The widespread SD propagation provides a novel mechanism for the severe neurological dysfunction during FHM1 attacks. Furthermore, cortical SDs propagated into striatum more consistently in S218L than R192Q mutants, and thalamic and hippocampal SDs were observed only in S218L mutants with an allele-dosage relationship. Hence, the propensity of SD to traverse barriers with low neuronal density and high white matter content corresponds to the strength of gain of function in S218L and R192Q mutant CaV2.1 channels (van den Maagdenberg et al., 2010).
Although striatal propagation of cortical SD has been reported in rats (Fifkova and Bures, 1964; Vinogradova et al., 1991), propagation into the hippocampus and the thalamus appears to be a manifestation of genetically enhanced SD susceptibility in FHM1 mutants. SD propagation requires high neuronal and synaptic density and is impeded by the presence of large extracellular space and an abundance of astrocytes and myelin (Bures et al., 1974; Somjen, 2001; Merkler et al., 2009). Consequently, white matter and areas with lower neuronal density (e.g., subiculum) serve as natural barriers against SD propagation. Whereas thick white matter bundles such as corpus callosum are impervious to SD, the extracellular K+ and glutamate surge may still permeate thinner white matter tracts and areas with low neuronal density to rekindle the process on the other side of the barrier. FHM1 mutations augment Cav2.1 current density after weak depolarization and enhance presynaptic Ca2+ influx and glutamate release, thereby lowering the depolarization threshold for SD induction (Tottene et al., 2005, 2009; Eikermann-Haerter et al., 2009; van den Maagdenberg et al., 2010). The S218L mutation causes larger Cav2.1 gain of function and a lower SD threshold than R192Q. The lower the SD threshold is, the more likely the ionic and neurotransmitter fluxes to evoke SD on the other side of the barrier and the more widespread the SD propagation, explaining both the allele-dosage effect and the hippocampal and thalamic spread exclusively in the S218L mutants. Also supporting a role for glutamate in subcortical spread, guanosine, which inhibits glutamatergic hyperexcitability by stimulating astrocytic glutamate uptake (Frizzo et al., 2001; Lara et al., 2001), suppressed hippocampal and thalamic spread in FHM1 mutants.
In our previous work using combined isoflurane and nitrous oxide anesthesia, subcortical SD propagation rates were much lower in both WT and FHM1 mutant mice (Eikermann-Haerter et al., 2009). Isoflurane together with nitrous oxide is known to suppress SD susceptibility (Kudo et al., 2008). To avoid this confound, in the present study we used pentobarbital anesthesia, which has the least suppressive effect on CSD compared with other anesthetics that have been tested (Kitahara et al., 2001; Kudo et al., 2008), and detected widespread subcortical propagation in the FHM1 mutants. However, compared with the awake state, pentobarbital may still increase SD threshold (Whieldon and Van Harreveld, 1951; Van Harreveld and Stamm, 1953). Therefore, in unanesthetized FHM1 mutants, SDs might propagate even farther and exhibit longer-lasting reentrant patterns between different cortical and subcortical structures.
Results using diffusion-weighted MRI and c-fos immunoreactivity as surrogates of SD propagation were consistent with the electrophysiological data and reflected SD propagation into striatum, hippocampus, and thalamus of S218L mutant mice. Diffusion-weighted MRI is an established method to map SD propagation in which transient ADC reductions spatiotemporally correlate with the electrophysiological changes during SD (de Crespigny et al., 1998). Enhanced expression of the c-fos also shows spatial correspondence to SD propagation (Moskowitz et al., 1993; Shimazawa et al., 1995). Although subcortical c-fos expression might reflect increased cortico-subcortical efferent activity during a cortical SD, more typically, cortical SD leads to suppression of glucose metabolism in subcortical structures (Shinohara et al., 1979), suggesting that suppression of neuronal activity (e.g., cortex) in the wake of SD has the opposite effect on efferent input to target structures.
Despite observing afterdischarges in the wake of an SD in the S218L mutant, we believe contiguous spread, rather than seizure activity, was responsible for subcortical SD occurrence. First, the SD latencies between cortical and subcortical structures were longer than expected if evoked synaptically via corticofugal axonal projections (Koroleva and Vinogradova, 1990). Second, ADC reductions were slow and contiguous between cortex and subcortical structures, which is not consistent with seizure activity (Engelhorn et al., 2007).
We observed transient BP elevations during SD in S218L HOM mice. Because the occurrence of these hypertensive episodes was tightly linked to thalamic SDs (i.e., never observed in the absence of a thalamic SD), and because hypothalamic nuclei showed diffuse c-fos upregulation, we speculate that BP transients were triggered by direct propagation of SD into the hypothalamus, rather than upstream modulation of hypothalamic autonomic output by SD in amygdala. It should be noted that although SD is limited to gray matter nuclei, the associated ionic shifts can influence conduction in white matter bundles embedded in gray matter, causing additional signs and symptoms unrelated to the involved gray matter (Bures and Hartman, 1967; Bures et al., 1967; Bures and Fifkova, 1968).
Probable subcortical propagation pathways
Amygdala is a likely path for cortico-subcortical SD propagation because of its lower proportion of myelin and direct gray matter contiguity with striatum, entorhinal and piriform cortices, and basal forebrain nuclei (Fifkova and Bures, 1964; Fifkova and Syka, 1964; Krivanek and Fifkova, 1965; Bures et al., 1974; DeLuca et al., 1975; Vinogradova et al., 1991). Subiculum may be an alternative pathway into hippocampus, despite its diminished neuronal density (Bures et al., 1974). Human brain, of course, has a greater abundance of white matter than rodents. Nevertheless, these anatomical relationships are preserved in mammalian brains. For example, human entorhinal cortex still forms a gray matter bridge between hippocampus and neocortex. Amygdala, phylogenetically the oldest part of basal ganglia, is immediately anterior and superior to hippocampus, in contiguity with striatum, claustrum, uncus, and subiculum. Therefore, we speculate that in highly susceptible FHM1 patients (e.g., S218L carriers), cortical SD may propagate into subcortical tissues and possibly reverberate within and/or among the cortex and gray matter nuclei, leading to a prolonged and severe clinical state including encephalopathy and coma. It is equally plausible that SD may originate within the susceptible subcortical nuclei in FHM1 patients de novo. Changes in glucose metabolism and ADC have been detected in subcortical structures using diffusion-weighted MRI and 18F-2-fluoro-2-deoxy-d-glucose positron emission tomography during FHM attacks (Chabriat et al., 2000; Gutschalk et al., 2002).
Speculations on subcortical SD and the clinical features of FHM
Many of the FHM1 aura symptoms and signs can be linked to dysfunction in subcortical structures. For example, hemiparesis can be caused by SD within the basal ganglia, as shown in experimental animals (Weiss and Fifkova, 1963; Trachtenberg et al., 1970; Jakobartl and Huston, 1977). One-third of FHM1 patients develop severe attacks with transient impairment of consciousness ranging from stupor to sometimes fatal coma, more commonly associated with a subset of mutations including S218L (Terwindt et al., 1998; Battistini et al., 1999; Ducros et al., 1999, 2001; Chabriat et al., 2000; Vahedi et al., 2000; Kors et al., 2001; Curtain et al., 2006; Stam et al., 2009). Thalamic SD may be the mechanism for the depressed level of consciousness in FHM1 patients, because bilateral thalamic dysfunction can cause stupor and coma (Saper, 1985), and we observed bilateral thalamic SD in S218L HOM mutants. Hippocampus is highly susceptible to SD (Bures et al., 1974), and SD has been observed in human hippocampus (Sramka et al., 1977; Avoli et al., 1991). Importantly, hippocampal SD can trigger seizure activity. Clinically, 20% of FHM patients carrying the S218L mutation develop seizures during attacks (Thomsen et al., 2002). Similarly, we observed generalized seizures in S218L HOM mice after a cortical SD (Eikermann-Haerter et al., 2009). Hippocampal dysfunction may be associated with amnesia, and limbic disturbances such as dysphoria, yawning, and fluid retention can occur during migraine attacks (Cutrer and Olesen, 2006). Amygdala may also play a role in the development of complex neuropsychiatric, autonomic, and neuroendocrine symptoms during an attack (Olesen, 2006).
Conclusion
Our data show that SD is a remarkable phenotype in FHM1 mutant mice. Both S218L and R192Q mutants show enhanced striatal SD as a possible cause for transient hemiplegia in FHM1. In addition, the S218L mutant shows widespread SD propagation in the forebrain, including thalamus and hippocampus, two findings that may explain coma and seizures during some attacks in patients carrying the S218L mutation. Therefore, subcortical SD, either arising de novo or propagating from cortex, might account for the severe attacks with hemiplegia, coma, and seizures in FHM1 patients.
Notes
Supplemental material for this article is available at http://www2.massgeneral.org/NCS/Supplemental_Online_Movie.mpeg. This material has not been peer reviewed.
Footnotes
This work was supported by the American Heart Association (Grants SDG2610275 and 0835451N), the National Institutes of Health (Grants NS061505, NS35611, and EB002066), the Netherlands Organization for Scientific Research (Grants 903-52-291 and Vici 918.56.602), European Union “EUROHEAD” (Grant LSHM-CT-2004-504837), and the Centre for Medical Systems Biology in the framework of the Netherlands Genomics Initiative. We thank Ludo Broos (Leiden University Medical Centre, Leiden, The Netherlands) for confirmatory genotyping.
The authors declare no competing financial interests.
References
- Avoli M, Drapeau C, Louvel J, Pumain R, Olivier A, Villemure JG. Epileptiform activity induced by low extracellular magnesium in the human cortex maintained in vitro. Ann Neurol. 1991;30:589–596. doi: 10.1002/ana.410300412. [DOI] [PubMed] [Google Scholar]
- Ayata C, Dunn AK, Gursoy OY, Huang Z, Boas DA, Moskowitz MA. Laser speckle flowmetry for the study of cerebrovascular physiology in normal and ischemic mouse cortex. J Cereb Blood Flow Metab. 2004;24:744–755. doi: 10.1097/01.WCB.0000122745.72175.D5. [DOI] [PubMed] [Google Scholar]
- Battistini S, Stenirri S, Piatti M, Gelfi C, Righetti PG, Rocchi R, Giannini F, Battistini N, Guazzi GC, Ferrari M, Carrera P. A new CACNA1A gene mutation in acetazolamide-responsive familial hemiplegic migraine and ataxia. Neurology. 1999;53:38–43. doi: 10.1212/wnl.53.1.38. [DOI] [PubMed] [Google Scholar]
- Bures J, Fifkova E. The effect of potassium ions liberated from nerve structures invaded by spreading depression on conduction in fibres of the adjacent white matter. Physiol Bohemoslov. 1968;17:405–410. [PubMed] [Google Scholar]
- Bures J, Hartman G. Conduction block in capsula interna fibres caused by striatal spreading depression in rats. Experientia. 1967;23:736–737. doi: 10.1007/BF02154146. [DOI] [PubMed] [Google Scholar]
- Bures J, Hartmann G, Lukyanova LD. Blockade of thalamocortical and pyramidal pathways by striatal spreading depression in rats. Exp Neurol. 1967;18:404–415. doi: 10.1016/0014-4886(67)90058-1. [DOI] [PubMed] [Google Scholar]
- Bures J, Buresova O, Krivanek J. New York: Academic; 1974. The mechanism and applications of Leao's spreading depression of electroencephalographic activity. [Google Scholar]
- Chabriat H, Vahedi K, Clark CA, Poupon C, Ducros A, Denier C, Le Bihan D, Bousser MG. Decreased hemispheric water mobility in hemiplegic migraine related to mutation of CACNA1A gene. Neurology. 2000;54:510–512. doi: 10.1212/wnl.54.2.510. [DOI] [PubMed] [Google Scholar]
- Chang JC, Shook LL, Biag J, Nguyen EN, Toga AW, Charles AC, Brennan KC. Biphasic direct current shift, haemoglobin desaturation and neurovascular uncoupling in cortical spreading depression. Brain. 2010;133:996–1012. doi: 10.1093/brain/awp338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtain RP, Smith RL, Ovcaric M, Griffiths LR. Minor head trauma-induced sporadic hemiplegic migraine coma. Pediatr Neurol. 2006;34:329–332. doi: 10.1016/j.pediatrneurol.2005.08.033. [DOI] [PubMed] [Google Scholar]
- Cutrer FM, Olesen J. Migraines with aura and their subforms. In: Olesen J, Goadsby PJ, Ramadan NM, Tfelt-Hansen P, Welch KMA, editors. The headaches. Ed 3. Philadelphia: Lippincott Williams and Wilkins; 2006. pp. 407–422. [Google Scholar]
- de Crespigny A, Rother J, van Bruggen N, Beaulieu C, Moseley ME. Magnetic resonance imaging assessment of cerebral hemodynamics during spreading depression in rats. J Cereb Blood Flow Metab. 1998;18:1008–1017. doi: 10.1097/00004647-199809000-00010. [DOI] [PubMed] [Google Scholar]
- DeLuca B, Shibata M, Brozek G, Bures J. Facilitation of Leao's spreading depression by a pyrrolopyrimidine derivative. Neuropharmacology. 1975;14:537–545. doi: 10.1016/0028-3908(75)90118-5. [DOI] [PubMed] [Google Scholar]
- Ducros A, Denier C, Joutel A, Vahedi K, Michel A, Darcel F, Madigand M, Guerouaou D, Tison F, Julien J, Hirsch E, Chedru F, Bisgard C, Lucotte G, Despres P, Billard C, Barthez MA, Ponsot G, Bousser MG, Tournier-Lasserve E. Recurrence of the T666M calcium channel CACNA1A gene mutation in familial hemiplegic migraine with progressive cerebellar ataxia. Am J Hum Genet. 1999;64:89–98. doi: 10.1086/302192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducros A, Denier C, Joutel A, Cecillon M, Lescoat C, Vahedi K, Darcel F, Vicaut E, Bousser MG, Tournier-Lasserve E. The clinical spectrum of familial hemiplegic migraine associated with mutations in a neuronal calcium channel. N Engl J Med. 2001;345:17–24. doi: 10.1056/NEJM200107053450103. [DOI] [PubMed] [Google Scholar]
- Eikermann-Haerter K, Dilekoz E, Kudo C, Savitz SI, Waeber C, Baum MJ, Ferrari MD, van den Maagdenberg AM, Moskowitz MA, Ayata C. Genetic and hormonal factors modulate spreading depression and transient hemiparesis in mouse models of familial hemiplegic migraine type 1. J Clin Invest. 2009;119:99–109. doi: 10.1172/JCI36059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhorn T, Hufnagel A, Weise J, Baehr M, Doerfler A. Monitoring of acute generalized status epilepticus using multilocal diffusion MR imaging: early prediction of regional neuronal damage. AJNR Am J Neuroradiol. 2007;28:321–327. [PMC free article] [PubMed] [Google Scholar]
- Fifkova E, Bures J. Spreading depression in the mammalian striatum. Arch Int Physiol Biochim. 1964;72:171–179. doi: 10.3109/13813456409105260. [DOI] [PubMed] [Google Scholar]
- Fifkova E, Syka J. Relationships between cortical and striatal spreading depression in rat. Exp Neurol. 1964;9:355–366. [Google Scholar]
- Frizzo ME, Lara DR, Dahm KC, Prokopiuk AS, Swanson RA, Souza DO. Activation of glutamate uptake by guanosine in primary astrocyte cultures. Neuroreport. 2001;12:879–881. doi: 10.1097/00001756-200103260-00051. [DOI] [PubMed] [Google Scholar]
- Frizzo ME, Schwalm FD, Frizzo JK, Soares FA, Souza DO. Guanosine enhances glutamate transport capacity in brain cortical slices. Cell Mol Neurobiol. 2005;25:913–921. doi: 10.1007/s10571-005-4939-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutschalk A, Kollmar R, Mohr A, Henze M, Ille N, Schwaninger M, Hartmann M, Hahnel S, Haberkorn U, Rupp A, Meyding-Lamade U. Multimodal functional imaging of prolonged neurological deficits in a patient suffering from familial hemiplegic migraine. Neurosci Lett. 2002;332:115–118. doi: 10.1016/s0304-3940(02)00940-0. [DOI] [PubMed] [Google Scholar]
- Jakobartl L, Huston JP. Circling and consumatory behavior induced by striatal and neocortical spreading depression. Physiol Behav. 1977;19:673–677. doi: 10.1016/0031-9384(77)90042-7. [DOI] [PubMed] [Google Scholar]
- Kitahara Y, Taga K, Abe H, Shimoji K. The effects of anesthetics on cortical spreading depression elicitation and c-fos expression in rats. J Neurosurg Anesthesiol. 2001;13:26–32. doi: 10.1097/00008506-200101000-00005. [DOI] [PubMed] [Google Scholar]
- Koroleva VI, Vinogradova LV. [Spreading depression in the thalamus, hippocampus and caudate nucleus of the rat during electrical stimulation of the parietal area of the cortex] Neirofiziologiia. 1990;22:36–44. [PubMed] [Google Scholar]
- Koroleva VI, Gorelova NA, Vinogradova LV. [The electrophysiological characteristics and behavioral manifestations of hippocampal and thalamic spreading depression] Zh Vyssh Nerv Deiat Im I P Pavlova. 1991;41:1019–1032. [PubMed] [Google Scholar]
- Kors EE, Terwindt GM, Vermeulen FL, Fitzsimons RB, Jardine PE, Heywood P, Love S, van den Maagdenberg AM, Haan J, Frants RR, Ferrari MD. Delayed cerebral edema and fatal coma after minor head trauma: role of the CACNA1A calcium channel subunit gene and relationship with familial hemiplegic migraine. Ann Neurol. 2001;49:753–760. doi: 10.1002/ana.1031. [DOI] [PubMed] [Google Scholar]
- Krivanek J, Fifkova E. The value of ultramicro-analysis of lactic acid in tracing the penetration of Leao's cortical spreading depression to subcortical areas. J Neurol Sci. 1965;2:385–392. doi: 10.1016/0022-510x(65)90020-1. [DOI] [PubMed] [Google Scholar]
- Kudo C, Nozari A, Moskowitz MA, Ayata C. The impact of anesthetics and hyperoxia on cortical spreading depression. Exp Neurol. 2008;212:201–206. doi: 10.1016/j.expneurol.2008.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara DR, Schmidt AP, Frizzo ME, Burgos JS, Ramirez G, Souza DO. Effect of orally administered guanosine on seizures and death induced by glutamatergic agents. Brain Res. 2001;912:176–180. doi: 10.1016/s0006-8993(01)02734-2. [DOI] [PubMed] [Google Scholar]
- Merkler D, Klinker F, Jurgens T, Glaser R, Paulus W, Brinkmann BG, Sereda MW, Stadelmann-Nessler C, Guedes RC, Bruck W, Liebetanz D. Propagation of spreading depression inversely correlates with cortical myelin content. Ann Neurol. 2009;66:355–365. doi: 10.1002/ana.21746. [DOI] [PubMed] [Google Scholar]
- Moskowitz MA, Nozaki K, Kraig RP. Neocortical spreading depression provokes the expression of c-fos protein-like immunoreactivity within trigeminal nucleus caudalis via trigeminovascular mechanisms. J Neurosci. 1993;13:1167–1177. doi: 10.1523/JNEUROSCI.13-03-01167.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olesen J. Sporadic and familial hemiplegic migraines. In: Olesen J, Goadsby P, Ramadan NM, Tfelt-Hansen P, Welch KM, editors. The headaches. Philadelphia: Lippincott Williams and Wilkins; 2006. pp. 577–585. [Google Scholar]
- Ophoff RA, Terwindt GM, Vergouwe MN, van Eijk R, Oefner PJ, Hoffman SM, Lamerdin JE, Mohrenweiser HW, Bulman DE, Ferrari M, Haan J, Lindhout D, van Ommen GJ, Hofker MH, Ferrari MD, Frants RR. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell. 1996;87:543–552. doi: 10.1016/s0092-8674(00)81373-2. [DOI] [PubMed] [Google Scholar]
- Saper CB. Organization of cerebral cortical afferent systems in the rat. II. Hypothalamocortical projections. J Comp Neurol. 1985;237:21–46. doi: 10.1002/cne.902370103. [DOI] [PubMed] [Google Scholar]
- Shimazawa M, Hara H, Watano T, Sukamoto T. Effects of Ca2+ channel blockers on cortical hypoperfusion and expression of c-Fos-like immunoreactivity after cortical spreading depression in rats. Br J Pharmacol. 1995;115:1359–1368. doi: 10.1111/j.1476-5381.1995.tb16624.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara M, Dollinger B, Brown G, Rapoport S, Sokoloff L. Cerebral glucose utilization: local changes during and after recovery from spreading cortical depression. Science. 1979;203:188–190. doi: 10.1126/science.758688. [DOI] [PubMed] [Google Scholar]
- Soares FA, Schmidt AP, Farina M, Frizzo ME, Tavares RG, Portela LV, Lara DR, Souza DO. Anticonvulsant effect of GMP depends on its conversion to guanosine. Brain Res. 2004;1005:182–186. doi: 10.1016/j.brainres.2004.01.053. [DOI] [PubMed] [Google Scholar]
- Somjen GG. Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiol Rev. 2001;81:1065–1096. doi: 10.1152/physrev.2001.81.3.1065. [DOI] [PubMed] [Google Scholar]
- Sramka M, Brozek G, Bures J, Nadvornik P. Functional ablation by spreading depression: possible use in human stereotactic neurosurgery. Appl Neurophysiol. 1977;40:48–61. [PubMed] [Google Scholar]
- Stam AH, Luijckx GJ, Poll-The BT, Ginjaar IB, Frants RR, Haan J, Ferrari MD, Terwindt GM, van den Maagdenberg AM. Early seizures and cerebral oedema after trivial head trauma associated with the CACNA1A S218L mutation. J Neurol Neurosurg Psychiatry. 2009;80:1125–1129. doi: 10.1136/jnnp.2009.177279. [DOI] [PubMed] [Google Scholar]
- Terwindt GM, Ophoff RA, Haan J, Vergouwe MN, van Eijk R, Frants RR, Ferrari MD. Variable clinical expression of mutations in the P/Q-type calcium channel gene in familial hemiplegic migraine. Dutch Migraine Genetics Research Group. Neurology. 1998;50:1105–1110. doi: 10.1212/wnl.50.4.1105. [DOI] [PubMed] [Google Scholar]
- Thomsen LL, Eriksen MK, Roemer SF, Andersen I, Olesen J, Russell MB. A population-based study of familial hemiplegic migraine suggests revised diagnostic criteria. Brain. 2002;125:1379–1391. doi: 10.1093/brain/awf132. [DOI] [PubMed] [Google Scholar]
- Tottene A, Fellin T, Pagnutti S, Luvisetto S, Striessnig J, Fletcher C, Pietrobon D. Familial hemiplegic migraine mutations increase Ca(2+) influx through single human CaV2.1 channels and decrease maximal CaV2.1 current density in neurons. Proc Natl Acad Sci U S A. 2002;99:13284–13289. doi: 10.1073/pnas.192242399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tottene A, Pivotto F, Fellin T, Cesetti T, van den Maagdenberg AM, Pietrobon D. Specific kinetic alterations of human CaV2.1 calcium channels produced by mutation S218L causing familial hemiplegic migraine and delayed cerebral edema and coma after minor head trauma. J Biol Chem. 2005;280:17678–17686. doi: 10.1074/jbc.M501110200. [DOI] [PubMed] [Google Scholar]
- Tottene A, Conti R, Fabbro A, Vecchia D, Shapovalova M, Santello M, van den Maagdenberg AM, Ferrari MD, Pietrobon D. Enhanced excitatory transmission at cortical synapses as the basis for facilitated spreading depression in Ca(v)2.1 knockin migraine mice. Neuron. 2009;61:762–773. doi: 10.1016/j.neuron.2009.01.027. [DOI] [PubMed] [Google Scholar]
- Trachtenberg MC, Hull CD, Buchwald NA. Electrophysiological concomitants of spreading depression in caudate and thalamic nuclei of the cat. Brain Res. 1970;20:219–231. doi: 10.1016/0006-8993(70)90290-8. [DOI] [PubMed] [Google Scholar]
- Vahedi K, Denier C, Ducros A, Bousson V, Levy C, Chabriat H, Haguenau M, Tournier-Lasserve E, Bousser MG. CACNA1A gene de novo mutation causing hemiplegic migraine, coma, and cerebellar atrophy. Neurology. 2000;55:1040–1042. doi: 10.1212/wnl.55.7.1040. [DOI] [PubMed] [Google Scholar]
- van den Maagdenberg AM, Pietrobon D, Pizzorusso T, Kaja S, Broos LA, Cesetti T, van de Ven RC, Tottene A, van der Kaa J, Plomp JJ, Frants RR, Ferrari MD. A Cacna1a knockin migraine mouse model with increased susceptibility to cortical spreading depression. Neuron. 2004;41:701–710. doi: 10.1016/s0896-6273(04)00085-6. [DOI] [PubMed] [Google Scholar]
- van den Maagdenberg AM, Haan J, Terwindt GM, Ferrari MD. Migraine: gene mutations and functional consequences. Curr Opin Neurol. 2007;20:299–305. doi: 10.1097/WCO.0b013e3281338d1f. [DOI] [PubMed] [Google Scholar]
- van den Maagdenberg AM, Pizzorusso T, Kaja S, Terpolilli N, Shapovalova M, Hoebeek FE, Barrett CF, Gherardini L, van de Ven RC, Todorov B, Broos LA, Tottene A, Gao Z, Fodor M, De Zeeuw CI, Frants RR, Plesnila N, Plomp JJ, Pietrobon D, Ferrari MD. High cortical spreading depression susceptibility and migraine-associated symptoms in Ca(v)2.1 S218L mice. Ann Neurol. 2010;67:85–98. doi: 10.1002/ana.21815. [DOI] [PubMed] [Google Scholar]
- Van Harreveld A, Stamm JS. Effect of pentobarbital and ether on the spreading cortical depression. Am J Physiol. 1953;173:164–170. doi: 10.1152/ajplegacy.1953.173.1.164. [DOI] [PubMed] [Google Scholar]
- Vinogradova LV, Koroleva VI, Bures J. Re-entry waves of Leao's spreading depression between neocortex and caudate nucleus. Brain Res. 1991;538:161–164. doi: 10.1016/0006-8993(91)90392-9. [DOI] [PubMed] [Google Scholar]
- Weiss T, Fifkova E. The effect of neocortical and caudatal spreading depression on “circling movements” induced from the caudate nucleus. Physiol Bohemoslov. 1963;12:332–338. [PubMed] [Google Scholar]
- Whieldon JA, Van Harreveld A. Drug effects on the results of minimal cortical stimulation. Electroencephalogr Clin Neurophysiol. 1951;3:31–35. doi: 10.1016/0013-4694(51)90052-1. [DOI] [PubMed] [Google Scholar]