

The homotypic fusion and protein sorting (HOPS) tethering complex of the yeast vacuole is involved in multiple fusion reactions. We demonstrate that HOPS has two binding sites for SNAREs and that binding to the minimal SNARE complex is necessary for HOPS-stimulated fusion. Our data highlight the dual role of HOPS in Rab-mediated tethering and SNARE-driven fusion.
Abstract
Membrane fusion within the endomembrane system follows a defined order of events: membrane tethering, mediated by Rabs and tethers, assembly of soluble N-ethylmaleimide-sensitive factor (NSF) attachment protein receptor (SNARE) complexes, and lipid bilayer mixing. Here we present evidence that the vacuolar HOPS tethering complex controls fusion through specific interactions with the vacuolar SNARE complex (consisting of Vam3, Vam7, Vti1, and Nyv1) and the N-terminal domains of Vam7 and Vam3. We show that homotypic fusion and protein sorting (HOPS) binds Vam7 via its subunits Vps16 and Vps18. In addition, we observed that Vps16, Vps18, and the Sec1/Munc18 protein Vps33, which is also part of the HOPS complex, bind to the Q-SNARE complex. In agreement with this observation, HOPS-stimulated fusion was inhibited if HOPS was preincubated with the minimal Q-SNARE complex. Importantly, artificial targeting of Vam7 without its PX domain to membranes rescued vacuole morphology in vivo, but resulted in a cytokinesis defect if the N-terminal domain of Vam3 was also removed. Our data thus support a model of HOPS-controlled membrane fusion by recognizing different elements of the SNARE complex.
INTRODUCTION
Protein and lipid transport between organelles of the endomembrane system requires a defined cascade of membrane-active proteins, which promote the budding of vesicles and the final fusion at their destination (Hurley et al., 2010). Fusion of membranes depends on the initial tethering of the two membranes, which is followed by SNARE complex assembly and bilayer mixing (Bröcker et al., 2010; Hughson and Reinisch, 2010). For most fusion events, multisubunit tethering complexes interact with Rab GTPases to bridge the two membranes, whereas the interaction and folding of SNAREs present on both membranes drive the final fusion event (Jahn and Scheller, 2006). In addition to Rabs, several multisubunit tethering complexes harbor binding sites for SNAREs, which could be necessary for tethering or regulation of membrane fusion. For instance, the exocyst that operates in exocytosis binds the SNARE Sec9 via its Sec6 subunit (Sivaram et al., 2005), and the Dsl complex interacts with the endoplasmic reticulum SNAREs Use1 and Sec20 via its subunits Dsl3 and Tip20 (Ren et al., 2009).
For fusion events at the yeast vacuole, several important details have been clarified. Vacuoles receive material via several pathways, including the endocytic pathway and fusion with Golgi-derived adaptor protein complex 3 (AP-3) vesicles and autophagosomes (Bowers and Stevens, 2005). The organelle also undergoes homotypic fusion, which has been reconstituted with isolated vacuoles and with proteoliposomes (Wickner, 2010). The general fusion machinery at vacuoles includes the Rab GTPase Ypt7, which binds to the HOPS (homotypic fusion and protein sorting) complex in an initial tethering event. A total of five SNAREs is required for these fusion reactions (Ungermann et al., 1999). For fusion of vesicles with vacuoles, the Q-SNAREs Vam3, Vam7, and Vti1 pair with the R-SNARE Ykt6, whereas homotypic fusion uses the R-SNARE Nyv1 instead (Dilcher et al., 2001). Q and R refer to the central conserved amino acid residue of the SNARE domain of the respective protein. Isolated vacuoles contain cis-SNARE complexes that are disassembled in an ATP-dependent reaction with the help of Sec17 (yeast α-SNAP) and Sec18 (yeast NSF)(Mayer et al., 1996; Ungermann et al., 1998). This activation liberates Vam7, which binds phosphatidylinositol-3-phosphate (PI-3-P) via its PX domain and associates with its remaining partner SNAREs to form the fusion-active trans-SNARE complex (Boeddinghaus et al., 2002). Importantly, recombinant Vam7 can bypass this activation process if added to isolated vacuoles, and thus drives fusion (Thorngren et al., 2004).
HOPS is a key factor in the fusion reaction. On isolated vacuoles, HOPS binds both Ypt7 and SNAREs (Price et al., 2000; Laage and Ungermann, 2001). Purified HOPS is necessary to promote SNARE assembly and fusion of proteoliposomes in a reconstituted reaction (Mima et al., 2008). Moreover, HOPS can bridge membranes in a SNARE-dependent manner, and may thus trigger tethering (Stroupe et al., 2009), although efficient tethering requires just the Rab Ypt7, HOPS, and liposomes with vacuole lipid composition (Hickey and Wickner, 2010). Recent data also suggest that HOPS not only promotes trans-SNARE assembly, but also protects the trans-SNARE complex from Sec17/18-mediated disassembly (Hickey and Wickner, 2010; Xu et al., 2010). Due to the homology to Sec1/Munc18 proteins, the Vps33 subunit of the HOPS complex is likely a candidate to bind to the SNARE complex and not just Vam3 as previously suggested (Dulubova et al., 2001). Indeed, mutations in Vps33 affect the hemifusion-to-fusion transition, presumably by blocking the interaction with the SNARE complex (Pieren et al., 2010).
In an attempt to understand the interplay of HOPS and SNAREs during fusion processes at the vacuole, we analyzed the interaction sites within the HOPS complex for the vacuolar/late endosomal SNAREs. Within our analysis, we identified two binding sites for SNAREs within the HOPS complex and addressed the functional relevance using the vacuole fusion assay and in vivo analyses. Our data indicate that the HOPS complex primarily recognizes the assembled SNARE complex and not the individual SNAREs to promote fusion progression.
RESULTS
HOPS binds a minimal Q-SNARE complex
To address how HOPS interacts with the SNARE complex, we first established protocols to purify the individual SNAREs (outlined in Figure 1A). Assembly of the SNAREs, using GST-tagged Vam3 as a bait, was specific and robust with either Ykt6 or Nyv1 as R-SNARE (Figure 1B). Purified HOPS complex could then be captured by GST pull-down by using the assembled SNARE complex as a bait (Figure 1C). The interaction between both complexes was strong enough to allow for isolation of the HOPS complex from yeast cells that overexpressed HOPS (Figure 1D).
FIGURE 1:

Interaction of HOPS and the SNARE complex. (A, left) Schematic view of the domain structures of Vam3, Vti1, Vam7, Ykt6, and Nyv1. Scissors indicate the truncations used in this study. Membrane-anchoring domains/motifs are shown in black, the SNARE domain in gray. (A, right) Model of the HOPS complex. Subunits are arranged according to our previous findings (Ostrowicz et al., 2010). (B) Assembly of the SNARE complex. GST-tagged Vam3 or GST (10 μg each) were centrifuged for 30 min at 100,000 × g to remove aggregates and then preincubated with 12 μg of His-tagged SNAREs, which were pretreated similarly, for 2 h at 4°C, washed with buffer, and eluted in SDS-sample buffer. Eluted proteins were resolved on SDS–PAGE gels and stained with Coomassie. (C and D) Interaction of HOPS with assembled SNAREs. HOPS was added either as a purified complex (C) or from lysate of a strain overexpressing all HOPS subunits (D) to the SNARE complex or to GST. In (D), SNARE complexes with either Ykt6 or Nyv1 as the respective R-SNARE were used. Samples were incubated for 30 min with HOPS, then washed and eluted by boiling in SDS sample buffer. Proteins were analyzed as in (B).
We then asked whether any single SNARE is sufficient to bind HOPS, and detected binding to Vam7 (Figure 2A, lane 3), but no significant interaction with Vam3 (lane 7). These observations are consistent with previous studies, which identified the Vam7 PX domain as a major HOPS binding site (Stroupe et al., 2006). We could confirm this (Figure 2B), and used GST-tagged Vam7 SNARE domain, which lacks the PX domain (Vam7 SD) as the template to assemble the SNARE complex or just the Q-SNAREs, and detected equally efficient HOPS interaction (Figure 3A). This observation suggested that the assembled Q-SNAREs alone provide one major binding platform for HOPS. Binding to the SNARE complex (Figure 3B) or the Q-SNAREs (Figure 3C) was also detected if both Vam3 and Vam7 lacked their N-terminal domains, consistent with our binding studies. Our data thus reveal that, in addition to the PX domain of Vam7, a Q-SNARE complex consisting of Vti1 and the SNARE domains of Vam3 and Vam7 is sufficient to bind the HOPS complex.
FIGURE 2:

Defining the interface between individual SNAREs and the HOPS complex. (A) Interaction of the individual SNAREs with HOPS. GST-tagged SNAREs and GST (12 μg) were incubated with or without purified HOPS complex and analyzed as before. (B) Interaction of Vam7 PX and the HOPS complex. Vam7, its SNARE domain (Vam7 SD), the PX domain (Vam7 PX), or GST was incubated with HOPS, and samples were processed as in Figure 1C. To estimate binding efficiency, 25 and 50% of the eluate were applied to the gel. It should be noted that binding of HOPS was most efficient to GST-Vam7 and GST-Vam7PX despite their lower protein amount on the beads.
FIGURE 3:
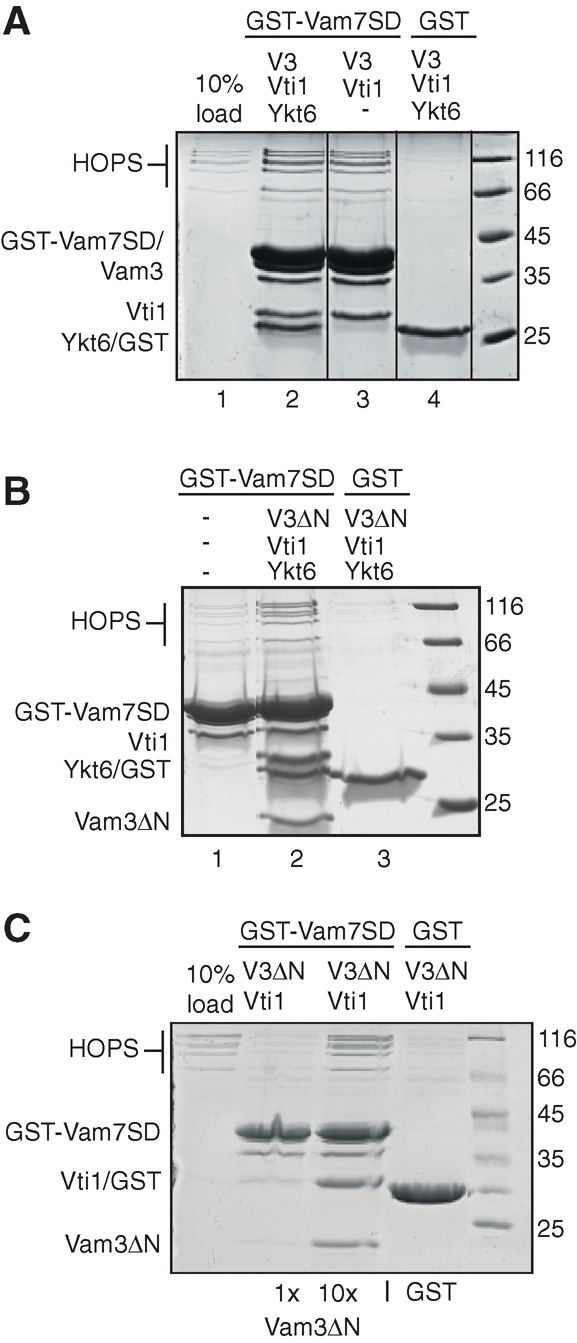
Identification of a minimal SNARE complex that binds HOPS. (A) Interaction of HOPS with the Q-SNARE complex. GST-Vam7 SD or GST was incubated with Vam3, Vti1, and, where indicated, with Ykt6. The assembled complexes were then incubated for 30 min with purified HOPS and were washed. The bound proteins were eluted by boiling in sample buffer, resolved by SDS–PAGE, and stained with Coomassie. (B) Binding of HOPS to a minimal SNARE complex. Assembly was as in (A), except that a truncated Vam3 lacking the Habc domain (V3ΔN) was used. (C) Interaction of HOPS with a minimal Q-SNARE complex. Vam3ΔN was added in two concentrations to the SNAREs Vti1, GST-Vam7SD, and HOPS. After an incubation of 30 min with GSH beads, the beads were washed and eluted by boiling. The reactions were analyzed as in (A).
HOPS subunits with SNARE binding properties
Within the HOPS complex, Vps33 is the most obvious interaction partner of the SNAREs (Pieren et al., 2010), although additional subunits might be involved in the binding to the entire Q-SNARE complex (reviewed by Bröcker et al., 2010). We overexpressed and purified individual HOPS subunits from yeast as before (Ostrowicz et al., 2010), and incubated them with purified GST-tagged Vam7, Vti1, Vam3, and Ykt6, as well as SNARE complexes of different composition (Figure 4). To follow assembly of a Q-SNARE complex lacking the N-terminal domains of Vam3 and Vam7 (Q-SNAREs truncated), we added two different concentrations of Vam3ΔN (1× and 10×) to the assembly reaction. Our data reveal a clear separation of SNARE binding within the HOPS complex. Neither Vps11, Vps41, nor Vps39 could interact with SNAREs (Figure 4A). These subunits are expected to occupy one end of the HOPS complex, and we showed before that, Vps41 and Vps39, which were isolated similarly, bind to Ypt7 (Ostrowicz et al., 2010) and highly curved membranes (Cabrera et al., 2010). We thus assume that all HOPS subunits retain their potential ability to interact with SNAREs if isolated as individual proteins. Vps16 and Vps18 could interact with Vam7 and the SNARE complex (Figure 4, B and C), whereas Vps33 bound only to SNARE complexes (Figure 4D). Our data thus indicate that the HOPS complex contains more than one SNARE binding site, which may facilitate recognition and subsequent fusion.
FIGURE 4:

Two distinct binding sites within the HOPS for vacuolar SNAREs. GST-tagged SNAREs were either assembled in a minimal complex (GST-Vam7 SD, Vti1, and either one- or 10-fold excess of Vam3ΔN), or provided as individual proteins. HOPS or its subunits were purified as overproduced and TAP-tagged proteins from yeast, using IgG sepharose and TEV cleavage (see Materials and Methods and Ostrowicz et al., 2010). The purified proteins were then added to the beads, incubated for 30 min at 4°C, washed four times, and eluted by boiling in SDS sample buffer. A fraction (10%) of the eluate was then analyzed by SDS–PAGE and Western blotting against the calmodulin peptide present in the remaining tag of the HOPS subunits. In (A), HOPS, Vps41, Vps39, and Vps11 are shown. In (B), purified Vps16, (C) Vps18, or (D) Vps33 were added. The GST-tagged proteins or complexes (80%) were visualized by SDS–PAGE and Coomassie staining, and are shown (bottom) for Vps33. Control SDS–PAGE gels for all other pull-down assays were similar (unpublished data).
Influence of SNARE truncations on fusion
To analyze the contribution of the different HOPS binding sites, we turned to the Vam7-dependent fusion assay. Vam7 (Thorngren et al., 2004) or just its SNARE domain (Fratti et al., 2007; Starai et al., 2008) can promote fusion even in the absence of ATP by assembling SNAREs of the vacuole membrane. Under these conditions, the HOPS function could be separated from the ATP-dependent priming reaction. As observed before (Laage and Ungermann, 2001; Pieren et al., 2010), vacuoles with Vam3 lacking the N-terminal domain (Vam3ΔN) fused inefficiently, most likely due to the impaired interaction of Vam3 and the SNARE complex with Vps33 (Laage and Ungermann, 2001). Both wild-type vacuoles and Vam3ΔN vacuoles were stimulated efficiently by Vam7 and to a lesser extent by the Vam7 SD, suggesting that HOPS binding to the assembled SNAREs can still support fusion (Figure 5A). Next we asked whether HOPS might sense the lack of one binding site. Previous studies indeed discovered that HOPS quenched fusion if the Vam7 SNARE domain was added to the fusion assay (Starai et al., 2008). As shown in Figure 5B, this can indeed be observed if HOPS is in excess. If HOPS is limiting, however, Vam7 SNARE domain nicely stimulates fusion in a dose-dependent manner (Figure 5B, black squares). Vam3 and Vam7 have similar concentrations on the vacuole (our observations), and Vam7 concentration in the fusion reaction is approximately 20 nM (Merz and Wickner, 2004). The amount of HOPS that stimulates fusion is thus at a similar concentration as the SNAREs present on the vacuole surface. This indicates that one HOPS complex per SNARE complex is sufficient to stimulate fusion. If the HOPS complex reaches higher concentrations, it can proofread the SNARE complex and thus inhibits Vam7 SNARE domain–triggered fusion. We therefore conclude that more than one HOPS complex per SNARE complex is necessary for proofreading.
FIGURE 5:

The two distinct HOPS binding sites can be distinguished in the vacuole fusion assay. (A) Analysis of wild-type and Vam3ΔN vacuoles in the fusion reaction. BJ and DKY vacuoles of the respective strains were purified and subjected to fusion as outlined in Materials and Methods. Reactions were carried out without ATP, but with increasing concentrations of Vam7 or Vam7 SNARE domain (Vam7 SD). Fusion reactions were developed after 90 min at 26°C. All fusion assays were carried out at least three times with similar results. (B) Cooperation of SNAREs and HOPS during fusion. Fusion of wild-type vacuoles was carried out in the presence of the indicated concentrations of Vam7 (0.7 and 0.176 μM) and Vam7 SNARE domain (SD; at 0.7, 0.35, and 0.175 μM) in the absence of ATP. HOPS was added in three different concentrations (35, 70, and 140 nM) to each Vam7-triggered fusion assay. (C–E) Competition of Q-SNAREs with HOPS. Q-SNAREs (GST-Vam7 SD, Vam3ΔN, and Vti1) were assembled on GSH beads, washed, and eluted with 15 mM reduced glutathione. Glutathione was removed from the complex in a SpinTrap G-25 column. One aliquot of the eluted SNAREs was incubated with HOPS for 2 h. A second aliquot was incubated with buffer as a control (D). HOPS, Q-SNAREs, or HOPS preincubated with Q-SNAREs (HOPS+Q-SNAREs) were then added at the indicated concentrations to wild-type vacuoles in the presence of Vam7 (0.7 μM) and absence of ATP (C). In (E), Vam7 SNARE domain (Vam7 SD) was added instead. All reactions were incubated for 90 min at 26°C and then developed.
To test whether the quenching of HOPS binding to the SNARE complex abolishes its activity, we purified the assembled minimal Q-SNARE complex, and preincubated it with equimolar amounts of HOPS (Figure 5D). We then analyzed the effect of the preincubated HOPS complex on vacuole fusion. To avoid a potential action of Sec18 and Sec17 to disassemble SNARE complexes, we used conditions without ATP and added suboptimal amounts of Vam7. Under these conditions, HOPS nicely stimulates fusion (Figure 5C, black triangles), whereas buffer was without effect (open triangles). Preincubation of HOPS with the minimal Q-SNARE complex, however, quenched this stimulatory effect of HOPS (black circles). The minimal fusion stimulation at low amounts of the HOPS-SNARE complex might be due to the presence of free GST-Vam7 SNARE domain, which was used to assemble the Q-SNARE complex (see also Figure 5D). The minimal Q-SNARE complex alone inhibited fusion in a dose-dependent manner (open circles). We then repeated the experiment, now using Vam7 SNARE domain as a stimulator in the fusion reactions, and observed the same effect (Figure 5E). The stimulation of fusion by HOPS was lost if HOPS was preincubated with the Q-SNARE complex. Thus, regardless of whether fusion was monitored in the presence of the complete Vam7 protein or just the Vam7 SNARE domain, the stimulation of fusion required HOPS with an unoccupied SNARE binding site. This site is most likely provided by Vps33.
In vivo function of the N-terminal domains of the vacuolar SNAREs
Our data are consistent with a major role of HOPS in recognizing the SNARE complex during fusion, whereas the binding to the N-terminal domains of Vam3 or Vam7 seems to have a regulatory function (Laage and Ungermann, 2001; Pieren et al., 2010). We therefore asked how the loss of the N-terminal domains of Vam7 and Vam3 would affect vacuole biogenesis, and initially focused on Vam7. To address this question, we searched for possibilities to maintain Vam7 localization despite loss of its PX domain. To localize Vam7 to vacuoles in the absence of its natural PX domain, we replaced its PX domain with the N-terminal 18 residues of Vac8 (Pal-Vam7ΔPX), a dually lipidated vacuolar fusion factor (Hou et al., 2009). Under these conditions, Vam7 was found on isolated vacuoles, and vacuole morphology was restored (Figure 6, A and B), indicating that the PX domain of Vam7 is a membrane-targeting domain that can be substituted by an artificial membrane anchor.
FIGURE 6:

Cooperation of the N-terminal domains of Vam3 and Vam7. (A) Vacuole morphology in cells lacking the Vam7 PX domain. Wild-type (wt) or vam7Δ cells with or without the indicated Vam7 variants were grown in YPD, stained with FM4–64, and visualized by fluorescence microscopy. In the bottom sample, Vam3 was truncated additionally in its N-terminal Habc domain. Scale bar = 10 μm. (B) Expression levels of Vam7 and Pal-Vam7. Proteins of wt, vam7Δ, Pal-Vam7ΔPX, and Pal-Vam7ΔPX Vam3ΔN cells were precipitated by trichloroacetic acid and separated by SDS–PAGE. Proteins were detected by Western blotting using specific antibodies. (C) In vivo fusion of cells with N-terminally truncated SNAREs. Wild-type cells and cells carrying Pal-Vam7ΔPX and Vam3ΔN were grown to logarithmic phase at 30°C, labeled with FM4–64, and then transferred to YPD with 0.4 M NaCl. After 10 min in salt, cells were reisolated and incubated in YPD for 1 h. Pictures were taken before salt stress (control), after 10 min in salt (0.4 M NaCl), and after 1 h in YPD (recovered). From 200 cells counted, the recovery rate of Pal-Vam7ΔPX Vam3ΔN cells was 95% of wild type. (D) Fusion of vacuoles with truncated SNAREs. Statistics of the fusion activity of wild-type vacuoles in comparison to mutants containing Vam3ΔN (V3ΔN), Pal-Vam7ΔPX (Pal-V7), or both are shown (n = 3). Optimal fusion conditions for all strains were used (0.7 μM Vam7 and 35 nM HOPS). (E) Expression level of selected vacuolar proteins. Purified vacuoles from the indicated strains were resolved on SDS–PAGE and blotted onto nitrocellulose membranes. Proteins were detected by Western blotting with the indicated antibodies. (F) Titration of Vam7 into the fusion reaction. Wild-type vacuoles and vacuoles with truncated Vam3 and Vam7 (V3ΔN Pal-V7) were incubated for 90 min at 26°C in the presence of 17.5 nM HOPS and increasing concentrations of full-length Vam7. ATP was added where indicated. Fusion reactions were then developed and analyzed as described in Materials and Methods. (G and H) Cells with N-terminally truncated Vam3 and Vam7 exhibit a cytokinesis defect. Cells were grown at 30°C in YPD and analyzed by microscopy using differential interference contrast (DIC) optics (G). Cells with defects in cytokinesis are indicated with arrowheads. Approximately 200 cells were counted for the statistical analysis (H). (I) Vacuole inheritance. The indicated cells were stained with 30 μM FM4–64 for 30 min, isolated, resuspended in fresh medium, and incubated for an additional 3 h. Vacuole staining in the daughter cell was scored as positive vacuole inheritance (n > 200 cells). (K) Localization of vacuole fusion proteins in mutant cells. The indicated proteins were N-terminally tagged with GFP and analyzed with fluorescence microscopy.
Studies of several syntaxins in yeast and mammalian cells indicate a critical role of the N-terminal Habc domain of Vam3 and the N-terminal peptide to interact with Sec1/Munc18 proteins (Rathore et al., 2010; Shen et al., 2010). Previously, we generated a Vam3 mutant lacking the entire Habc domain, which showed reduced binding to the HOPS complex on isolated vacuoles, but resulted in round vacuoles (Laage and Ungermann, 2001). We therefore asked whether removal of the N-terminal domains of Vam3 and Vam7 would result in an enhanced defect. Surprisingly, we still obtained round vacuoles in this mutant (Figure 6A, bottom). To ask whether these vacuoles are fusion competent in vivo, we subjected cells to osmotic salt stress, which induces fragmentation, and observed the vacuoles after removal of the salt (Figure 6C). Under these conditions, vacuoles from both strains showed round vacuoles after recovery with almost the same efficiency (∼90% of wild-type fusion). We noticed, however, that some vacuoles remained fragmented in the mutant cells (Figure 6C). To quantify a potential fusion defect, we turned to the in vitro vacuole fusion assay. As previously reported (Laage and Ungermann, 2001), vacuoles lacking the Vam3 Habc domain fused with approximately half the fusion activity of wild-type vacuoles (Figure 6D). Surprisingly, vacuoles from cells expressing the palmitoylated and truncated Vam7 construct were inactive, unless the wild-type Vam7 protein and HOPS were added to the fusion reaction (Figure 6, D and F). We then analyzed several fusion proteins of the vacuole membrane. Whereas Nyv1, Vti1, or Vac8 was present at wild-type concentrations, Vam3 and Pho8 were reduced. Pho8 activity, as determined by an in vitro activation assay, was equally reduced to 65% (for Pal-Vam7) and 55% (for the double mutant) of wild type. The combined deficiency could account for a 50% reduction in the fusion signal. Even under optimal conditions in the presence of purified HOPS and Vam7, however, low fusion was observed, and this reaction was not influenced by ATP addition (Figure 6F). Fusion was equally poor if Vam3 was also truncated (Figure 6, D and F). We consider it likely that the fusion defect caused by the additional truncation of Vam3 in the double mutant is underestimated, as differences at this low fusion activity may not be significant. We then analyzed whether the coexpression of Pal-Vam7ΔPX would affect vacuole fusion equally it were present on wild-type vacuoles, but we did not observe any effect on fusion (unpublished data). We therefore conclude that the artificial targeting of Vam7 SNARE domain, which restricts its natural mobility (Boeddinghaus et al., 2002), results in an in vitro fusion defect.
We finally asked whether the lack of the N-terminal domains and the restricted mobility of Vam7 would affect the cellular physiology. We indeed observed that cells lacking both the Vam3 and Vam7 N terminus have a cytokinesis defect (Figure 6, G and H). At least 20% of the cells were unable to separate properly during the cell cycle (Figure 6H). Some cells had multiple buds; many initiated the next cell division without completing the previous one (Figure 6G). Cells nevertheless did not have a significant vacuole inheritance phenotype (Figure 6I), and the HOPS subunits Vps41 and Vps39 were still localized as in wild-type cells (Figure 6K). The localization of Vps41 and Vps39 indicates that the defect may be a result of poor coordination of the tethering complex with SNAREs rather than its localization. In agreement with this proposal, no major defect was observed for trafficking pathways, such as the AP-3 pathway or the endosomal pathway, to the vacuole (unpublished data). For instance, Nyv1 and Yck3 localized to vacuoles in the double mutant (Figure 6, E and K). If the AP-3 pathway were defective, Yck3 had been found on the plasma membrane (Sun et al., 2004; Cabrera et al., 2010). Our data therefore suggest that the function of SNAREs at the vacuole requires the N-terminal domains of Vam3 and Vam7 and potentially their recognition by the HOPS complex in vivo.
DISCUSSION
In this article, we provide evidence for defined binding interfaces between the HOPS complex and the vacuolar SNAREs, which likely control fusion fidelity. Our data reveal that HOPS binds the SNAREs via its subunits Vps18, Vps16, and Vps33. Whereas Vps16 and Vps18 seem to recognize the N-terminal PX domain of Vam7, Vps33 interacts only with assembled SNAREs. Loss of the N-terminal domain of Vam7 does not block HOPS- stimulated fusion at low HOPS concentrations, whereas high concentrations of HOPS lead to a quenching of the fusion reaction, consistent with earlier findings (Starai et al., 2008). Our data are in agreement with a model in which HOPS recognizes the assembled SNAREs and their N-terminal domains during the fusion reaction.
How do the N-terminal domains of Vam7 and Vam3 cross-talk with HOPS during membrane fusion? The PX domain targets Vam7 to PI-3-P–positive domains (Cheever et al., 2001) but also interacts with the HOPS complex (Stroupe et al., 2006). Even if Vam7 is added exogenously to the vacuole fusion reaction, its binding to membranes still depends on PI-3-P (Boeddinghaus et al., 2002; Thorngren et al., 2004), suggesting that the PI-3-P binding site does not coincide with the HOPS binding site. Interestingly, we could target Vam7 to vacuoles without its PX domain and rescue vacuole morphology in vivo, although in vitro fusion was successful only if additional full-length Vam7 was added. This observation already indicated that the PX domain is not only a targeting device, but also directly participates in fusion as indicated by this study and earlier work (Stroupe et al., 2006; Fratti et al., 2007; Starai et al., 2008). In support of this idea, we observed that HOPS can also stimulate fusion if just the Vam7 SNARE domain is added (Figure 5B) but loses its ability to stimulate if its SNARE binding site was previously quenched. This stimulation was, however, restricted to HOPS concentrations that were in the range of the endogenous SNARE concentrations on isolated vacuoles (Merz and Wickner, 2004), whereas higher HOPS concentrations clearly block fusion (Starai et al., 2008). On the basis of these data we speculate that low concentrations of HOPS can stimulate fusion, because only one HOPS complex per SNARE complex is necessary for the stimulation. Higher HOPS concentrations may induce proofreading, because more than one HOPS complex will be needed to proofread the assembled SNARE complex and inhibit fusion.
Sec1/Munc18 (SM) proteins like Vps33 are thought to act on SNAREs in a dual recognition mode, which includes the N-terminal peptide and the SNARE domain (Bracher and Weissenhorn, 2002; Carpp et al., 2006; Togneri et al., 2006; Furgason et al., 2009). At least the neuronal syntaxin protein can also form a closed conformation through the interaction of the Habc and SNARE domain, which then interacts with the SM protein Munc18 (Südhof and Rothman, 2009). Our data would argue that HOPS recognizes the SNARE complex via Vps33, whereas the interaction of Vam7 PX domain with HOPS is independent of Vps33.
Vps16 and Vps18 provide an additional binding site for HOPS (Figure 4, B and C). The two subunits may support the Vps33-mediated SNARE interaction. This interaction could also be important to recruit HOPS to Vam7-positive membranes. Alternatively, it is possible that HOPS helps to target Vam7 to the right fusion site, as Vam7 is dynamically distributed between cytosol and membrane (Figure 7; Boeddinghaus et al., 2002). Thus, HOPS has extended its binding surface to SNAREs beyond the SM protein, which could be important to control fusion fidelity and efficiency (Xu et al., 2010).
FIGURE 7:

Model of the interplay of HOPS and SNAREs in the fusion cycle. Fusion is subdivided into five steps for simplicity: (1) Disassembly and activation of the assembled cis-SNAREs occurs in an ATP-dependent reaction. Under these conditions, Vam7 is released from the membrane. (2) Tethering of the vesicle with the target membrane. HOPS recruits Vam7 from the cytosol to the membrane and supports SNARE assembly. HOPS also protects the preassembled Q-SNAREs from disassembly by Sec18/Sec17. (3) Tethering of the vesicles or late endosome via the interaction of HOPS with Ypt7 and assembled SNAREs. (4) Fusion of the vesicle with the target membrane, which is mediated by trans-SNARE complexes. (5) Release of HOPS. SNAREs remain assembled and inactive as a cis-SNARE complex in the same membrane.
Our in vivo analysis of Vam7 agrees with this interpretation. We could replace the N-terminal PX domain of Vam7 by an N-terminal SH4-palmitoylation sequence, which is sufficient to target green fluorescent protein (GFP) to the vacuole (Hou et al., 2005). In the presence of Pal-Vam7ΔPX, we still observed round vacuoles that were able to fuse efficiently in vivo (Figure 6, A and C). Our data thus indicate that the main function of the PX domain is to target Vam7 to membranes, although it may not be the only one. As mentioned earlier in the text, Pal-Vam7ΔPX vacuoles required exogenously added full-length Vam7 and HOPS for optimal in vitro fusion (Figure 6, D and F). It is possible that the dynamics of Pal-Vam7ΔPX is sufficient in vivo, but not in vitro. The fusion protein is also not a preferred binding partner of the vacuolar SNAREs as additionally expressed Pal-Vam7ΔPX did not affect wild-type vacuole fusion (unpublished data).
Interestingly, loss of the N-terminal domain of Vam3 in addition to Vam7'7 PX domain resulted in a cytokinesis defect (Figure 6, G and H). Neither N-terminal truncation alone affects cell physiology to this extent, although Vam3 lacking the N-terminal domain bound poorly to the HOPS complex if isolated from cell lysates (Laage and Ungermann, 2001). Neither vacuole inheritance nor localization of the HOPS-specific subunits Vps41 and Vps39 was affected (Figure 6, I and K). Nevertheless, the cross-talk between HOPS and SNAREs may be required for efficient fusion, which may indirectly affect cytokinesis. Interestingly, both vacuole SNAREs have been linked to several components of the septin ring by genetic interactions (Costanzo et al., 2010).
Our data contribute to the understanding of HOPS as a multifunctional tethering complex that binds the Rab Ypt7 on one side, primarily via Vps41 (Brett et al., 2008; Ostrowicz et al., 2010), and SNAREs on the opposite side, where Vps16 and Vps33 are likely located (Figure 7) (Ostrowicz et al., 2010). It is thus conceivable that HOPS binds the SNARE complex, potentially together with the Vam7 PX domain. Considering the strong stimulation of fusion by HOPS and its proofreading activity (Stroupe et al., 2006; Mima et al., 2008; Starai et al., 2008; Ostrowicz et al., 2010; Xu et al., 2010), a model emerges in which HOPS combines tethering and fusion-promoting activities (Figure 7). For tethering, HOPS may bind both SNAREs and the Rab Ypt7 (Stroupe et al., 2009), even though a recent study suggests that the primary tethering activity of HOPS requires only Ypt7 and lipids (Hickey and Wickner, 2010). HOPS binding to SNAREs may then be important to promote and control SNARE assembly. A dissection of the molecular interfaces combined with functional in vivo and in vitro assays will be an important next step.
MATERIALS AND METHODS
Yeast strains and molecular biology
Yeast strains used are listed in Supplemental Table S1. Yeast strains were cultured in yeast peptone dextrose (YPD) medium, except the strains for the overexpression of HOPS or HOPS subunits, which were grown in galactose-containing yeast peptone galactose (YPG) medium. Deletions and tagging of genes with GFP or tandem affinity purification (TAP) tags were performed using homologous recombination of PCR-amplified fragments (Puig et al., 2001; Janke et al., 2004). For the expression of Pal-VAM7ΔPX under the VAM3 promoter, the PX domain was replaced by the sequence coding for the first 18 amino acid residues of VAC8, and the corresponding PCR fragment was cloned into a pRS406 vector containing the VAM3 promoter (see Table 1). The resulting plasmid was transformed into vam7Δ strains.
TABLE 1:
Vectors used in this study.
| Name | Proteinresidues | Vector | Insertion sites | Source |
|---|---|---|---|---|
| pET28a-VAM7 | Vam71–316 | pET28a | BamHI and EcoRI | provided by D. Langosch (TU Munich) |
| pET28a-VAM3ΔTMD | Vam31–264 | pET28a | BamHI and HindIII | This study |
| pET28a-VAM3ΔNΔTMD | Vam3145–264 | pET28a | BamHI and XhoI | This study |
| pET28a-VTI1ΔTMD | Vti1aa1–189 | pET28a | BamHI and XhoI | This study |
| pETHIS-YKT6 | Ykt61–200 | pET-HIS | BamHI and HindIII | Meiringer et al., 2008 |
| pGEX-KT-VAM7 | Vam71–316 | pGEX-KT | BamHI and EcoRI | Merz et al., 2004 |
| pGEX-KT-VAM7ΔPX | Vam7126–316 | pGEX-KT | BamHI and EcoRI | This study |
| pGEX-2tk-VAM7SD | Vam7190–316 | pGEX-2tk | BamHI and EcoRI | This study |
| pGEX-KT-VAM7PX | Vam71–129 | pGEX-KT | BamHI and EcoRI | Merz et al., 2004 |
| pGEX-KT-VAM3ΔTMD | Vam31–260 | pGEX-2tk | BamHI and EcoRI | Dulubova et al., 2001 |
| pGEX-2tk-NYV1ΔTMD | Nyv11–260 | pGEX-2tk | BamHI and XhoI | This study |
Plasmids and protein purification
The soluble part of Vti1, Vam7, Vam3, and Ykt6 were amplified by PCR from Saccharomyces cerevisiae genomic DNA with the help of the Pfu polymerase, and cloned into pET28a, pETHIS, pET32H, or pGEX-2tk expression vectors (see Table 1). The plasmids were transformed into Escherichia coli BL21 (DE3) Rosetta cells and induced at OD600 ∼ 0.8 with 0.75 mM isopropyl-b-d-thiogalactopyranoside (IPTG) overnight at 16°C. After harvesting, cells were lysed in a multiflex in buffer containing 50 mM HEPES, 150 mM NaCl, 1 mM phenylmethylsulfonyl fluoride (PMSF) and 0.5-fold PIC (protease inhibitor cocktail; 1× = 0.1 mg/ml of leupeptin, 1 mM o-phenanthroline, 0.5 mg/ml of pepstatin A, 0.1 mM Pefabloc), pH 7.5. Lysates were then centrifuged for 15 min at 30,000 × g. The supernatants were incubated for 1 h either with glutathione sepharose fast flow beads (GE Healthcare, Munich, Germany) for GST-tagged proteins, or Ni-NTA agarose (Qiagen, Hildesheim, Germany) for His-tagged proteins. Beads were washed with 15 ml of buffer containing 15 mM imidazole but lacking PMSF and PIC. His-tagged proteins were eluted from beads with buffer containing 0.3 M imidazole. GST-tagged proteins were eluted with buffer containing 15 mM reduced glutathione. For storage at –80°C, the buffer was exchanged to storage buffer containing 10% glycerol using a PD-10 column.
Tandem affinity purification (TAP)
Culture (4 l) was grown at 30°C to OD600 ∼ 4, and cells were harvested by centrifugation (10 min, 3000 × g at 4°C). Cells were mechanically disrupted with glass beads, using lysis buffer (50 mM HEPES/NaOH, pH 7.4, 300 mM NaCl, 0.15% NP-40 [Igepal CA-630; Sigma-Aldrich, St. Louis, MO], 1.5 mM MgCl2, 1× FY protease inhibitor mix [Serva, Heidelberg, Germany], 0.5 mM PMSF, and 1 mM dithiothreitol [DTT]). Lysates were centrifuged for 1.5 h at 100,000 × g, and supernatants were incubated with IgG sepharose beads for 1 h at 4°C on a nutator. Beads were then isolated by centrifugation (800 × g, 5 min, 4°C) and washed with 15 ml of lysis buffer containing 0.5 mM DTT. TAP-tagged proteins were eluted by tobacco etch virus (TEV) cleavage (on a turning wheel; 16°C, 1 h), and TEV eluates were analyzed by SDS–PAGE. If proteins were used in the fusion assay, NP-40 was excluded from the purification procedure.
SNARE assembly
All purified SNAREs were stored at –80°C. After thawing, proteins were centrifuged for 30 min at 100,000 × g to remove any aggregates. For SNARE assembly, 10 μg of the GST-tagged bait protein were preincubated with 12 μg of His-tagged SNAREs for 2 h at 4°C on a nutator. GSH beads were washed once with TAP buffer containing 150 mM NaCl and 3.5% bovine serum albumin and four times with the same buffer lacking bovine serum albumin. The sample volume was increased to 200 μl by adding 15 μl of GSH beads and TAP buffer with 150 mM NaCl and 1 mM DTT, and the sample was incubated at 4°C on a nutator. The beads were then washed four times with 500 μl of buffer, and were eluted by boiling in sample buffer. To examine the interaction of HOPS or HOPS subunits with assembled SNAREs, the TAP-purified proteins were added additionally to the single SNAREs during the preincubation.
Vacuole fusion
Vacuoles from the two tester strains, BJ3505 and DKY6281 (see Table 2), were obtained by diethylaminoethyl cellulose (DEAE)-dextran lysis and Ficoll density gradient flotation (Cabrera and Ungermann, 2008). Isolated vacuoles were diluted to 0.3 mg/ml in PS (10 mM PIPES/KOH, pH 6.8, 200 mM sorbitol) buffer containing 0.1× PIC (protease inhibitor cocktail). Vacuole fusion was measured by using a biochemical complementation assay as described previously (Haas, 1995). Standard fusion reactions containing 3 μg each of BJ and DKY vacuoles were incubated for 90 min at 26°C in 30 μl of PS buffer supplemented with salts (5 mM MgCl2 and 125 mM KCl), 10 μM CoA, 0.01 μg His-Sec18, with or without an ATP-regenerating system. p-nitrophenyl phosphate, a colorimetric substrate of alkaline phosphatase, was added to detergent-solubilized reactions, and the alkaline phosphatase activity was determined by measuring the absorbance of the generated nitrophenol at 400 nm.
TABLE 2:
Strains used in this study.
| Strain | Genotype | Reference |
|---|---|---|
| BJ3505 | MATa pep4Δ::HIS3 prb1-Δ1.6R lys2–208 trp1Δ101 ura3–52 gal2 | Haas, 1995 |
| DKY6281 | MATalpha leu2–3 leu2–112 ura3–52 his3-Δ200 trp1-Δ101 lys2–801 suc2-Δ9 PHO8::TRP1 | Haas, 1995 |
| BY4732 | MATa his3Δ200 leu2Δ0 met15Δ0 trp1Δ63 ura3Δ0 | Brachmann et al., 1998 |
| BY4727 | MATalpha his3Δ200 leu2Δ0 lys2Δ0 met15Δ0 trp1Δ63 ura3Δ0 | Brachmann et al., 1998 |
| BY 4720 | MATalpha lys2Δ0 trp1Δ63 ura3Δ0 | Brachmann et al., 1998 |
| CUY270 | BJ3505 vam7Δ::TRP1 | This study |
| CUY291 | BJ3505 VAM3::VAM3ΔN | Laage et al., 2001 |
| CUY292 | DKY6281 VAM3::VAM3ΔN | Laage et al., 2001 |
| CUY2152 | BY 4732 VPS41pr::TRP1-GAL1pr VPS41::TAP-URA3 | Ostrowicz et al., 2010 |
| CUY2675 | BY4732xBY4727 VAM41::TRP1-GAL1pr VAM41::TAP-URA3 VAM39::KanMX-GAL1pr VPS33::HIS3-GAL1pr VPS11::HIS3-GAL1Pr VPS16::natNT2-GAL1Pr VPS18::kanMX-GAL1Pr-3HA VPS33::HIS3-GAL1pr | Ostrowicz et al., 2010 |
| CUY2839 | BY4732 VPS33pr::TRP1-GAL1pr VPS33::TAPUra3 | Ostrowicz et al., 2010 |
| CUY2858 | BY4732 VPS16::KanMX-Gal1Pr VPS16::TAP-Ura3 | This study |
| CUY3210 | BY 4732 VPS18pr::KanMX-GAL1Pr VPS18::TAP-URA3 | Ostrowicz et al., 2010 |
| CUY3588 | BY4732xBY4720 VAM39::KanMX- Gal1Pr VAM39::URA3-TAP | This study |
| CUY4291 | BY4727 VPS11pr::HIS3-GAL1pr VPS11::TAP-URA3 | Ostrowicz et al., 2010 |
| CUY5506 | BJ3505 vam7Δ::TRP1 URA::pRS406-VAM3pr-Pal-VAM7ΔPX | This study |
| CUY5531 | BJ3505 vam7Δ::TRP1 VAM3::NOP1pr-VAM3ΔN-kanMX | This study |
| CUY5579 | BJ3505 vam7::TRP1 VAM3::NOP1pr-VAM3ΔN-kanMX URA::pRS406-VAM3pr-Pal-VAM7ΔPX | This study |
| CUY5642 | BJ3503 VPS39::natNT2-ADHpr-GFP | This study |
| CUY5643 | BJ3503 VPS41::natNT2-ADHpr-GFP | This study |
| CUY5644 | BJ3503 YCK3::natNT2-ADHpr-GFP | This study |
| CUY5645 | BJ3503 vam7Δ::TRP1 VAM3::NOP1pr-VAM3ΔN-kanMX URA::pRS406-VAM3pr-Pal-VAM7ΔPX VPS39::natNT2-ADHpr-GFP | This study |
| CUY5646 | BJ3503 vam7Δ::TRP1 VAM3::NOP1pr-VAM3ΔN-kanMX URA::pRS406-VAM3pr-Pal-VAM7 VPS41::natNT2-ADHpr-GFP | This study |
| CUY5647 | BJ3503 vam7Δ::TRP1 VAM3::NOP1pr-VAM3ΔN-kanMX URA::pRS406-VAM3pr-Pal-VAM7 YCK3::natNT2-ADHpr-GFP | This study |
| CUY5674 | DKY6281 vam7Δ::natNT2 ura3::pRS406-Pal-Vam7ΔPX | This study |
| CUY5693 | DKY6281 vam7Δ::hphNT1 VAM3::NOP1pr-VAM3ΔN-kanMX URA::pRS406- VAM3pr-Pal-VAM7 YCK3::natNT2-ADHpr-GFP | This study |
| CUY6385 | BJ3503 URA::pRS406- VAM3pr-Pal-Vam7ΔPX | This study |
| CUY6386 | DKY6281 URA::pRS406- VAM3pr-Pal-Vam7ΔPX | This study |
Microscopy
Cells (see Table 2) were grown to mid-logarithmic phase in YPD, harvested by centrifugation, washed once with phosphate-buffered saline, and analyzed by using fluorescence microscopy at room temperature. For labeling of vacuoles with the lipophilic dye FM4–64 (Invitrogen, Carlsbad, CA), cells were grown to an OD600 of 0.5 and incubated in YPD medium containing 30 μM FM4–64 for 30 min. After being washed with YPD medium, cells were incubated in the same medium without dye for 1 h. Cells were imaged using an epifluorescence microscope (DM5500B; Leica, Wetzlar, Germany) equipped with differential interference contrast optics, an HCX PL APO 100× 1.46 or 1.4–0.7 oil objective lens, a camera (DFC 350 FX; Leica), and filter sets for GFP (excitation D480/30, emission D535/40, and beamsplitter 505 dichroic long pass) and FM4-64 (excitation D535/50, emission E590LPv2, and beamsplitter 565 dichroic long pass; Chroma Technology, Bellows Falls, VT). Images were captured using image acquisition and analysis software (LAS-AF; Leica), and brightness and contrast were adjusted with Photoshop CS3 (Adobe).
Acknowledgments
We thank Christine Boeddinghaus for the generation of some of the Vam7 truncation constructs and all coworkers for discussions throughout this study. This work has been supported by the Deutsche Forschungsgemeinschaft (DFG) (UN111/4-2), the Sonderforschungsbereich (SFB) 431 and 944, and the Hans-Mühlenhoff Foundation (to C.U.).
Abbreviations used:
- AP-3
adaptor protein complex 3
- DTT
dithiothreitol
- GFP
green fluorescent protein
- HOPS
homotypic fusion and protein sorting
- PI-3-P
phosphatidylinositol-3-phosphate
- PMSF
phenylmethylsulfonyl fluoride
- SNARE
soluble N-ethylmaleimide-sensitive factor (NSF) attachment protein receptor
- TAP
tandem affinity purification
- SM
Sec1/Munc18
- TEV
tobacco etch virus
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E11-02-0104) on May 25, 2011.
REFERENCES
- Boeddinghaus C, Merz AJ, Laage R, Ungermann C. A cycle of Vam7p release from and PtdIns 3-P-dependent rebinding to the yeast vacuole is required for homotypic vacuole fusion. J Cell Biol. 2002;157:79–90. doi: 10.1083/jcb.200112098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers K, Stevens TH. Protein transport from the late Golgi to the vacuole in the yeast Saccharomyces cerevisiae. Biochim Biophys Acta. 2005;1744:438–454. doi: 10.1016/j.bbamcr.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Bracher A, Weissenhorn W. Structural basis for the Golgi membrane recruitment of Sly1p by Sed5p. EMBO J. 2002;21:6114–6124. doi: 10.1093/emboj/cdf608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, Boeke JD. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14:115–132. doi: 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Brett CL, Plemel RL, Lobinger BT, Vignali M, Fields S, Merz AJ. Efficient termination of vacuolar Rab GTPase signaling requires coordinated action by a GAP and a protein kinase. J Cell Biol. 2008;182:1141–1151. doi: 10.1083/jcb.200801001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bröcker C, Engelbrecht-Vandré S, Ungermann C. Multisubunit tethering complexes and their role in membrane fusion. Curr Biol. 2010;20:R943–R952. doi: 10.1016/j.cub.2010.09.015. [DOI] [PubMed] [Google Scholar]
- Cabrera M, Ungermann C. Purification and in vitro analysis of yeast vacuoles. Methods Enzymol. 2008;451:177–196. doi: 10.1016/S0076-6879(08)03213-8. [DOI] [PubMed] [Google Scholar]
- Cabrera M, et al. Phosphorylation of a membrane curvature-sensing motif switches function of the HOPS subunit Vps41 in membrane tethering. J Cell Biol. 2010;191:845–859. doi: 10.1083/jcb.201004092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpp LN, Ciufo LF, Shanks SG, Boyd A, Bryant NJ. The Sec1p/Munc18 protein Vps45p binds its cognate SNARE proteins via two distinct modes. J Cell Biol. 2006;173:927–936. doi: 10.1083/jcb.200512024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheever ML, Sato TK, de Beer T, Kutateladze TG, Emr SD, Overduin M. Phox domain interaction with PtdIns(3)P targets the Vam7 t-SNARE to vacuole membranes. Nat Cell Biol. 2001;3:613–618. doi: 10.1038/35083000. [DOI] [PubMed] [Google Scholar]
- Costanzo M, et al. The genetic landscape of a cell. Science. 2010;327:425–431. doi: 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilcher M, Kohler B, von Mollard GF. Genetic interactions with the yeast Q-SNARE VTI1 reveal novel functions for the R-SNARE YKT6. J Biol Chem. 2001;276:34537–34544. doi: 10.1074/jbc.M101551200. [DOI] [PubMed] [Google Scholar]
- Dulubova I, Yamaguchi T, Wang Y, Südhof TC, Rizo J. Vam3p structure reveals conserved and divergent properties of syntaxins. Nat Struct Biol. 2001;8:258–264. doi: 10.1038/85012. [DOI] [PubMed] [Google Scholar]
- Fratti RA, Collins KM, Hickey CM, Wickner W. Stringent 3Q.1R composition of the SNARE 0-layer can be bypassed for fusion by compensatory SNARE mutation or by lipid bilayer modification. J Biol Chem. 2007;282:14861–14867. doi: 10.1074/jbc.M700971200. [DOI] [PubMed] [Google Scholar]
- Furgason ML, MacDonald C, Shanks SG, Ryder SP, Bryant NJ, Munson M. The N-terminal peptide of the syntaxin Tlg2p modulates binding of its closed conformation to Vps45p. Proc Natl Acad Sci USA. 2009;106:14303–14308. doi: 10.1073/pnas.0902976106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas A. A quantitative assay to measure homotypic vacuole fusion in vitro. Methods Cell Sci. 1995;17:283–294. [Google Scholar]
- Hickey CM, Wickner W. HOPS initiates vacuole docking by tethering membranes before trans-SNARE complex assembly. Mol Biol Cell. 2010;21:2297–2305. doi: 10.1091/mbc.E10-01-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou H, John Peter AT, Meiringer C, Subramanian K, Ungermann C. Analysis of DHHC acyltransferases implies overlapping substrate specificity and a two-step reaction mechanism. Traffic. 2009;10:1061–1073. doi: 10.1111/j.1600-0854.2009.00925.x. [DOI] [PubMed] [Google Scholar]
- Hou H, Subramanian K, LaGrassa TJ, Markgraf D, Dietrich LE, Urban J, Decker N, Ungermann C. The DHHC protein Pfa3 affects vacuole-associated palmitoylation of the fusion factor Vac8. Proc Natl Acad Sci USA. 2005;102:17366–17371. doi: 10.1073/pnas.0508885102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughson FM, Reinisch KM. Structure and mechanism in membrane trafficking. Curr Opin Cell Biol. 2010;22:454–460. doi: 10.1016/j.ceb.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley JH, Boura E, Carlson LA, Rozycki B. Membrane budding. Cell. 2010;143:875–887. doi: 10.1016/j.cell.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn R, Scheller RH. SNAREs – engines for membrane fusion. Nat Rev Mol Cell Biol. 2006;7:631–643. doi: 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- Janke C, et al. A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast. 2004;21:947–962. doi: 10.1002/yea.1142. [DOI] [PubMed] [Google Scholar]
- Laage R, Ungermann C. The N-terminal domain of the t-SNARE Vam3p coordinates priming and docking in yeast vacuole fusion. Mol Biol Cell. 2001;12:3375–3385. doi: 10.1091/mbc.12.11.3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer A, Wickner W, Haas A. Sec18p (NSF)-driven release of Sec17p (alpha-SNAP) can precede docking and fusion of yeast vacuoles. Cell. 1996;85:83–94. doi: 10.1016/s0092-8674(00)81084-3. [DOI] [PubMed] [Google Scholar]
- Merz AJ, Wickner WT. Resolution of organelle docking and fusion kinetics in a cell-free assay. Proc Natl Acad Sci USA. 2004;101:11548–11553. doi: 10.1073/pnas.0404583101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mima J, Hickey CM, Xu H, Jun Y, Wickner W. Reconstituted membrane fusion requires regulatory lipids, SNAREs and synergistic SNARE chaperones. EMBO J. 2008;27:2031–2042. doi: 10.1038/emboj.2008.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrowicz CW, Bröcker C, Ahnert F, Nordmann M, Lachmann J, Peplowska K, Perz A, Auffarth K, Engelbrecht-Vandré S, Ungermann C. Defined subunit arrangement and rab interactions are required for functionality of the HOPS tethering complex. Traffic. 2010;11:1334–1346. doi: 10.1111/j.1600-0854.2010.01097.x. [DOI] [PubMed] [Google Scholar]
- Pieren M, Schmidt A, Mayer A. The SM protein Vps33 and the t-SNARE H(abc) domain promote fusion pore opening. Nat Struct Mol Biol. 2010;17:710–717. doi: 10.1038/nsmb.1809. [DOI] [PubMed] [Google Scholar]
- Price A, Seals D, Wickner W, Ungermann C. The docking stage of yeast vacuole fusion requires the transfer of proteins from a cis-SNARE complex to a Rab/Ypt protein. J Cell Biol. 2000;148:1231–1238. doi: 10.1083/jcb.148.6.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig O, Caspary F, Rigaut G, Rutz B, Bouveret E, Bragado-Nilsson E, Wilm M, Seraphin B. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods. 2001;24:218–229. doi: 10.1006/meth.2001.1183. [DOI] [PubMed] [Google Scholar]
- Rathore SS, Bend EG, Yu H, Hammarlund M, Jorgensen EM, Shen J. Syntaxin N-terminal peptide motif is an initiation factor for the assembly of the SNARE-Sec1/Munc18 membrane fusion complex. Proc Natl Acad Sci USA. 2010;107:22399–22406. doi: 10.1073/pnas.1012997108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Y, Yip CK, Tripathi A, Huie D, Jeffre PD, Walz T, Hughson FM. A structure-based mechanism for vesicle capture by the multisubunit tethering complex Dsl1. Cell. 2009;139:1119–1129. doi: 10.1016/j.cell.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Rathore SS, Khandan L, Rothman JE. SNARE bundle and syntaxin N-peptide constitute a minimal complement for Munc18–1 activation of membrane fusion. J Cell Biol. 2010;190:55–63. doi: 10.1083/jcb.201003148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivaram MV, Saporita JA, Furgason ML, Boettcher AJ, Munson M. Dimerization of the exocyst protein Sec6p and its interaction with the t-SNARE Sec9p. Biochemistry. 2005;44:6302–6311. doi: 10.1021/bi048008z. [DOI] [PubMed] [Google Scholar]
- Starai VJ, Hickey CM, Wickner W. HOPS proofreads the trans-SNARE complex for yeast vacuole fusion. Mol Biol Cell. 2008;19:2500–2508. doi: 10.1091/mbc.E08-01-0077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroupe C, Collins KM, Fratti RA, Wickner W. Purification of active HOPS complex reveals its affinities for phosphoinositides and the SNARE Vam7p. EMBO J. 2006;25:1579–1589. doi: 10.1038/sj.emboj.7601051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroupe C, Hickey CM, Mima J, Burfeind AS, Wickner W. Minimal membrane docking requirements revealed by reconstitution of Rab GTPase-dependent membrane fusion from purified components. Proc Natl Acad Sci USA. 2009;106:17626–17633. doi: 10.1073/pnas.0903801106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC, Rothman JE. Membrane fusion: grappling with SNARE and SM proteins. Science. 2009;323:474–477. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun B, Chen L, Cao W, Roth AF, Davis NG. The yeast casein kinase Yck3p is palmitoylated, then sorted to the vacuolar membrane with AP-3-dependent recognition of a YXXPhi adaptin sorting signal. Mol Biol Cell. 2004;15:1397–1406. doi: 10.1091/mbc.E03-09-0682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorngren N, Collins KM, Fratti RA, Wickner W, Merz AJ. A soluble SNARE drives rapid docking, bypassing ATP and Sec17/18p for vacuole fusion. EMBO J. 2004;23:2765–2776. doi: 10.1038/sj.emboj.7600286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togneri J, Cheng YS, Munson M, Hughson FM, Carr CM. Specific SNARE complex binding mode of the Sec1/Munc-18 protein, Sec1p. Proc Natl Acad Sci USA. 2006;103:17730–17735. doi: 10.1073/pnas.0605448103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungermann C, Nichols BJ, Pelham HR, Wickner W. A vacuolar v-t-SNARE complex, the predominant form in vivo and on isolated vacuoles, is disassembled and activated for docking and fusion. J Cell Biol. 1998;140:61–69. doi: 10.1083/jcb.140.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungermann C, von Mollard GF, Jensen ON, Margolis N, Stevens TH, Wickner W. Three v-SNAREs and two t-SNAREs, present in a pentameric cis-SNARE complex on isolated vacuoles, are essential for homotypic fusion. J Cell Biol. 1999;145:1435–1442. doi: 10.1083/jcb.145.7.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickner W. Membrane fusion: five lipids, four SNAREs, three chaperones, two nucleotides, and a Rab, all dancing in a ring on yeast vacuoles. Annu Rev Cell Dev Biol. 2010;26:115–136. doi: 10.1146/annurev-cellbio-100109-104131. [DOI] [PubMed] [Google Scholar]
- Xu H, Jun Y, Thompson J, Yates J, Wickner W. HOPS prevents the disassembly of trans-SNARE complexes by Sec17p/Sec18p during membrane fusion. EMBO J. 2010;29:1948–1960. doi: 10.1038/emboj.2010.97. [DOI] [PMC free article] [PubMed] [Google Scholar]