

Abstract
Background
Only 29 cases of constitutional 9q22 deletions have been published and all have been sporadic. Most associate with Gorlin syndrome or nevoid basal cell carcinoma syndrome (NBCCS, MIM #109400) due to haploinsufficiency of the PTCH1 gene (MIM *601309).
Methods and Results
We report two mentally retarded female siblings and their cognitively normal father, all carrying a similar 5.3 Mb microdeletion at 9q22.2q22.32, detected by array CGH (244 K). The deletion does not involve the PTCH1 gene, but instead 30 other gene,s including the ROR2 gene (MIM *602337) which causing both brachydactyly type 1 (MIM #113000) and Robinow syndrome (MIM #268310), and the immunologically active SYK gene (MIM *600085). The deletion in the father was de novo and FISH analysis of blood lymphocytes did not suggest mosaicism. All three patients share similar mild dysmorphic features with downslanting palpebral fissures, narrow, high bridged nose with small nares, long, deeply grooved philtrum, ears with broad helix and uplifted lobuli, and small toenails. All have significant dysarthria and suffer from continuous middle ear and upper respiratory infections. The father also has a funnel chest and unilateral hypoplastic kidney but the daughters have no malformations.
Conclusions
This is the first report of a familial constitutional 9q22 deletion and the first deletion studied by array-CGH which does not involve the PTCH1 gene. The phenotype and penetrance are variable and the deletion found in the cognitively normal normal father poses a challenge in genetic counseling.
Background
The first constitutional deletion of 9q22 was published in 1978 by Turleau [1] and since then only 29 patients have been reported, including two terminated fetuses. All have been sporadic (Table 1). In recent years, array comparative genomic hybridization (array CGH) has enabled more detailed reports on the genetic basis of 9q22 deletions. Reported deletion sizes vary from 2.3 Mb to more than 18 Mb and no recurrent breakpoints have been observed (Table 1). While most reported deletions are sporadic, three balanced parental chromosomal rearrangements involving 9q have been detected and two of these have likely predisposed to the deletion in the descendants [2,3] while one deletion lies outside the parental rearrangement [4]. Parental origin of the deleted chromosome has been traced in eleven cases of which eight were paternal [2 (patient 3), 3 (patient 1), 5-8,] and three maternal [3 (patient 2), 9, 10] (Table 1). Characteristic to all these deletions are non-recurrent breakpoints leading to variable gene composition and an inconsistent phenotype. Most of them, however, span the PTCH1 gene (MIM *601309) and associate with Gorlin syndrome or nevoid basal cell carcinoma (NBCCS, MIM #109400) due to haploinsufficiency of PTCH1.
Table 1.
Summary of clinical and molecular features of previously reported patients with constitutional 9q22 deletions and the present ones.
| Reference | Patient's age and gender | Postnatal height | Postnatal OFC | CNS features | Malformations | Dysmorphic features | Clinical Gorlin syndrome | Method of detection | Locus of the deletion | Parental origin and/or Parental chromosomal rearrangement | Size of deletion | PTCH1 | ROR2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| [28] | 1y11m male | + 3,8SD | > p 90 | global delay hypotonia no falx calcification |
right hydronephrosis left multicystic kidney left hand preaxial polydactyly |
hypertelorism frontal bossing epicanthi low nasal bridge low-set ears auricular pits long philtrum high palate short, webbed neck sacral dimple |
with reservations to young age | aCGH (Agilent 180 chip) and FISH | 9q22.3 de novo | 2,44 Mb | deleted | nr | |
| [14] | 2 y 3 m male | > p97 | p75-97 | moderate MR, wide cranial sutures, open posterior fontanelle |
submucous cleft palate, pectus excavatum |
epicanthic folds, wide and short neck, low nuchal hairline, wide nasal bridge, low-set and posteriorly rotated ears, micrognathia, widely spaced nipples, small teeth, deep plantar grooves |
with reservations to young age | aCGH at 1 Mb resolution | 9q22.32q31.1 de novo | 6,54-8,12 Mb | deleted | nr | |
| [5] | 3 y 9 m female | +2,2SD | +3,5SD | normal development, hypotonia, spasticity, | cleft lip-palate, pigmented cyst on shoulder (ectopic meninx) ASD |
frontal bossing epicanthic folds, broad eyebrows, synophrys, c-a-l spot on legs and arm |
yes palmar & plantar pits |
aCGH (Agilent 105A chip) | 9q22.32 de novo | paternal | 2.3 Mb | deleted | nr |
| [26, patient 1] | 12 y male | -1,1SD | +1,7SD | moderate MR, seizures/epilepsy |
epicanthi mouth small upper lip thin strabismus |
yes falx calcification, frontal ganglioglioma, rib anomalies, odontogenic ceratocysts, palmar and plantar pits |
FISH with BAC clones | 9q21.33q22.33 de novo | 15,33-16,04 Mb | deleted | nr | ||
| [26, patient 2] | 23 y male | +1,9SD | +2,6SD | severe MR, seizures, trigonocephaly, craniosynostosis, |
cleft lip-palate retinal detachement, cataract, glaucoma double urethra |
hypertelorism | yes odonto-genic ceratocysts, thyroid adenocarcinoma |
FISH with BAC clones | 9q21.33q31.1 de novo | 18,08-18,54 Mb | deleted | nr | |
| [27, restudy of patient 1 originally presented in 33] | 50 y female | normal | macrocephaly | mild MR, hypotonia, |
"rib and bone anomalies" kidney problems, one eye blind |
frontal bossing, palpebral fissures slant down, epicanthi, maxillary prognatism,, dense eyebrows, dental anomalies, delayed dental eruption |
yes basocellular carcinomas, jaw cysts, intracranial calcification, palmoplantar pits |
quantitative multiplex fluorescent PCR, polymorphic markers, long-range PCR, sequencing | 9q22.32q22.33 de novo | 4,5 Mb | deleted | nr | |
| [25, patient G10] | 8 y male | nr | nr | severe MR, epilepsy, hydrocephalus |
inguinal hernia, polydactyly scoliosis |
hypertelorism | yes palmar & plantar pits |
HR microarray | inv(9)(q21.2q33.1) = 9q22.32 92,934,973/92,945,040-98,137,216/98,141,889 | 5 Mb | deleted | nr | |
| [16, patient G5, restudy of patient originally presented in 34] | 12 y male | nr | >p97 | severe MR, epilepsy, dilated lateral ventricles, thin corpus callosum, hydrocephalus |
hydronephrosis, scoliosis | hypertelorism epicanthi webbed neck |
yes basal cell carcinoma, palmar and plantar pits, multiple jaw cysts odonto-genic ceratocysts, calcification of falx and tentorium cerebelli |
HR microarray | 9q21.31q22.31 de novo 88,656,506/88,656,835-99,686,554/99,687,352 | 11 Mb | deleted | deleted | |
| [17] | 12 y female | nr | normal | severe MR, brain atrophy | laryngeal stenosis, pulmonary valve stenosi, pectus excavatum, kyphoscoliosis, hypoplastic clavicles | down-slanting palpebral fissures, epicanthi, prognatism, asymmetric palpebral fissures, broad eyebrows, synophrys high forehead, pointed chin short neck, c-a-l-spots |
yes | aCGH | 9q22.1q22.32 de novo | 7,7 Mb | deleted | deleted | |
| [6] | 13 y female | nr | nr | MR, ventriculomegaly |
mild macroglossia | hypertelosim, frontal bossing, epicanthi, ears posteriorly rotated, teeth small, prominent gingivae, toenails hypoplastic, mild hemihypertrophy |
yes mandibular cysts, plantar and palmar spots, rhabdomyosarcoma, Wilms tumor |
karyotype,polymorphic markers at the PTCH1 region | 9q22q32 de novo | paternal | nr | deleted | nr |
| [7, patient 1] | 5 y male | +2,5SD | +2SD | severe MR | umbilical hernia, pectus excavatum, trigonocephaly- craniosynostosis |
epicanthi mouth small upper lip thin ear pits ears low set ear lobules uplifted hyperlaxity, short neck, |
with reservations to young age | aCGH at 1 Mb resolution | 9q22.32q22.33 de novo | paternal | < 6,5 Mb | not tested | nr |
| [7, patient 2] | 8 y female | +2SD | >+3SD | severe MR, seizures trigonocephaly ventriculomegaly thin corpus callosum |
thyroglossal cyst with sternal fistula, no dentition, umbilical hernia, pectus excavatum, kyphosis |
epicanthi, palpebral fissures slant down, small mouth, thin upper lip, thick ears indentation of ear lobules. short neck |
with reservations to young age | aCGH at 1 Mb resolution | 9q22.32q22.33 de novo | paternal | < 6,5 Mb | not tested | nr |
| [27] | 5 m male | p75-90 | p90-97 | MR, hypotonia seizures hydfrocephalus caused by compression by cerebellar vermis |
inguinal hernias, PDA, undescended testes, high arched palate, postaxial polydactyly of left foot, cervical ribs |
hypertelorism, prognatism, broad face and forehead, broad nasal bridge, supraorbital ridges well developed |
yes | HR karyotype, aCGH, FISH | 9q22.32q31.3 de novo | paternal | 12 Mb | deleted | nr |
| [15] | 21 y male | +0,2SD | -1SD | mild MR | kyphosis, postaxial polydactyly, mild pulmonary valve stenosis, inguinal hernias, undescended testes, hypodontia of permanent teeth, palate high arched, uvula bifid, bilateral nasal stenosis, taurodonty of 2nd molars |
frontal bossing epicanthi, palperbral fissures slant down, prognatism, synophrys, hypotelorism, excess nuchal skin, ears low-set and posteriorly rotated, nares anteverted, lips thick, face high |
yes calcification of cella turgica and falx cerebri, basal cell nevus carcinomas, jaw cysts |
HR karyotype, FISH with BAC clones | 9q21.3q31 de novo | 15.3-15.6 MB | deleted | deleted | |
| [32] | 29 y female | short | nr | poor vision, telangiectatic nodule on skin hemivertebra T5, scoliosis, elongated clavicle |
hypotelorism, ulnar deviation of hands |
yes multiple basaliomas, calcification of falx, tentorium cerebri and cella turcica, mandibular cysts |
karyotype | 9q22.1q31.2 | parents not available | nr | assumed deleted | nr | |
| [4] | 12 y female | nr | nr | mild MR, bridging of cella turcica, broad cavum septi pellucid, dilated cerebral ventricles |
hyperopia, deverticulum of the renal calyx, occult spina bifida L5-S4, pectus excavatum, bilateral patellar dysplasia, unsual clavicles, exostosis of distal phalanx of thumb thumbs, abnormal hypoplasia of maxilla |
hypertelorism, biparietal bossing, epicanthi, palpebral fissures slant down, prognatism, synophrys, webbed neck, synophrys and broad eyebrows, low midface, broad nose tip, low set, posteriorly rotated ears |
yes basalioma basal cell nevi trichoepithelioma |
HR karyotype, FISH | 9q22.32q33.2 de novo outside the maternal translocation | familial t(9;17)(q34.1p11.2)mat | nr | deleted | nr |
| [30, patient A] | age nr, female | nr | nr | MR | nr | karyotype, aCGH | 9q21q22.1 | nr | nr | deleted | |||
| [30, patient B] | age nr, female | nr | nr | MR | nr | karyotype, aCGH | 9q22.1q31.2 | nr | nr | deleted | |||
| [8] | 6 y male | p50 | >p97 | severe MR hypotony |
PDA, severe scoliosis, fingers slender, 5th finger camptodactyly, palate high arched, short metacarpals and distal phalanges | frontal bossing, epicanthi, palpebral fissures slant down, ears low-set, hypoplastic nostrils, micrognarhia, small nails |
with reservations to young age increasing nr of nevi, sole pits |
HR karyotype, FISH, genotyping | 9q22.31q31.2 de novo | paternal | D9S303 ->D9S930 | deleted | deleted |
| [3, patient 1] | 15 y female | <p3 | p75 | MR, hydrocephalus with shunt, corpus callosum agenesis | inguinal hernias, bilateral conductive hearing loss, ectopic eruption of incisors, occult spina bifida T2-T3, scoliosis |
frontal bossing, synophrys, prognathism |
yes palmoplantar pits, bifid ribs |
HR karyotype, RLFP polymorphisms | 9q22q22 de novo | paternal | 10-16 cM | deleleted | deleted |
| [3, patient 2] | 26 y female | short | microcephaly | MR | PDA, CoA, anomalous right subclavian artery, bilat conductive hearing loss |
frontal bossing, hypertelorism, prognatism, prominent supraorbital ridges, high palate |
yes multiple basal cell carcinomas, leiomyoma coli, multiple bifid ribs odontogenic ceratocysts, ameloblastoma |
HR karyotype, RLFP polymorphisms FISH | 9q22q32 de novo | maternal t(ins[9][p22q32q22];16)(p22;q21) mat | 22-39 cM | deleted | deleted |
| [9] | 14 y male | <p3 | <p3 | severe MR brachycephaly, dilated ventricles |
undescended testes, left pes equinovarus, I partial I-IV toe syndactyly, |
frontal bossing, hyperteloris, epicanthi, palpebral fissures slant down, broad eyebrows, wide mouth, thick lips, irregular dentition, ears small, no ear lobes, short neck, wide internipple distance, hypoplastic genitalia, tapering fingers, small toes, medial deviation of toes |
nr | karyotype | 9q22q32 de novo | maternal | nr | nr | nr |
| [21] | 15 m male | p25 | p75 | severe MR, partial aplasia of corpus callosum, dilatation of ventricles |
laryngomalacia, cleft palate, PDA, abnormal aortic valve, epiglottic dysplasia, abnormal vocal cords |
frontal bossing, palpebral fissures slant down, hypertrichosis, long eyelashes, epicanthus inversus, nose short, brodge depressed, nares upturned, long philtrum, small mouth, thin upper lip, receding chin, ears large and low set, large lobules, loose skin on cheeks |
nr | karyotype (500 bands) | 9q22q2207 de novo | nr | nr | nr | |
| [2, patient 3] | 7 m female | p90 | p75 | mild to moderate MR, brachycephaly, hydrocephalus |
VSD, PDA hallux valgus |
hypertelorism,epicanthi, synophrys, ptosis, philtrum short ears posteriorly rotated |
nr | HR karyotype | 9q22.3q31.1 de novo | paternal dir ins(4;9)(q33;q22.3q31.1) pat | nr | nr | nr |
| [35] | infant male | nr | nr | asymmetric ventricles, partial fusion of cerebellar hemispheres, polymicrogyria, delayed cerebellar neuronal migration, enlarged massa intermedia | cryptorchidism, focal glomerulosclerosis, accessory spleen, partial fusion of vertebrae D2 and D3, pectus excavatum, irregular ribs interphalangeal ankylosis |
nr | karyotype | 9q22q32 de novo | nr | nr | nr | ||
| [10] | 3 m male | nr | nr | death at 3 months, seizures | duodenal atresia, malrotation, Meckel diverticulum, multilobulated spleen, accessory spleens renal dysplasia, hydroureter polydactyly of hand, syndactyly of feet angulated clavicles thorax asymmetric |
palpebral fissures slant down, epicanthi, hypotelorism, hirsutism, short palpebral, fissures, depressed nasal bridge, auditory canals narrow, philtrum long |
nr unusual ribs, |
karyotype, Q, C and R bands | 9q22q32 de novo | maternal | nr | nr | nr |
| [1, patient 1] | 14 y male | -1SD | 0SD | severe MR, epilepsy |
hypertelorism, palpebral fissures slant down, nose short, nares anteverted, philtrum long, mild micrognathia |
nr | karyotype with R bands | 9q11q22 de novo | nr | nr | nr | ||
| Present case 3 | 37 y male | -1SD | -0,25SD | normal cognition, dysarthria |
grade IV vesicoureteral reflux, hydroureter and hypoplastic left kidney. funnel chest, three lower-most costal cartilages broadly fused | deepset and small toenails, palpebral fissures slant down, high bridged nose, narrow nares, long deep furrowed philtrum, ears with broad helices and uplifted lobuli, short 2nd finger nails | no | aCGH | 9q22.2q22.31 de novo | 5,3Mb | not deleted | deleted | |
| Present case 1 | 8,5 y female | 0,1SD | +0,5SD | moderate MR, dysarthria |
short neck, slight ptosis on the right, downward slant of the palpebral fissures, narrow nose, small nares, long philtrum with a narrow deep groove, tented upper lip, ears with broad helices and uplifted lobuli. toe nails II-V bilaterally small | no | aCGH | 9q22.2q22.31 | paternal | 5,3 Mb | not deleted | deleted | |
| Present case 2 | 4 y female | -0,2SD | +1SD | moderate MR, dysarthria |
down slanting palpebral fissures, mild left side ptosis, narrow nares, uplifted ear lobuli and thick helices, long philtrum with a deep furrow, thin and tented upper lip. | no | aCGH | 9q22.2q22.31 | paternal | 5,3Mb | not deleted | deleted | |
All values given as reported in each original paper.
Abbreviations: nr = no report, y = year, m = month, OFC = head circumference; CNS = central nervous system; MR = mental retardation; p = percentile; SD = standard deviation, aCGH = array comparative genomic hybridization, HR = high resolution.
We describe a family where two mentally retarded siblings and their cognitively normal father share an identical 5,3 Mb deletion at 9q22.2q22.32, not including PTCH1, and discuss genotype-phenotype correlation in these patients.
Methods
Cytogenetic analysis
Chromosome metaphase spreads of patients 1, 3, and the healthy mother were analyzed by standard G-banding karyotype analysis (400 band resolution). Additional subtelomere-fluoresence in situ hybridization (FISH) analysis was conducted on patient 1 according to standard protocols. Targeted FISH-analysis of lymphocyte cells of patient 3 was performed using BAC-probe RP11-30L4.
Molecular karyotyping
Mental retardation in addition to subtle but undisputable dysmorphic features in patients 1 and 2 were indications to perform additional high-resolution analysis by comparative genomic hybridization (CGH) and single nucleotide polymorphism (SNP) arrays. Standard molecular karyotyping was performed using a 244 K CGH array (Agilent Technologies, Santa Clara, CA, USA), as previously described [11].
Array CGH results were confirmed by SNP array analysis of patients 1 and 3 and the paternal grandparents, using the Genome-Wide Human SNP array 6.0 according to manufacturer instructions (Affymetrix, Santa Clara, CA, USA). The SNP 6.0 array contains 906,000 SNP probes and 946,000 copy number probes and has an average resolution of 0.7 kb. Data was extracted using the Genotyping console software V.3.0.2 with default settings, including the BRLMM-P-Plus algorithm and the Hidden-Markow Model for smoothing the copy number data. As a reference set we used data from 90 Caucasian HapMap samples. The extracted data was further analyzed using the Chromosome Analysis Suite software V.1.0. Copy number changes were called and filtered based on reference data and a minimum amount of 10 consecutive probes. All changes that were called were further compared to the Database of Genomic Variants (DGV, http://projects.tcag.ca/variation) as well as in-house data (unpublished material). All array data is stored in and available from the CanGEM Database (http://www.cangem.org). Array CGH and SNP data were analyzed using the reference genome build 18 (NCBI 36).
Clinical report
The family has four children and in addition the mother has a history of four first trimester miscarriages. Patients 1 and 2 are the 2nd and 3rd born children in the family. The psychomotor development of the eldest daughter has been within normal limits, but she suffers from short attention span and need special support at school. The youngest boy is considered healthy at 10 months of age. The paternal grandparents have neither learning problems nor congenital abnormalities and the family history is unremarkable (Figure 1).
Figure 1.
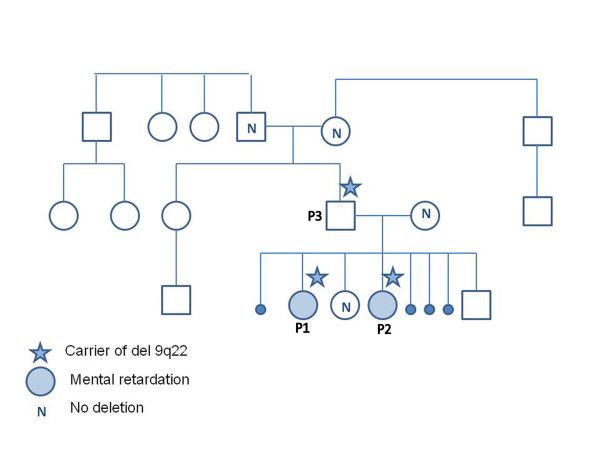
Pedigree of the family Patients 1, 2, and 3 all have the deletion of 9q22, as indicated by the star. The healthy mother, sister, and paternal grandparents, do not have the deletion, as indicated by N. The healthy brother was not tested for the deletion. Only patients 1 and 2 (P1, P2) have mental retardation.
Patient 1, a girl, was born after an uneventful pregnancy. She had normal birth size (3450 g, 49 cm, OFC 35 cm) and Apgar score was 10 at 1 and 5 minutes. A drop in blood hemoglobin from 172 g/l to 127 g/l was observed during the first day and she received a red cell transfusion. No evidence for bleeding, infection, hemoglobin abnormality, or immunization were found. She was discharged in good condition at the age of one week. She has suffered from recurrent middle ear infections since 1 year of age and repeated insertion of grommets and prophylactic antibiotics were not helpful. She developed bronchial asthma at the age of four years. At 2 and 3 years she had febrile convulsions but EEG recordings were normal.
She learned to walk independently at 20 months and spoke her first words at 15 months. At 4 years neuropsychological examination showed mild mental retardation, defective linguistic development and clumsy fine motor performance. At 9 years she attends a school for developmentally handicapped children and has learned elements of reading and writing. She needs help in basic daily skills. She speaks using sentences, but her speech is dysarthric. The vicious circle of ear infections continues. She needs treatment for asthma and uses melatonin for sleep problems.
During the first two years her height was at +2SD and has thereafter approached +0.5SD (target height -0.5SD). She has overweight (BMI 24) since 2 years of age. The OFC grows steadily at -0.5SD. She has normal body proportions, short neck, slight ptosis on the right, downward slant of the palpebral fissures, narrow nose, small nares, long philtrum with a narrow deep groove, tented upper lip, ears with broad helices and uplifted lobuli. Her toe nails II-V are small on both feet; otherwise feet, toes, hands, fingers and fingernails are normal (Figures 2 and 3).
Figure 2.
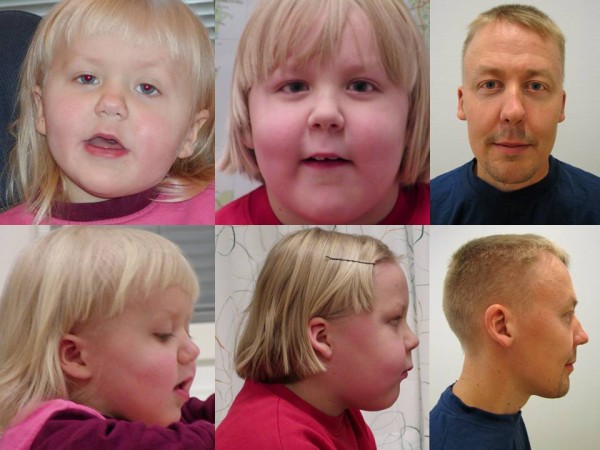
Facial features of the patients Facial features of patients 1 to 3 from left to right. Note short neck, slight unilateral ptosis, downward slant of the palpebral fissures, narrow nose, small nares, long philtrum with a narrow deep groove, tented upper lip, ears with broad helices and uplifted lobuli.
Figure 3.
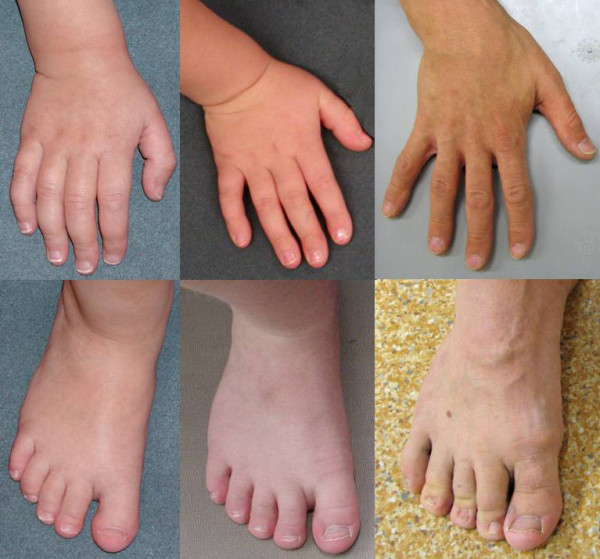
Hands and feet of the patients Hands and feet of patients 1 to 3 from left to right. Note that patient 1 has small toe nails II-V on both feet and patient 3 has small and deep-set toenails and his 2nd finger nails are short.
Blood lymphocyte counts have repeatedly been normal. She has normal immunoglobulin levels including IgG subclasses, C3 and C4 and alpha-1-antitrypsin and no IgE related allergies have been found. Urine metabolic screen was negative an no vacuolated lymphocytes were detected. The brain MRI and US of kidneys and abdominal organs were normal at the age of 4.5 years. X-rays of the thorax showed no skeletal anomalies. Chromosome analysis (400 bands), subtelomere-FISH, 22q11 FISH (TUPLE1 probe), and FRAXA mutation analysis were normal.
Patient 2 is the younger sister of patient 1. She was born at term after an uneventful pregnancy. The mother was treated with acyclovir for herpes simplex type1 infection from the 17th week up until the delivery and with hydroxysitsin for her itching. Her birth size was normal (3770 g, 52 cm, OFC 35 cm) At the age of three weeks she was admitted to hospital for apneas caused by pulmonary and middle ear infections and needed mechanical ventilation for four days. The causative microorganism was not identified. EEG recording showed right sided spikes and phenobarbital treatment was started. At 6 months the medication was discontinued due to absence of epileptic symptoms and normal EEG. Brain MRI, cardiac conduction examinations, and cardiac ultrasound were normal.
Psychomotor developmental delay was obvious since the age of 6 months. She learned to walk independently at 2.5 years, spoke words at 3 years and short sentences at 5 years of age. At 5 years her speech is dysarthric, she has problems of falling asleep in the evening and treatment with melatonin has shown modest success. She has sudden attacks of aggression and abrupt standstills when she is withdrawn from contact and stares or cries and holds her head between her hands. EEG recordings show no epileptic activity. Her ability of reciprocal contact is very defective. She also suffers from abundant ear infections and from a tendency to asthmatic bronchitis. During the 1st year her height increased from +1SD to +2SD and then has declined to -0,5SD (her target height). Her OFC has grown at 0SD during the first 6 months of life and thereafter has remained at +1SD. She shows down slanting palpebral fissures, mild left side ptosis, narrow nares, uplifted ear lobuli and thick helices, long philtrum with a deep furrow, thin and tented upper lip (Figures 2 and 3). Her immunoglobulin levels are normal, but IgE related food allergy has been suspected.
Patient 3 is the father of patients 1 and 2. He was born term after a normal pregnancy. His birth size was normal (53 cm, 4090 g) and Apgar score was 9 at 1 and 10 at 5 minutes. At the age of 2.5 months he had a febrile pyelonephritis and urological examinations revealed grade IV vesicoureteral reflux, hydroureter and hypoplastic left kidney. In adult age his renal function has been normal. He suffered from recurrent middle ear infections and secretory otitis up to his teens. Surgery to correct his funnel chest was attempted when he was 6 years old. The surgery report describes the three lower-most costal cartilages being broadly fused. The immediate postoperative X-ray could be traced and it showed normal claviculae, ribs and thoracic vertebrae.
His psychomotor development during childhood was considered normal except speech development and he had speech therapy up to his teens. At adult age he still suffers from dysarthria. His school and military service history are unremarkable. Presently he works as a mailman and studies to become a nurse's aide.
At 1 year of age his height was 80 cm (+1SD). His adult height is 171,5 cm (-1SD), weight 68 kg (BMI 23) and OFC 57 cm (-0,8SD). He has partial upper denture after removal of decayed front teeth. The thoracic cage shows sequelae of childhood surgery and the sternum appears short but body-limb proportions are normal. He has down-slanting palpebral fissures, high nasal bridge, narrow nares, long philtrum with deep furrow, and the ears have broad helices and uplifted lobule. The 2nd finger nails are short and he has deep set toe nails. (Figures 2 and 3).
Results
Karyotype analysis of patients 1, 3, and the mother, were normal. Sub-telomere FISH analysis of patient 1 was also normal.
Both array CGH and SNP array analysis revealed an identical 5.3 Mb deletion of chromosome 9q22.2q22.32 in all three patients, ranging from basepair 91,523,558 to 96,858,929 (Agilent probes A_16P18688441-A_16_P02135862), covering 30 genes (Figure 4). The average log2-ratio was -1, indicating a one-copy deletion.
Figure 4.

Array CGH results revealed a deletion at 9q22 in all three patients Array CGH results of patients 1, 2 and 3 revealed a 5.3 Mb deletion in all patients at 9q22.2q22.32. The shaded area indicates the deleted area with an average log2-ration of -1, indicating loss of one copy of the genomic segment.
The parents of patient 3 and the older sister of patients 1 and 2 were studied for the microdeletion with normal results. Targeted FISH-analysis of at least 80 metaphase spreads, using BAC-probe RP11-30L4, was conducted in an effort to detect mosaicism 9q22 in patient 3. All metaphases showed the deletion of 9q22. Although these results do not exclude germline mosaicism of patient 3, the likelihood is small.
Discussion
The clinical and molecular findings of the previously reported 27 live patients and our 3 patients with constitutional 9q22 deletions are summarized in Table 1. Two prenatally terminated fetuses are not included in the analysis [12,13]. Based on available breakpoint information, at least eight reported patients have an overlapping deletion with the one detected in this family (Figure 5) [5,7,14-17].
Figure 5.

Overlapping deletions at 9q22 The figure depicts the genomic segment deleted in the patients of this study, as displayed in the UCSC Genome Browser (http://genome.ucsc.edu). Reported deletions of 9q22 that overlap the deletion found in this study are marked by black bars; a) Redon et al. [7], patients 1 and 2, b) Shimojima et al. [5], c.1) Fujii et al. [16] patient G5, c.2) Fujii et al. [16] patient G10, d) Nowakowska et al. [17], e) de Ravel et al. [14], f) Boonen et al. [15], g) Kosaki et al. [28]. Arrows on the black bar indicate that the deletion starts or ends outside the area shown in the picture.
The deletion found in our three patients leaves the PTCH1 gene intact. Instead, according to DECIPHER database (the DECIPHER consortium, http://decipher.sanger.ac.uk/), three genes in the deleted area, viz. SYK, IARS and ASPN, are scored as likely haploinsufficient [18]. SYK is involved in several important biological processes as discussed below. An allelic variant of ASPN has been implicated in osteoarthritis [19] and lumbar disc degeneration [20]. Neither of these features was present in our patients. IARS encodes an isoleucyl-tRNA synthetase protein and deletions of this gene have not been associated with human disease. In addition, three clustered micro-RNAs (miRNA) called hsa-let-7a, hsa-let-7f and hsa-let-7d are found in the deleted area.. All of these miRNAs have a common primary target gene, HMGA2, as scored by Target Scan (http://www.targetscan.org/) and miRBase (http://www.mirbase.org/), indicating that they all function as repressors of this gene. The deletion is flanked by some short segmental duplications. These, however, do not lie at the breakpoints but further up-/downstream and thus it is unclear whether the rearrangement could initially have been due to non-allelic homologous recombination (NAHR).
Almost all patients published so far have been reported to have dysmorphic facial features. However, no unique facial gestalt does emerge. All three patients described here had similar mild dysmorphic features: downward slanting palpebral fissures, high bridged narrow nose with small nares, deep grooved long philtrum, tented and thin upper lip, ears with broad helices and uplifted lobuli. In addition patients 1 and 3 have small toenails.When compared with the previous reports, only the patient reported by Olivieri et al. (2003) has facial features closely resembling those of our patients [8].
A significant common feature in our three patients is incessant middle ear and upper respiratory tract infections. No major immunological deficiency, however, was found to cause the vicious circle of infections, and in the father this symptom disappeared at his teens. Only three previously reported patients suffered from recurrent respiratory infections [14,15,21]. Interestingly, the region deleted in our patients includes the gene Spleen Tyrosine Kinase (SYK) (MIM *600085) which is widely expressed in hematopoietic cells and other cells of epithelial origin. SYK plays an important role in regulation of innate immunity and inflammatory response, and is involved in several human diseases such as allergy, autoimmunity, and haematological malignancies (reviewed by Mócsai et.al [22]). The infections seen in our patients could be linked to reduced SYK activity and thereby reduced, albeit not lacking, ability to activate the inflammatory response. Recent studies in mice show that a SYK-deletion reduces their antibacterial host defence [23]. SYK is deleted in the patients with recurrent respiratory infections reported by Pfeiffer et al., and Boonen et al. but not de Ravel et al. [14,15,21]. The relevance of its haploinsufficiency to human recurrent early-age upper airway infections might be a subject for further studies.
Mental retardation among the reported 9q22 deletion patients is common and usually moderate to severe. Only the 4-year-old patient of Shimojima et al. with a 2,3 Mb deletion is reported to have normal development [5]. The intelligence of the father in our family is within normal limits although a formal neuropsychological testing has not been performed. Instead, his two daughters carrying a similar deletion are moderately retarded. In genetic counseling a similar deletion found both in patients with mental retardation and in a family member with normal cognitive function understandably poses difficulties. The cause of the variable expression remains to be found. According to the Database of Genomic Variants (http://projects.tcag.ca/variation) the deletion described here has not been found in healthy controls. Reduced penetrance was not found to be due to mosaicism admitting the fact that in addition to lymphocytes no other tissue like semen or buccal cells were examined. The parents did not consider further studies for mosaicism important and thus it was not pursued.
Another explanation for variable expression would be two simultaneous genomic hits acting either independently or by affecting the same signaling pathway [24]. In our family the first hit would be the deletion, but the second hit would need other methods to be detected. The paternal microdeletion could also reveal a maternal allelic recessive defect and the consequent autosomal compound heterozygosity in the two daughters would be the cause of their cognitive impairment. No obvious candidate gene is situated in the deleted region. Microdeletions are increasingly frequently encountered associated with the phenomenon of variable penetrance and even nonpenetrance, 15q13.3 microdeletion syndrome being one example [25]. The deleted region is not known to be affected by imprinting that could cause cognitive differences among the patients.
A common developmental problem of our three patients is dysarthria, which is severe and making even the speech of the cognitively normal father difficult to comprehend. A similar problem has not been described in the previously published patients and further studies are needed to explore whether a locus for dysarthria resides in the region defined by the deletion.
Both daughters in our family had symptoms suspected to be epileptic although the diagnosis could not be confirmed. Eight patients from the literature suffer from epilepsy, but no characteristic seizure type emerged [1, 7, 10, 26 (two patients), 16 (two patients), 27].
Patient 3 had unilateral hypoplastic kidney and vesicoureteral reflux. Also three previous patients had a renal problem; hydronephrosis, renal dysplasia or hydroureter [10,16,28]. Interestingly, patient 3 had a congenital chest deformity similar to one of the diagnostic criteria of NBCCS [29]. Additional X-rays could have clarified the presence of possible other NBCCS related skeletal signs. The 9q22 deletion detected did not, however, include PTCH1 gene and since our patient otherwise had no indications for further X-ray examinations, it was considered clinically inappropriate to take them. The patient's chest deformity can be independent of the deletion since patients 1 and 2 do not have thoracic anomalies. Yet, one is tempted to speculate that it is a sign that the deletion might effect the expression of PTCH1.
Another clinically important gene included in the deletion is ROR2. Gain of function mutations of one allele are known to cause brachydactyly type B1 while a mutation of both alleles cause Robinow Syndrome [30,31]. However, haploinsufficiency of ROR2 has no effect on phenotype as shown by Oldridge et al., who described two unrelated patients with de novo deletions at 9q22 [30]. Accordingly, none of the six previous patients, nor our patients, whose deletion includes ROR2, have either brachydactyly type B1 or features of Robinow Syndrome [[3]3 (restudied in 15), [8,15-17]] (Table 1).
Shimojima et al. suggested that del 9q22 might be a novel overgrowth syndrome by paternal imprinting [5]. Among the previous patients eight carry a deletion in the paternally inherited chromosome [[3] (patient 1), [5,7] (patients 1 and 2), [8,28]] and three are maternally deleted [[3] (patient 2), [9,10]]. These small numbers are naturally liable to bias. Indeed, the paternally deleted patients seem to have a large neonatal size. Among those with data on postnatal growth 3/5 are mildly tall and 4/6 have macrocephaly. Growth data of the three maternally deleted patients is scanty but none of them seems to be tall or to have macrocephaly. The maternal deletions reported appear to be much larger which implies a more significant genomic imbalance and could alone explain differences in phenotype (Table 1).
Recently Kosaki et al. [28] narrowed the region of the proposed overgrowth factor closer to PTCH1 gene by their patient's deletion in relation to that of the patients published by Shimojima et al. and Redon et al. [5,7]. Our family's deletion helps to bring it even closer to PTCH1. On the other hand tallness and macrocephaly are also inherent features of NBCCS [32]. In a thorough survey of a large series of NBCCS patients Kimonis et al. observed no imprinting effect regarding any feature of the syndrome including macrocephaly [29]. Overgrowth like macrocephaly observed in del 9q22 patients could thus simply reflect the simultaneous haploinsufficiency of PTCH1.
Our family underlines the importance to try and include both parental samples in array molecular karyotyping. In a more usual situation with only one developmentally retarded child in the family one might for several reasons be tempted to accept the analysis of the child alone or with only one parent and unknowingly thus get misleading information of the situation. Microdeletion 9q22 adds to the increasing number genomic imbalances that challenge genetic counseling.
Conclusions
In conclusion, 9q22 deletions are rare and both phenotypically and molecularly unique. In the majority the deletion contains the PTCH1 gene, which signifies that the patients in addition to the common mental retardation also develop the tumor proneness syndrome of Gorlin, which is to be taken into account in counseling and follow-up. We present the first familial 9q22 deletion, a father and his two developmentally delayed daughters. Their deletion leaves the PTCH1 gene intact. The father does not have significant cognitive problems but has renal and thoracic cage malformations while the daughters do not have congenital malformations. Dysarthric speech and prolonged tendency to ear and upper respiratory infections are common to all three. Major differences in psychomotor development warrant cautiousness in genetic counseling in patients with similar deletions.
Consent
Written informed consent was obtained from the father, the mother and the paternal grandparents for publication of this case report and accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
LS carried out the molecular genetic studies, participated in the design of the study and drafted the manuscript. MP carried out the clinical studies, participated in the design of the study and drafted the manuscript. MS carried out the sample collection. TM composed the photos for publication. TY and KS performed the FISH analysis and helped to draft the manuscript. JI critically revised the manuscript. SK participated in the study design and coordination and critically revised the manuscript. All authors read and approved the final manuscript.
Contributor Information
Linda Siggberg, Email: linda.siggberg@helsinki.fi.
Maarit Peippo, Email: maarit.peippo@vaestoliitto.fi.
Marjatta Sipponen, Email: marjatta.sipponen@vaestoliitto.fi.
Taina Miikkulainen, Email: taina.miikkulainen@vaestoliitto.fi.
Keiko Shimojima, Email: keiko.shimojima@twmu.ac.jp.
Toshiyuki Yamamoto, Email: toshiyuki.yamamoto@twmu.ac.jp.
Jaakko Ignatius, Email: jaakko.ignatius@tyks.fi.
Sakari Knuutila, Email: sakari.knuutila@helsinki.fi.
Acknowledgements
The authors thank the family for participating in this study. Dr. Eino Marttinen from the Department of Medical Imaging of the Helsinki University Hospital is acknowledged for important discussions about the radiological findings in patient 3. The Department of Medical Genetics, The Family Federation of Finland, is funded by Finland's Slot Machine Association (RAY). LS was funded by the Rinnekoti Research Foundation, Medicinska Understödsföreningen Liv och Hälsa r.f., and the State Appropriations of the Helsinki University Central Hospital.
References
- Turleau C, de Grouchy J, Chabrolle JP. Intercalary deletions of 9q. Ann Genet. 1978;21:234–236. [PubMed] [Google Scholar]
- Farrell SA, Siegel-Bartelt J, Teshima I. Patients with deletions of 9q22q34 do not define a syndrome: three case reports and a literature review. Clin Genet. 1991;40:207–214. doi: 10.1111/j.1399-0004.1991.tb03078.x. [DOI] [PubMed] [Google Scholar]
- Shimkets R, Gailani MR, Siu VM, Yang-Feng T, Pressman CL, Levanat S, Goldstein A, Dean M, Bale AE. Molecular analysis of chromosome 9q deletions in two Gorlin syndrome patients. Am J Hum Genet. 1996;59:417–422. [PMC free article] [PubMed] [Google Scholar]
- Midro AT, Panasiuk B, Tumer Z, Stankiewicz P, Silahtaroglu A, Lupski JR, Zemanova Z, Stasiewicz-Jarocka B, Hubert E, Tarasow E, Famulski W, Zadrozna-Tolwinska B, Wasilewska E, Kirchhoff M, Kalscheuer V, Michalova K, Tommerup N. Interstitial deletion 9q22.32-q33.2 associated with additional familial translocation t(9;17)(q34.11;p11.2) in a patient with Gorlin-Goltz syndrome and features of Nail-Patella syndrome. Am J Med Genet A. 2004;124A:179–191. doi: 10.1002/ajmg.a.20367. [DOI] [PubMed] [Google Scholar]
- Shimojima K, Adachi M, Tanaka M, Tanaka Y, Kurosawa K, Yamamoto T. Clinical features of microdeletion 9q22.3 (pat) Clin Genet. 2009;75:384–393. doi: 10.1111/j.1399-0004.2008.01141.x. [DOI] [PubMed] [Google Scholar]
- Cajaiba MM, Bale AE, Alvarez-Franco M, McNamara J, Reyes-Mugica M. Rhabdomyosarcoma, Wilms tumor, and deletion of the patched gene in Gorlin syndrome. Nat Clin Pract Oncol. 2006;3:575–580. doi: 10.1038/ncponc0608. [DOI] [PubMed] [Google Scholar]
- Redon R, Baujat G, Sanlaville D, Le Merrer M, Vekemans M, Munnich A, Carter NP, Cormier-Daire V, Colleaux L. Interstitial 9q22.3 microdeletion: clinical and molecular characterisation of a newly recognised overgrowth syndrome. Eur J Hum Genet. 2006;14:759–767. doi: 10.1038/sj.ejhg.5201613. [DOI] [PubMed] [Google Scholar]
- Olivieri C, Maraschio P, Caselli D, Martini C, Beluffi G, Maserati E, Danesino C. Interstitial deletion of chromosome 9, int del(9)(9q22.31-q31.2), including the genes causing multiple basal cell nevus syndrome and Robinow/brachydactyly 1 syndrome. Eur J Pediatr. 2003;162:100–103. doi: 10.1007/s00431-002-1116-4. [DOI] [PubMed] [Google Scholar]
- Kroes HY, Tuerlings JH, Hordijk R, Folkers NR, ten Kate LP. Another patient with an interstitial deletion of chromosome 9: case report and a review of six cases with del(9)(q22q32) J Med Genet. 1994;31:156–158. doi: 10.1136/jmg.31.2.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying KL, Curry CJ, Rajani KB, Kassel SH, Sparkes RS. De novo interstitial deletion in the long arm of chromosome 9: a new chromosome syndrome. J Med Genet. 1982;19:68–70. doi: 10.1136/jmg.19.1.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siggberg L, Ala-Mello S, Jaakkola E, Kuusinen E, Schuit R, Kohlhase J, Böhm D, Ignatius J, Knuutila S. Array CGH in molecular diagnosis of mental retardation - a study of 150 patients. Am J Med Genet A. 2010;152A:1398–1410. doi: 10.1002/ajmg.a.33402. [DOI] [PubMed] [Google Scholar]
- Paoloni-Giacobino A, Floris E, Dahoun SP. Fetus with a 9q22q34 interstitial deletion and hygroma. Prenat Diagn. 2000;20:855–856. doi: 10.1002/1097-0223(200010)20:10<855::AID-PD919>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- L'Hermine AC, Aboura A, Simon-Bouy B, Robin F, Audibert F, Strouk N, Capron F, Frydman R, Tachdjian G. Female pseudohermaphroditism in a fetus with a deletion 9(q22.2q31.1) Prenat Diagn. 2002;22:652–655. doi: 10.1002/pd.353. [DOI] [PubMed] [Google Scholar]
- de Ravel TJ, Ameye L, Ballon K, Borghgraef M, Vermeesch JR, Devriendt K. Early detection of chromosome 9q22.32q31.1 microdeletion and the nevoid basal cell carcinoma syndrome. Eur J Med Genet. 2009;52:145–147. doi: 10.1016/j.ejmg.2009.02.002. [DOI] [PubMed] [Google Scholar]
- Boonen SE, Stahl D, Kreiborg S, Rosenberg T, Kalscheuer V, Larsen LA, Tommerup N, Brondum-Nielsen K, Tumer Z. Delineation of an interstitial 9q22 deletion in basal cell nevus syndrome. Am J Med Genet A. 2005;132A:324–328. doi: 10.1002/ajmg.a.30422. [DOI] [PubMed] [Google Scholar]
- Fujii K, Ishikawa S, Uchikawa H, Komura D, Shapero MH, Shen F, Hung J, Arai H, Tanaka Y, Sasaki K, Kohno Y, Yamada M, Jones KW, Aburatani H, Miyashita T. High-density oligonucleotide array with sub-kilobase resolution reveals breakpoint information of submicroscopic deletions in nevoid basal cell carcinoma syndrome. Hum Genet. 2007;122:459–466. doi: 10.1007/s00439-007-0419-y. [DOI] [PubMed] [Google Scholar]
- Nowakowska B, Kutkowska-Kazmierczak A, Stankiewicz P, Bocian E, Obersztyn E, Ou Z, Cheung SW, Cai WW. A girl with deletion 9q22.1-q22.32 including the PTCH and ROR2 genes identified by genome-wide array-CGH. Am J Med Genet A. 2007;143A:1885–1889. doi: 10.1002/ajmg.a.31845. [DOI] [PubMed] [Google Scholar]
- Huang N, Lee I, Marcotte EM, Hurles ME. Characterising and predicting haploinsufficiency in the human genome. PLoS Genet. 2010;6:e1001154. doi: 10.1371/journal.pgen.1001154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song JH, Lee HS, Kim CJ, Cho YG, Park YG, Nam SW, Lee JY, Park WS. Aspartic acid repeat polymorphism of the asporin gene with susceptibility to osteoarthritis of the knee in a Korean population. Knee. 2008;15:191–195. doi: 10.1016/j.knee.2007.11.005. [DOI] [PubMed] [Google Scholar]
- Song YQ, Cheung KM, Ho DW, Poon SC, Chiba K, Kawaguchi Y, Hirose Y, Alini M, Grad S, Yee AF, Leong JC, Luk KD, Yip SP, Karppinen J, Cheah KS, Sham P, Ikegawa S, Chan D. Association of the asporin D14 allele with lumbar-disc degeneration in Asians. Am J Hum Genet. 2008;82:744–747. doi: 10.1016/j.ajhg.2007.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer RA, Lachmann E, Schreyer W, Volleth M. Deletion of 9q22: a new observation suggesting a specific phenotype. Ann Genet. 1993;36:167–170. [PubMed] [Google Scholar]
- Mocsai A, Ruland J, Tybulewicz VL. The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat Rev Immunol. 2010;10:387–402. doi: 10.1038/nri2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Ziffle JA, Lowell CA. Neutrophil-specific deletion of Syk kinase results in reduced host defense to bacterial infection. Blood. 2009;114:4871–4882. doi: 10.1182/blood-2009-05-220806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girirajan S, Rosenfeld JA, Cooper GM, Antonacci F, Siswara P, Itsara A, Vives L, Walsh T, McCarthy SE, Baker C, Mefford HC, Kidd JM, Browning SR, Browning BL, Dickel DE, Levy DL, Ballif BC, Platky K, Farber DM, Gowans GC, Wetherbee JJ, Asamoah A, Weaver DD, Mark PR, Dickerson J, Garg BP, Ellingwood SA, Smith R, Banks VC, Smith W, McDonald MT, Hoo JJ, French BN, Hudson C, Johnson JP, Ozmore JR, Moeschler JB, Surti U, Escobar LF, El-Khechen D, Gorski JL, Kussmann J, Salbert B, Lacassie Y, Biser A, McDonald-McGinn DM, Zackai EH, Deardorff MA, Shaikh TH, Haan E, Friend KL, Fichera M, Romano C, Gecz J, DeLisi LE, Sebat J, King MC, Shaffer LG, Eichler EE. A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat Genet. 2010;42:203–209. doi: 10.1038/ng.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masurel-Paulet A, Andrieux J, Callier P, Cuisset JM, Le Caignec C, Holder M, Thauvin-Robinet C, Doray B, Flori E, Alex-Cordier MP, Beri M, Boute O, Delobel B, Dieux A, Vallee L, Jaillard S, Odent S, Isidor B, Beneteau C, Vigneron J, Bilan F, Gilbert-Dussardier B, Dubourg C, Labalme A, Bidon C, Gautier A, Pernes P, Pinoit JM, Huet F, Mugneret F, Aral B, Jonveaux P, Sanlaville D, Faivre L. Delineation of 15q13.3 microdeletions. Clin Genet. 2010;78:149–161. doi: 10.1111/j.1399-0004.2010.01374.x. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Yoshihashi H, Furuya N, Adachi M, Ito S, Tanaka Y, Masuno M, Chiyo H, Kurosawa K. Further delineation of 9q22 deletion syndrome associated with basal cell nevus (Gorlin) syndrome: report of two cases and review of the literature. Congenit Anom (kyoto) 2009;49:8–14. doi: 10.1111/j.1741-4520.2008.00212.x. [DOI] [PubMed] [Google Scholar]
- Chen CP, Lin SP, Wang TH, Chen YJ, Chen M, Wang W. Perinatal findings and molecular cytogenetic analyses of de novo interstitial deletion of 9q (9q22.3-->q31.3) associated with Gorlin syndrome. Prenat Diagn. 2006;26:725–729. doi: 10.1002/pd.1496. [DOI] [PubMed] [Google Scholar]
- Kosaki R, Fujita H, Ueoka K, Torii C, Kosaki K. Overgrowth of prenatal onset associated with submicroscopic 9q22.3 deletion. Am J Med Genet A. 2011;155:903–905. doi: 10.1002/ajmg.a.33835. [DOI] [PubMed] [Google Scholar]
- Kimonis VE, Goldstein AM, Pastakia B, Yang ML, Kase R, DiGiovanna JJ, Bale AE, Bale SJ. Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am J Med Genet. 1997;69:299–308. doi: 10.1002/(SICI)1096-8628(19970331)69:3<299::AID-AJMG16>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Oldridge M, Fortuna AM, Maringa M, Propping P, Mansour S, Pollitt C, DeChiara TM, Kimble RB, Valenzuela DM, Yancopoulos GD, Wilkie AO. Dominant mutations in ROR2, encoding an orphan receptor tyrosine kinase, cause brachydactyly type B. Nat Genet. 2000;24:275–278. doi: 10.1038/73495. [DOI] [PubMed] [Google Scholar]
- Schwarzer W, Witte F, Rajab A, Mundlos S, Stricker S. A gradient of ROR2 protein stability and membrane localization confers brachydactyly type B or Robinow syndrome phenotypes. Hum Mol Genet. 2009;18:4013–4021. doi: 10.1093/hmg/ddp345. [DOI] [PubMed] [Google Scholar]
- Haniffa MA, Leech SN, Lynch SA, Simpson NB. NBCCS secondary to an interstitial chromosome 9q deletion. Clin Exp Dermatol. 2004;29:542–544. doi: 10.1111/j.1365-2230.2004.01590.x. [DOI] [PubMed] [Google Scholar]
- Musani V, Cretnik M, Situm M, Basta-Juzbasic A, Levanat S. Gorlin syndrome patient with large deletion in 9q22.32-q22.33 detected by quantitative multiplex fluorescent PCR. Dermatology. 2009;219:111–118. doi: 10.1159/000219247. [DOI] [PubMed] [Google Scholar]
- Sasaki K, Yoshimoto T, Nakao T, Minagawa K, Takahashi Y, Watanabe Y, Tanabe C. A nevoid basal cell carcinoma syndrome with chromosomal aberration. No To Hattatsu. 2000;32:49–55. [PubMed] [Google Scholar]
- Robb LJ, Vekemans M, Richter A, Meagher-Villemure K, Neilsen K, Der Kaloustian VM. Interstitial deletion of the long arm of chromsome 9 [abstract] Am J Hum Genet. 1991;49(suppl):274. [Google Scholar]