

Abstract
Normal function of the p53 pathway is ubiquitously lost in cancers either through mutation or inactivating interaction with viral or cellular proteins. However, it is difficult in clinical studies to link p53 mutation status to cancer treatment and clinical outcome, suggesting that the p53 pathway is not fully understood. We have recently reported that the human p53 gene expresses not only 1 but 12 different p53 proteins (isoforms) due to alternative splicing, alternative initiation of translation, and alternative promoter usage. p53 isoform proteins thus contain distinct protein domains. They are expressed in normal human tissues but are abnormally expressed in a wide range of cancer types. We have recently reported that p53 isoform expression is associated with breast cancer prognosis, suggesting that they play a role in carcinogenesis. Indeed, the cellular response to damages can be switched from cell cycle arrest to apoptosis by only manipulating p53 isoform expression. This may provide an explanation to the hitherto inconsistent relationship between p53 mutation, treatment response, and outcome in breast cancer. However, the molecular mechanism is still unknown. Recent reports suggest that it involves modulation of gene expression in a p53-dependent and -independent manner. In this review, we summarize our current knowledge about the biological activities of p53 isoforms and propose a molecular mechanism conciliating our current knowledge on p53 and integrating p63 and p73 isoforms in the p53 pathway.
Keywords: splice, promoter, tumor, apoptosis, cell cycle
Introduction
p53 is a major tumor suppressor inactivated in almost all cancer types. p53 prevents cancer formation by regulating multiple pathways including the 2 most described p53-mediated cellular functions, which tilt the balance in favor of life (cell cycle arrest) or cell death. p53 is a transcription factor that binds directly and specifically as a tetramer to target sequences of DNA through p53-responsive elements (p53REs), thereby regulating gene expression.1,2 For example, p53 induces cell cycle arrest by transactivating genes such as the cyclin-dependent kinase inhibitor, p21, or the microRNA, miR34. Alternatively, p53 induces apoptosis by transactivating proapoptotic genes such as Bax, Puma, Scotin, and Fas and repressing the antiapoptotic gene Bcl2.3 However, one of the main unanswered questions is how p53 “decides” to trigger the prosurvival or cell death responses. It has been documented that depending on the tissue and cell type, the nature and intensity of the stress signal, and the extent of cellular damage, p53 would favor one response to another. In other words, the question that remains to be answered is how one protein, p53, can integrate all these variables to yield a coordinated and defined cellular response. Additionally, the molecular mechanisms involved in such decision making are still unclear (reviews4-6). Indeed, in clinical studies, it has been difficult to link p53 mutation status to therapeutic response and clinical outcome, suggesting that additional factors may affect the p53 pathway.
The p53 Family
Two p53-related genes, p63 and p73, exhibit, like p53, the 3 typical domains of a transcription factor: the amino-terminal transactivation domain (TAD), the DNA-binding domain (DBD), and the carboxy-terminal oligomerization domain (OD).7,8 These 2 p53-related proteins share significant structural and functional homologies with p53, particularly in the DBD, including conservation of all essential DNA contact residues (review9). The p63 gene was shown to express at least 3 alternatively spliced C-terminal isoforms (α, β, γ), while the p73 gene expresses at least 7 alternatively spliced C-terminal isoforms (α, β, γ, δ, ε, ζ, η) and 4 alternatively spliced N-terminal isoforms.9 Furthermore, both p63 and p73 genes can be transcribed from 2 different promoters: one upstream of exon 1 (the distal promoter) and another located within intron 3 (the internal promoter). The distal promoter leads to the expression of TAp63 and TAp73, respectively, while the internal promoter leads to the expression of isoforms deleted of the N-terminal domain, ΔNp63 and ΔNp73, respectively. Therefore, the p63 gene expresses 6 mRNA variants that code for 6 different p63 protein isoforms, while the p73 gene expresses at least 35 mRNA variants that would encode 29 different p73 protein isoforms.9
Genetic experiments on mice have shown that p63 is essential for epidermal morphogenesis and limb development. p63-null mice display severe deformities of the limbs and fail to develop a stratified epidermis and most epithelial tissues. In humans, 6 rare autosomal dominant developmental diseases involving limb and ectodermal development are due to germline mutations throughout the p63 gene. Each syndrome can result from mutations in the p63 gene affecting different p63 isoforms. For instance, EEC (ectrodactyly–ectodermal dysplasia–cleft) and ADULT (acro-dermato-ungual-lacrimal-tooth) syndromes result from missense mutations in the DBD of p63 affecting all p63 isoforms, whereas AEC (ankyloblepharon–ectodermal dysplasia–cleft) syndrome is caused by missense mutations in exon 13 mutating only TAp63α and ΔNp63α isoforms. This delineates the specific biological and biochemical activities that each p63 isoform could have (review9).
Regarding p73, mice deficient for all p73 isoforms are born without severe deformities but with profound defects in neurogenesis. They also develop chronic infections and inflammation and have abnormalities in pheromone sensory pathways, without increased susceptibility to spontaneous tumorigenesis. Contrary to p63, no human genetic disorders have been associated yet with germline mutations of the p73 gene10 (review9).
The recent work of Mak and colleagues confirmed the tumor suppressor function of TAp73 by generating mice deficient for TAp73 but retaining the ΔNp73 isoforms.11 Although the developmental defects of these mice are less severe than their p73−/− counterparts, TAp73−/− mice defined a role for TAp73 in the development of the central nervous system, as these mice show hippocampal dysgenesis.11 More importantly, they are more prone to spontaneous and DMBA-induced tumors (7,12-dimethylbenz[a]anthracene), recapitulating the tumor-prone phenotype of the p73+/− mice11 (review12).
More recently, 2 groups have also generated mice that are selectively deficient for the ΔNp73 isoforms.13,14 These mice are viable like the TAp73−/− mice. The developmental defects of the ΔNp73−/− mice are less severe than their p73−/− counterparts, but they display signs of neurodegeneration, supporting the role of ΔNp73 in neuronal survival.13,14 Mak and colleagues also reported a tumor inducer function of ΔNp73, as ΔNp73−/− mice show an impairment of tumor formation.14 Altogether, studies from human developmental diseases and genetic experiments on mice have highlighted specific biological and biochemical activities that each p63/p73 isoform could have (review9).
The human p53 gene was first thought to have a much simpler organization than the p63 and p73 genes, with transcription being initiated from 2 distinct sites upstream of exon 1 (P1 and P1′). Our laboratory has assessed the structure of the human p53 gene, using a PCR-based technique that specifically amplifies capped mRNAs.15 This showed that, similarly to p63 and p73, 12 distinct human p53 protein isoforms (p53** when we mention p53, we refer to FLp53) (full-length p53 or FLp53, p53β, p53γ, Δ40p53α, Δ40p53β, Δ40p53γ, Δ133p53α, Δ133p53β, Δ133p53γ, Δ160p53α, Δ160p53β, Δ160p53γ) can be produced through alternative initiation of translation, usage of an internal promoter, and alternative splicing (reviews16,17) (Fig. 1). Therefore, the dual gene structure, that is, the gene being transcribed from 2 distinct promoters, a distal and an internal promoter, is conserved for the 3 p53 family members (p53, p63, and p73). This suggests that the internal promoters play essential roles in the biological activities of the p53 gene family. Furthermore, our laboratory and others have shown that the dual gene structure of the p53 gene is conserved across different species, including the Drosophila and zebrafish p53 genes. Indeed, Chen and colleagues reported that the zebrafish p53 gene contains an internal promoter leading to the expression of Δ113p53, an amino-terminally truncated p53 protein initiated at codon 113 and homologous to human Δ133p53.18,19
Figure 1.
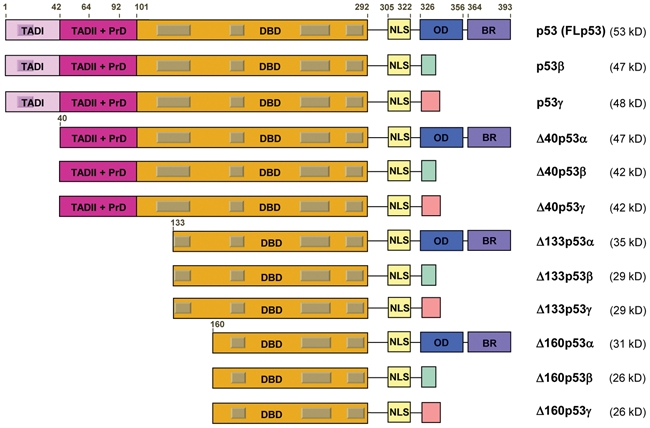
The human p53 gene expresses 12 distinct p53 protein isoforms. Schematic representation of the domains of human p53 isoform proteins including the 2 transactivation domains (TADI [light purple] and TADII [dark purple]), the DNA-binding domain (orange), the C-terminal domain comprised of the nuclear localization signal (NLS [yellow]), the oligomerization domain (OD [blue]), and the basic region (BR [violet]). The gray boxes represent the 5 highly conserved regions defining the p53 protein family. The amino acid positions defining the different p53 domains are indicated. The C-terminal domains of p53β (DQTSFQKENC) and p53γ (MLLDLRWCYFLINSS) are indicated with a green and pink box, respectively. The molecular weight of each p53 isoform protein is indicated.
Biological Activities of the p53 Isoforms
Δ133p53α
The first genetic evidence demonstrating that endogenous p53 isoforms have crucial biological activities came from gene silencing experiments after injection of p53 morpholino RNA (antisense oligonucleotides used to knock down gene expression) into zebrafish embryos. The zebrafish model provides numerous advantages over other mammalian models including the mouse model, in that it is cheaper and assays can be performed more rapidly. p53 isoforms have been identified in zebrafish and were found to be induced in response to developmental defects in zebrafish embryos.18,20 In collaboration with the laboratory of J. Peng (A-star, Singapore), we have recently reported that, in response to DNA-damaging agents and embryonic defects, the zebrafish p53 internal promoter is induced by p53, leading to Δ113p53 protein expression, which prevents p53-mediated apoptosis.19 Zebrafish embryos depleted of Δ113p53 expression after Δ113p53 morpholino injection were extremely sensitive to ionizing radiation and embryonic defects. Restoration of Δ113p53 induces p53 target gene expression including MDM2, cyclin G, p21, and Bcl-xL, thus inhibiting apoptosis and rescuing the development of embryos.19 Moreover, we have shown that Δ113p53 regulates gene expression in a promoter- and stress-dependent manner as well as in a p53-dependent and -independent manner. Therefore, Δ113p53 does not simply act in a dominant-negative manner toward p53 but rather modulates p53 response by differentially regulating the expression profile of p53 responsive genes at the physiological level in zebrafish. These data clearly demonstrate that the Δ113p53 isoform has intrinsic biological activity at the physiological level in zebrafish embryos. Δ113p53 prevents p53-mediated apoptosis in response to developmental defects and DNA damage.
In human cells, we have shown that ectopic expression of Δ133p53α inhibits p53-mediated apoptosis after transient transfection in H1299 cells, which are devoid of p53 expression.15 We have recently reported the regulation of Δ133p53α expression and its physiological role in modulating the cellular response to DNA damage.21 We showed that the human p53 internal promoter is directly transactivated by p53 in response to genotoxic stress, leading to Δ133p53α protein induction. The induced Δ133p53α differentially regulates gene expression in a p53-dependent and -independent manner, inhibiting then p53-dependent apoptosis and G1 arrest, without inhibiting p53-dependent G2 arrest in U2OS cells. This indicates that Δ133p53α can differentially regulate p53-dependent biological activities and that Δ133p53α does not act exclusively by inactivating p53. Moreover, it demonstrates that the cellular response to DNA damage could be regulated by modulating Δ133p53α expression.21
Consistent with this, our laboratory has recently reported in collaboration with the group of Professor C. Harris (National Institutes of Health, Bethesda, MD, USA) that expression of Δ133p53α isoform inhibits p53-mediated replicative senescence and promotes cellular proliferation of normal human fibroblasts by inhibiting p21 expression. Importantly, Δ133p53α concomitantly represses the expression of miR-34a, showing, for the first time, interplay between Δ133p53α and miR-34a to regulate p53-mediated senescence.22
Altogether, these results indicate that Δ133p53α modulates cell proliferation and cell fate outcome in response to DNA damage and developmental defect in a p53-dependent and -independent manner by differentially regulating the expression of microRNAs and protein-coding genes. Our findings may have profound significance to further our understanding of the mechanisms by which p53 exerts its tumor suppressor activity.
Δ133p53β and Δ133p53γ
p53 mRNAs are transcribed from the internal promoter (in intron 4) and are also subject to alternative splicing of exon 9b (in intron 9), producing, in addition to the Δ133p53α isoform, 2 other isoforms, Δ133p53β and Δ133p53γ.15 In H1299 cells, coexpression of Δ133p53β or Δ133p53γ with p53 does not alter p53 transcriptional activity on the p21 and Bax promoters or p53-mediated apoptosis.9,15 Δ133p53β and Δ133p53γ are expressed in normal human tissues. However, several clinical studies have shown that Δ133p53β and Δ133p53γ are abnormally expressed in tumors, suggesting that they play a role in carcinogenesis.15,23
Δ160p53α, Δ160p53β, and Δ160p53γ
Δ160p53α, Δ160p53β, and Δ160p53γ are p53 isoforms that we have recently reported to lack the first 159 amino acids of p53.17 Δ160p53α, Δ160p53β, and Δ160p53γ are encoded by Δ133p53α, Δ133p53β, and Δ133p53γ mRNAs, respectively, through alternative initiation of translation at ATG 160, which lies within a sequence environment matching Kozak’s consensus criteria (GCC GCC (A/G)CC ATG G).17 Intriguingly, ATG 160 is conserved in all mammalian p53 genes, while ATG 133 is conserved only in primates. Endogenous expression of Δ160p53 protein was detected in U2OS, T47D, and K562 cells.17 Interestingly, K562 cells were described as devoid of p53 expression because of a base insertion at codon 135, leading to a frameshift and a premature stop codon, which prevents p53 protein expression.24 However, we show that K562 cells express Δ160p53α and Δ160p53β both at the mRNA and protein levels. Moreover, upon hemin-induced erythroid differentiation of K562 cells, only Δ160p53β expression was reduced, while Δ160p53α expression was stable, suggesting that Δ160p53β plays a role in erythroid differentiation.17
p53β
It was shown that intron 9 (exon 9b) of the p53 gene can be alternatively spliced to produce 2 different truncated human p53 proteins, p53β (previously described as p53i9) and p53γ, terminating with 10 or 15 additional amino acids, respectively.15,25,26 Both p53β and p53γ isoforms lack the oligomerization domain of p53 because of a stop codon contained in exon 9b.
When p53β (or p53i9) was first described, it was shown to fail to bind DNA in vitro and to lack transcriptional activity in normal cells.26 However, using an antibody specific for the β peptide of p53 in chromatin immunoprecipitation (ChIP) assay on MCF7 cell extracts, we showed that endogenous p53β can bind specifically to p53-responsive promoters in a promoter-dependent manner. p53β preferentially binds to the Bax promoter but poorly to the MDM2 promoter, while p53 preferentially binds to the MDM2 promoter but poorly to the Bax promoter.15 Our laboratory determined that p53β can induce the PG13 promoter, an artificial promoter containing 25 adjacent p53REs upstream of a minimal promoter and driving a luciferase reporter gene, indicating that p53β has a residual intrinsic transcriptional activity on p53REs.9 Moreover, p53β enhances p53 transcriptional activity on the p21 promoter but has no effect on the Bax promoter in the absence of cellular stress. In addition, expression of p53β induces apoptosis independent of p53, albeit with a lower efficiency than p53. Furthermore, p53β enhances p53-mediated apoptosis after cotransfection with p53 in H1299 cells. This effect can be due to a direct interaction of p53β with p53 because endogenous p53β can form a protein complex with p53.15 However, we and others could not coimmunoprecipitate p53β with p53 after transient transfection in p53-null H1299 cells, suggesting that the interaction between p53 and p53β involves other p53 isoforms or proteins. It was also reported that p53β cooperates with p53 to accelerate replicative cellular senescence of human normal fibroblasts by increasing p53-dependent induction of p21 and miR-34a.22
Altogether, these data suggest that p53β regulates p53 tumor suppressor activity by modulating its transcriptional activity in a promoter-dependent manner. However, we should not rule out the possibility that p53β can regulate gene expression independent of p53, as p53β can bind p53REs and induce apoptosis independent of p53.
p53γ
p53γ is a p53 protein truncated of the last 60 amino acids of p53 and contains 15 additional amino acids (peptide γ).15 p53γ is expressed in normal human tissues through alternative splicing of exon 9b (in intron 9). The C-terminal domain of p53γ is hydrophobic, a feature that makes it difficult to raise an antibody specific for this isoform. The subcellular localization of p53γ is different from the one of p53β. p53β is mostly localized in the nucleus, while p53γ is either localized in the nucleus or the cytoplasm, suggesting that p53γ shuttles between the nucleus and the cytoplasm.15 By luciferase reporter assay, we determined that p53γ can transactivate the internal promoter of p53, independent of p53, and can enhance p53 transcriptional activity on the Bax promoter but not on the p21 promoter.15 In addition, we have attempted to generate stable U2OS and MCF7 cell lines constitutively expressing p53γ. However, we were not successful in generating cells with functional p53γ, as in the few clones that were able to grow, p53γ was localized exclusively in the cytoplasm, suggesting that p53γ was sequestered and thus inactivated. This led us to the conclusion that p53γ is cytotoxic.
However, it is important to note that these results are not consistent with those of Graupner and colleagues, who have reported that p53β and p53γ have no effect on senescence, apoptosis, and transcription.27 The report from Graupner and colleagues is not consistent with several publications demonstrating that p53β binds DNA, regulates transcription, forms a protein complex with p53, and regulates apoptosis, cell cycle progression, and senescence.9,15,22 The results of Graupner and colleagues can be explained by the experimental conditions used. The authors transfected cells with flag-tagged p53, flag-tagged-p53β, or flag-tagged p53γ and selected cells for several weeks that were able to proliferate in the presence of neomycin, a potent inducer of cell death. Hence, they obtained clones constitutively overexpressing p53, p53β, or p53γ. However, when Graupner and colleagues assessed the sensitivity of the clones to several cytotoxic treatments, they did not detect any differences with the parental cells and concluded that p53β and p53γ have no cytotoxic activities. A more likely interpretation is that by selecting cells resistant to neomycin, Graupner and colleagues have generated cells that have acquired a strong resistance to the cytotoxic effect of p53, p53β, and p53γ.
Δ40p53α, Δ40p53β, and Δ40p53γ
Δ40p53 (also named p47 or ΔNp53) is another p53 isoform that lacks the first transactivation domain of p53 (TADI) and that was shown to be obtained in humans by alternative splicing of intron 2 of p53 mRNA, leading to a p53I2 transcript.28 Recent studies have demonstrated the presence of internal ribosome entry site (IRES) sequences in p53 mRNA, which allow p53 mRNA translation in conditions where the cap-dependent initiation of translation is inhibited, such as cytotoxic and endoplasmic reticulum stresses.29-31
Consistently, the presence of the IRES sequences in p53 mRNA upstream of AUG 40 contributes to the alternative initiation of translation at codon AUG 40 (instead of AUG 1) and therefore to Δ40p53 expression.29,32,33 It was reported that the p53 IRES sequences are regulated by several IRES transacting factors (ITAFs) including polypyrimidine tract binding protein (PTB), dyskerin, and hnRNP C1/C2, modulating the expression of p53 and Δ40p53 proteins and thus p53 tumor suppressor activity.34-37 Δ40p53 isoform was also shown to be obtained by alternative initiation of translation at codon 40 located in exon 4 of the human p53 gene and lying within a sequence environment matching Kozak’s consensus criteria (GCC GCC (A/G)CC ATG G).28,32,33
It was further demonstrated that in addition to Δ40p53 obtained by classic splicing of the C-terminus (Δ40p53α), the p53 gene can produce alternatively spliced C-terminal Δ40p53 isoforms (Δ40p53β and Δ40p53γ). Although endogenous Δ40p53β and Δ40p53γ proteins are detected in several cell lines, their biological activities have not been investigated yet.
Although Δ40p53α lacks the TADI, it retains the second transactivation domain (TADII) and is therefore capable of regulating gene expression after transfection.38 Also after transfection, Δ40p53 can act in a dominant-negative manner toward p53, inhibiting its transcriptional activity and impairing p53-mediated growth suppression28,33 (review39).
The level of expression of p53 can be controlled through its degradation. However, it can also be controlled by regulating its mRNA translation, as determined in conditions of cellular stresses such as endoplasmic reticulum stress (ER stress).31,40 It has been suggested that Δ40p53 expression is controlled by alternative mechanisms of mRNA translation initiation via an IRES sequence present in the 5′UTR of p53 mRNA (upstream of codon 40). This would allow Δ40p53 to be expressed under conditions where the cap-dependent initiation of translation is inhibited, such as the G2/M transition of the cell cycle and ER stress. Following ER stress, Δ40p53 expression is increased (caused by PERK-mediated stimulation of Δ40p53 mRNA translation), and Δ40p53 homo-oligomers are formed, which bind to the 14-3-3σ promoter and mediate G2 cell cycle arrest or Δ40p53-induced apoptosis.31,40
p53 Isoforms in Animal Models
The first report revealing that the mouse p53 gene codes for more than one functional protein was published by Rotter and colleagues in 1985, describing the presence of another p53 variant in transformed mouse fibroblasts.41 This p53 variant was reported to be generated by alternative splicing using a cryptic 3′ splicing site in intron 10, located 96 bp upstream of the regular 3′ splicing site of exon 11, thereby naming it p53AS (alternatively spliced).42 A stop codon in the 96-bp insert from intron 10 leads to the production of a truncated p53 protein, in which the last 26 amino acids of mouse p53 are missing and are replaced by 17 new amino acids homologous to human β peptide of p5341,42 (Fig. 2). Like p53β, p53AS can form hetero-oligomers with p53.43 p53AS was shown to be expressed in normal mouse tissues as well as in normal epidermal and carcinoma cells and to localize to the nucleus.44-47
Figure 2.

p53AS: protein homology with p53β and biochemical activities. (A) Protein homology between human p53b ((h)p53b) and murine p53AS ((m)p53AS) isoforms. Conserved amino acids are highlighted in red. (B) Biochemical properties of mouse FLp53 and p53AS. (a) The intracellular localization, DNA binding, oligomerization, and (b) gene transactivation capacities (determined by luciferase assay) are highlighted. ND = not done; + = positive effect; ++ = stronger effect. Information obtained from Arai et al.,42 Kulesz-Martin et al.,45 Wu et al.,43 Miner and Kulesz-Martin,49 Wu et al.,47 Almog et al.,53 Wolkowicz et al.,48 Almog et al.,54 Almog et al.,55 and Huang et al.102.
Several groups have shown that p53 and p53AS bind specifically to p53REs but have distinct biochemical activities and are functionally different. Contrary to p53, p53AS protein is constitutively active for DNA binding.43,48,49 This is in accordance with in vivo results showing that deletion of the last 30 amino acids of p53 C-terminus or binding to the monoclonal antibody PAb421 (whose epitope lies between residues 370-378) activate the sequence-specific DNA binding and the transcriptional activities of p53.50-52 Additionally, p53 and p53AS can form hetero-oligomers, resulting in inactivation of p53AS DNA-binding activity.43,48
In stably transfected myeloid cells, p53AS is able to induce apoptosis, albeit with much slower kinetics (a 12-hour delay) when compared to that induced by p53, accompanied with a delay in Bax induction and Bcl-2 repression.53 Indeed, p53AS is a less potent transactivator of Bax and p21 promoters, although p53AS has a stronger affinity to p53REs.54
Interestingly, coexpression of p53 and p53AS in stably transfected myeloid cells or in transiently transfected p53-null H1299 cells results in an inhibition of p53 transcriptional activity and p53-mediated apoptosis.54 The p53AS inhibitory effect on p53-mediated apoptosis is MDM2 dependent, as p53AS is able to induce higher levels of the MDM2 protein than p53.55 Although p53AS was shown to regulate the expression of several genes including p21, Bax, and Bcl-2, Kulesz-Martin and colleagues reported that p53AS is more effective in transcription repression than p53, highlighting a role of p53 C-terminal domain in this effect.44
Mouse Δ40p53 isoform (also named ΔNp53 or p44) is expressed in normal tissue through alternative initiation of translation at codon 41. Genetically modified mice overexpressing Δ40p53 have been generated. These mice present an increased cellular senescence, a slower growth rate, memory loss, neurodegeneration, and premature aging phenotype.56-58 A ratio of p53/Δ40p53 has been proposed to regulate the aging program by modulating insulin-like growth factor 1 receptor (IGF-1R) signaling.57,58
Interestingly, heterozygote p53 mice (p53/Δ40p53) are less susceptible to cancer than heterozygote p53+/− mice. However, homozygote mice (Δ40p53/Δ40p53) are as cancer prone as p53−/− mice and do not show any accelerated aging phenotype, indicating that the accelerated aging phenotype is dependent on the interplay between Δ40p53 on wild-type p53.56,57
Altogether, these results highlight an important role for mouse p53 isoforms in regulating p53 transcriptional activity, especially under stress conditions. They also emphasize the importance of the ratio of p53 isoforms for regulating cell fate outcome.
Interestingly, a Δ40p53 isoform (also named ΔNp53) has recently been described in zebrafish. Δ40p53 was shown to be obtained by alternative splicing of intron 2 of the zebrafish p53 gene and not by alternative initiation of translation because codon 40 is not conserved in zebrafish p53 mRNA. Zebrafish Δ40p53 protein lacks part of the transactivation domain, but unlike human Δ40p53, it contains additional amino acids encoded by intron 2.20 Interestingly, ionizing radiation induces Δ40p53 and Δ113p53 mRNA transcript levels in zebrafish embryos, while p53 mRNA level is stable. Injection of zebrafish Δ40p53 mRNA induces lethality in approximately 30% of the embryos within 5 to 7 days, associated with hypoplastic and malformed heads, eyes, and somites in the surviving embryos.20 Depletion of p21 expression by injection of p21 morpholino into zebrafish embryos rescues developmental defects associated with Δ40p53 overexpression. Contrary to Δ40p53, injection of Δ113p53 mRNA into zebrafish embryos has no deleterious effect on embryo development.19,20 Altogether, these data indicate that Δ40p53 and Δ113p53 have distinct intrinsic activities.
Injection of zebrafish p53 mRNA induces lethality in approximately 80% of the embryos preceded by multiple morphological aberrations.20 The experiments performed by Davidson and colleagues further confirm that Δ40p53 and Δ113p53 have distinct intrinsic activities. Indeed, co-injection of zebrafish p53 and Δ113p53 mRNAs totally rescues p53-associated lethality, while co-injection of zebrafish p53 and Δ40p53 mRNAs partially rescues p53-associated lethality.
Although Δ40p53 can form a protein complex with Δ113p53, co-injection of Δ113p53 mRNA with Δ40p53 mRNA into zebrafish embryos does not rescue Δ40p53-associated developmental defects, suggesting that contrary to the inactivating effect of Δ113p53 on p53, Δ113p53 does not inactivate Δ40p53. Therefore, Δ40p53 and Δ113p53 are not equivalent and are likely to have specific p53-dependent and -independent biological activities.
p53 Isoforms and Cancer
Despite 30 years of research on p53 demonstrating the key role of p53 in cancer treatment and prevention of cancer formation, it is still difficult in clinical studies to link p53 mutation status to cancer prognosis and cancer treatment. The uncertainties around the link between p53 mutation, therapeutic response, and outcome in cancer suggest that additional factors may be involved. We believe that p53 isoforms could provide an explanation to this question.
Several clinical studies have reported that p53 isoforms are abnormally expressed in different types of human cancers (breast tumors, acute myeloid leukemia [AML], head and neck tumors [HNSCCs], melanoma, renal cell carcinoma, and colon, ovarian, and lung tumors), suggesting that abnormal expression of the p53 isoforms could contribute to cancer formation and cancer progression.15,23,59-64
Fujita and colleagues determined that Δ133p53α inhibits senescence while p53β promotes senescence in normal human fibroblasts. They analyzed the expression of Δ133p53α and p53β in a cohort of colon adenomas and carcinomas and reported an association of the senescent phenotype of colon adenoma with reduced Δ133p53α and increased p53β expression. Interestingly, Δ133p53α expression was increased, while p53β isoform expression was decreased in colon carcinomas, suggesting that deregulation of p53β and Δ133p53α may contribute to adenoma to carcinoma progression.22
Hofstetter and colleagues have recently analyzed p53 isoform expression in a cohort of 245 primary ovarian cancers in relation to clinical marker and clinical outcome. They reported that p53β expression was associated with adverse clinicopathological markers (serous and poorly differentiated cancers) and correlated with worse recurrence-free survival in patients expressing functionally active wild-type p53. Moreover, they reported frequent mutations in splicing sites of p53 that lead to the expression of tumor-specific p53 splice variant mRNA, including one named p53δ, which was associated with poor response to primary platinum-based chemotherapy. Consistently, p53δ mRNA expression was independently associated with poor prognosis. It remains to be shown whether p53δ mRNA leads to p53δ protein expression and whether p53δ has oncogenic activities.
We have recently reported the analysis of p53β and p53γ mRNA expression in relation to clinical outcome and clinical markers in a cohort of 127 primary breast tumors. We determined that p53β and p53γ are not randomly expressed in breast cancer. Indeed, p53β is associated with p53γ expression, and p53γ is associated with p53 gene mutation, while p53β is associated with estrogen receptor expression (ER).63 Interestingly, mutant p53 breast cancer patients expressing the p53γ isoform have low cancer recurrence and an overall survival as good as wild-type p53 breast cancer patients, independent of ER status. Conversely, mutant p53 breast cancer patients devoid of p53γ expression have a particularly poor prognosis. We did not observe any significant difference in wild-type p53 breast cancer patients whether they expressed p53β/p53γ or not.
Therefore, the determination of p53γ expression allows the identification of 2 populations of mutant p53 breast cancer patients with different prognoses, independent of ER status and cancer treatment. Indeed, mutant p53 breast cancer patients expressing p53γ have a prognosis as good as wild-type p53 breast cancer patients, suggesting that they may respond better to treatment. On the other hand, mutant p53 breast cancer patients not expressing p53γ have a particularly poor prognosis probably because they poorly respond to treatment. p53γ isoform may provide an explanation of the hitherto inconsistent relationship between p53 mutation, treatment response, and outcome in breast cancer.
In conclusion, the above clinical data report the expression of p53 isoforms in several types of cancer, confirming that p53 isoforms are expressed both at the mRNA and protein levels. Because p53 isoforms can regulate cell proliferation (cell cycle progression, senescence, and apoptosis) and are abnormally expressed in different cancer types, it suggests that their differential expression may disrupt the p53 response and contribute to tumor formation. Therefore, p53 isoforms may provide an explanation to the difficulties in many clinical studies to link p53 status to cancer prognosis and treatment. In cancer cells, restoration of p53β/p53γ or abolition of Δ133p53 expression would impair tumor cell growth by inducing senescence or cell death and therefore may represent novel therapeutic targets.
Biochemical Activities
Our results on Δ133p53α reveal the importance of p53-induced Δ133p53α in modulating the cellular response to DNA damage. The latest reports define Δ133p53α as an essential component of the p53 pathway involved in the cell fate decision and demonstrate that Δ133p53α does not act exclusively as a dominant-negative regulator of p53 activities but rather differentially modulates gene expression at both transcriptional and posttranscriptional levels.21,22 However, we cannot rule out the possibility that Δ133p53α has biological activities independent of p53, as revealed by the zebrafish embryo studies.19
It is well established that p53 transcriptional activity is required for its biological activities. p53 is a transcription factor that binds specifically p53RE DNA sequences as a tetramer. Δ133p53α lacks the L1 loop of the p53 DBD (residues 117-142), which is essential for the binding of p53 to p53RE. Consistently, Marcel and colleagues have demonstrated by gel shift assay that Δ133p53α does not bind the consensus p53RE without p53.65 However, it is important to note that although Δ133p53 lacks the L1 loop, it retains the highly conserved helix (H2) of the DBD (residues 270-286), which binds to the major groove of DNA and the C-terminal domain required for the linear diffusion of p53 along DNA.66,67 Therefore, we shall not rule out the possibility that Δ133p53α specifically binds to DNA using either a different DNA consensus sequence or different p53REs.
Δ133p53α could regulate transcription through direct interaction with transcription factors. Indeed, we showed that Δ133p53α can form a protein complex with p53, which suggests that the interaction of Δ133p53α with p53 could either enable Δ133p53α to bind p53RE or impair the binding of p53 to p53RE.21 Indeed, Marcel and colleagues have demonstrated by gel shift assay that Δ133p53α inhibits the binding of p53 on an oligonucleotide containing a p53RE sequence, suggesting that the hetero-oligomer p53:Δ133p53α is unable to bind this p53RE.65 Moreover, we determined by luciferase assay that cotransfection of Δ133p53α with p53 inhibits p53 transcriptional activity on the Bax and p21 promoters.15 Depletion of endogenous Δ133p53α after transfection of siRNA specific for Δ133p53 significantly increases the expression of p21 mRNA in response to DNA damage in U2OS cells.21
Altogether, these data suggest that Δ133p53α can inhibit p53 transcriptional activity on the p21 promoter by inhibiting its DNA-binding activity through direct inactivating interaction. However, further studies will determine whether Δ133p53α can regulate p21 or Bax promoter activity through other mechanisms.
Importantly, our results indicate that Δ133p53α does not act exclusively by inhibiting p53 because overexpression of Δ133p53α does not inhibit p53-dependent G2 cell cycle arrest in U2OS cells in response to DNA damage. Moreover, Δ133p53α induces Bcl-2 expression and contributes to p53-mediated induction of HDM2 in response to DNA damage.21 Further experiments are required to elucidate the regulation of HDM2 expression by Δ133p53α because the HDM2 gene contains 3 promoters (P1 upstream of exon 1, P2 in intron 1, and P3 in intron 3), 2 of which are responsive to p53; P2 is induced by p53, while P3 is repressed by p53.68,69
Δ133p53α-mediated induction of Bcl2 in response to stress seems to be p53 independent because depletion of p53 by siRNA (targeting specifically FLp53) does not induce Bcl2 expression.21 The Bcl2 gene contains 2 promoters, which have been reported to be repressed by p53. However, the regulatory regions responsive to p53 have not been clearly defined.70-72 Further experiments will allow the identification of the promoter region responsive to Δ133p53α. Altogether, this suggests that, despite its low protein expression level, Δ133p53α regulates gene expression in a promoter- and stress-dependent manner by inhibiting the binding of p53 to p53RE, by enhancing p53 transcriptional activity on promoters, or by regulating gene expression independent of p53.
Regarding p53β, it has been reported that this p53 isoform can bind directly p53-responsive promoters independent of p53, albeit p53β preferentially binds the Bax and p21 promoters rather than the HDM2 promoter, suggesting that p53β would bind only a subset of p53-responsive promoters.15 Endogenous p53β can form a protein complex with p53, as demonstrated by coimmunoprecipitation assay. By luciferase gene reporter assay, we reported that p53β can simultaneously enhance and inhibit p53-dependent transcriptional activity in a promoter- and stress-dependent manner.9,15,22 Of note, it is likely that p53β regulates gene expression independent of p53 because it can bind specifically to DNA in the absence of p53.
Therefore, based on our current knowledge, we would like to propose a speculative molecular mechanism for p53 isoforms. We will focus the model on the regulation of p53 transcriptional activity by p53β and Δ133p53 isoforms. However, it is worth mentioning that they may have transcriptional activities independent of p53 and may interact with other transcription factors.
It is well documented that depending on the tissue type, the nature of the cellular damages (such as DNA damage, damage to the cytoplasmic membrane, the endoplasmic reticulum, mitochondria), due to a variety of stress inducers (such as ultraviolet, hypoxia, ionizing radiation, nutrient deprivation, viral infection), and the intensity of the damages (acute or chronic) and the extent of the damages, the transcription factor p53 would trigger differentiation, senescence, and cell death or promote a prosurvival response by inducing cell cycle arrest and cell/organ repair, in order to rapidly restore cell and organ functions. Such key biological activities are complex to put in place, as they require the orchestrated regulation of multiple molecular pathways and the coordinated involvement of different cell types within a damaged tissue to rapidly restore its function.
Interestingly, p53 is involved in a vast number of molecular pathways (energy metabolism, antioxidant response, DNA repair, endoplasmic reticulum stress, lipid synthesis, nucleotide synthesis, ROS scavenger, nutrient supply, angiogenesis, cell motility, inflammation, antiviral response, among others) (review3). Thus, the key questions are the following: how can p53 protein be activated by almost any type of damage? How can p53 protein be involved in so many different pathways leading to one defined cellular response, adapted to the damages and the tissue type?
It is well established that p53 transcriptional activity is required for its biological activities. It thus implies that p53 differentially regulates gene expression. p53 can regulate gene expression at the transcriptional level by direct binding to DNA on p53RE or by interacting with other transcription factors. We will focus the model on the transcriptional activity of p53 upon binding to p53RE.
The DNA p53 consensus sequence of p53RE is degenerated and is written RRRCWWGYYY, where R is G/A, W is A/T, and Y is C/T. Several studies have shown that p53RE can have up to 3 discrepancies compared to the p53 consensus sequence. The discrepancies occur very rarely on C and G at positions 4 and 7, respectively.2,73-75 Therefore, there are 28 (or 256) different ways to write a sequence RRRCWWGYYY with no discrepancy, 2,048 different sequences with 1 discrepancy, 7,168 different sequences with 2 discrepancies, and 14,336 sequences with 3 discrepancies to the p53 consensus sequence. Hence, there are a total of 23,808 different ways to write a sequence RRRCWWGYYY with 0 to 3 discrepancies to the consensus sequence, without variation on C and G at positions 4 and 7 (Fig. 3). The nucleotide sequence of the p53RE as well as the number of p53REs present in a given promoter have been shown to influence promoter selectivity and the responsiveness to p53 (reviews5,6).
Figure 3.

Total number of p53RE sequences that can be written RRRCWWGYYY with 0 to 3 errors.
Several other factors have been shown to influence p53 promoter selectivity and thus the cell fate outcome. In the first part of the review, we reported that the cell fate outcome can be switched in response to a defined stress from p53-mediated prosurvival to p53-induced cell death by only manipulating p53 isoform expression (Fig. 4). Therefore, we consider that p53 isoforms play essential roles in modulating p53 biological activities and are essential to the coordination of the multiple molecular pathways involved.
Figure 4.
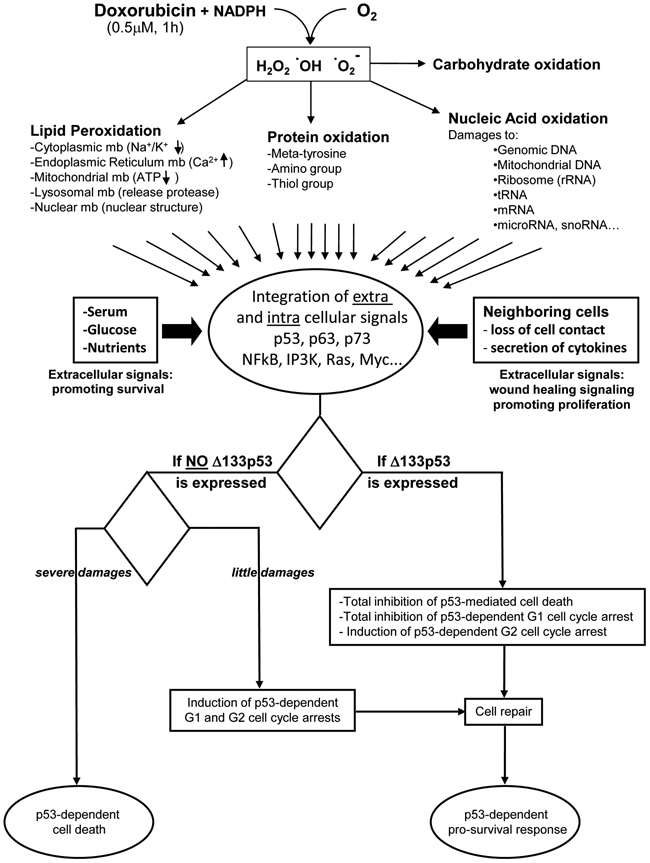
Extracellular and intracellular signals integrated by a single cell in response to a low dose of doxorubicin. Oxidation of doxorubicin generates free radicals that react with lipids, carbohydrates, proteins, and nucleic acids. Hence, doxorubicin causes lipid peroxidation, thus releasing the toxic content from the different subcellular organelles and causing loss of ATP production by mitochondria. Doxorubicin also causes loss of enzymatic activity and protein aggregation through thiol group oxidation, amino group oxidation, and formation of metatyrosine. Doxorubicin inhibits topoisomerase II activity, inducing DNA double-strand breaks. Moreover, doxorubicin binds covalently to DNA, inhibiting transcription and replication. Free radicals break all nucleic acids including mitochondrial DNA, ribosomal RNA, tRNA, mRNA, and microRNA. The damages are related to the concentration and duration of incubation with doxorubicin. The damaged cell is also in contact with surrounding cells, which secrete cytokines in response to doxorubicin treatment. If the neighboring cells die, the damaged cell loses its contact with the neighboring cells, triggering wound healing signaling. This is reinforced by the presence of nutrients, glucose, and growth factors. The extracellular and intracellular signals are integrated by cellular proteins (including p53, p63, and p73 isoforms), which will induce, in relation to the damages and the cell type, either prosurvival pathways (cell cycle arrest followed by cell repair, leading to proliferation or senescence) or cell death. The cell fate outcome is different if Δ133p53α is expressed or not. When Δ133p53α is expressed, p53-mediated apoptosis and G1 arrest are inhibited, while the p53-mediated G2 cell cycle arrest is promoted, allowing cell repair.
In addition, p63 and p73 isoform proteins, belonging to the p53 protein family, can also bind p53RE and induce the expression of p53 target genes, such as p21, Bax, PUMA, and NOXA, causing cell cycle arrest or apoptosis in response to cellular stress74,76,77 (reviews78,79). p63 and p73 were shown to oligomerize and form homotetramers and heterotetramers, but not with p53.80,81 However, p53/p63/p73 can assemble on p53RE of p53 target gene promoters.74,76,82 Flores and colleagues have shown in mouse models that the combined loss of p63 and p73 genes, abolishing the expression of all p63 or p73 isoforms, results in the failure of cells containing functional p53 to undergo apoptosis in response to DNA damage.83 In addition, they highlighted the synergistic effects of the p53 family in tumor suppression, with mice heterozygous for mutations in both p53 and p63 or p53 and p73 displaying higher tumor burden and metastasis, compared to p53+/− mice.84 This indicates that p63/p73 isoforms can complement some of the biological activities of p53. Hence, p63, p73, and p53 isoforms integrate the stress signals and modulate synergistically p53/p63/p73 target gene expression to orchestrate the cellular response according to the nature, intensity, and extent of the damages.
Several genetic animal studies emphasized the importance of a fine ratio of the different p63/p73 isoforms to balance and fine tune the cellular response. Indeed, ΔNp73−/− mice (expressing TAp73) have a defect in DNA repair and are more sensitive to apoptosis, while TAp73−/− mice (expressing ΔNp73) have a defect in maintaining genome integrity and are cancer prone and sterile.11,14 Moreover, TAp73 ensures normal adult neurogenesis by promoting the long-term maintenance of neural stem cells.85 Regarding p63, it has been shown that TAp63 can induce senescence and tumor suppression in vivo or prevent premature aging by promoting adult stem cell maintenance.86,87 In addition, TAp63 suppresses metastasis through coordinated regulation of Dicer and miRNAs.88 Specific depletion of ΔNp63 isoforms in mouse epidermis causes severe epidermal defects, leading to the development of severe skin erosions indistinguishable from that of AEC patients.89,90 As p53, p63, and p73 isoforms are expressed in a tissue-dependent manner, it may explain why different human cell types respond differently to identical cellular damages.91,92
The promoter selectivity and activation by p53, p63, and p73 are also regulated by cofactors such as ASPP1/ASPP2/iASPP and posttranslational modifications such as phosphorylation and acetylation93-96 (reviews97,98). p63 and p73 and particularly p53 proteins are extensively modified by posttranslational modifications so that they are not expressed as one major unique protein but as different posttranslationally modified p53, p63, and p73 protein isoforms (review98).
Therefore, we would consider the transcription factor p53 as a multi-subunit protein complex composed of different amounts of p53, p63, and p73 protein isoforms that assemble on p53RE in response to stress. Hence, the binding and assembling/dismantling of p53, p63, and p73 isoforms into the transcription factor p53, as well as the interaction of the assembled transcription factor p53 with cofactors and the transcriptional machinery, would be regulated by posttranslational modifications.
A large majority of p53-responsive promoters contain clusters of RRRCWWGYYY sequences (p53RE), which enable the assembling of multiple isoforms of p53, p63, and p73 on promoters.2 The assembled transcription factor p53 bound to p53RE could thus have a different isoform composition depending on the promoter, the damages, and the cell and tissue types. Hence, several types of the multi-subunit transcription factor p53 could coexist within a cell. Thus, the large diversity of p53/p63/p73 isoforms would enable the integration and translocation of many and different stress signals to a p53-responsive promoter. Indeed, the vast diversity of p53RE would match the vast diversity of stress signals, enabling the translation of the diverse stress signals into an adapted cell response. It is worth mentioning that some p53 target genes contain several clusters of p53RE.99 It has been shown that the induction of a p53-responsive promoter containing a cluster of p53RE close to the TATA box is increased 25-fold in the presence of a distal cluster of p53RE. This is due to the fact that p53 proteins bound on distal and proximal clusters of p53REs interact and form a DNA loop, maximizing the integration and translocation of stress signals to p53-responsive promoters.99-101
Hence, we speculate that p53-inducible proapoptotic genes would contain clusters of p53RE with low affinity for p53, p63, and p73 isoforms so that a maximum of stress signals has to be integrated and translocated to promoters by the p53 protein family to induce the expression of deadly proapoptotic genes. Reciprocally, one would predict that promoters of genes involved in cell cycle arrest contain p53RE with high affinity for a wide range of p53, p63, and p73 isoforms so that they are the first induced in response to a vast diversity of stress signals. Therefore, such promoters would be little affected by the loss of p53, p63, or p73 isoform expression and/or aberrant cell signal transduction. Reciprocally, we can expect that some p53-responsive genes would require a specific composition of posttranslationally modified p53/p63/p73 isoforms to be transactivated. Such genes would thus be sensitive to mutation/polymorphism of their p53RE, unbalanced expression of p53/p63/p73 isoforms, or aberrant cell signal transduction. Hence, this model could provide an explanation to the maintenance of p53/p63/p73-mediated prosurvival response in tumor cells, while the p53/p63/p73-induced cell death is compromised.
In conclusion, the study of the p53 isoforms is still in its infancy, but the increasing number of publications indicates that p53 isoforms modulate gene expression and thus cell fate outcome in response to developmental defects and cell damages. p53 isoforms can modulate gene expression in a p53-dependent and -independent manner in response to stress. Several studies have shown that p53 isoforms are abnormally expressed in a wide range of cancers, suggesting that they play a role in carcinogenesis. As a molecular mechanism, we propose that the transcription factor p53 is in fact a multi-subunit protein composed of posttranslationally modified p53/p63/p73 isoforms. Hence, the large diversity of p53/p63/p73 isoforms enables the integration of a large diversity of stress signals to p53/p63/p73-responsive promoters. The large diversity of p53RE sequences as well as the number of p53REs enables the accommodation of the binding of a large diversity of p53/p63/p73 isoforms to translocate a maximum of different stress signals to responsive promoters. This would allow the orchestration of a defined and adapted cell response to the damages.
We focused our model on the modulation of gene expression by transcription. However, we expect p53/p63/p73 isoforms to modulate cell response in a transcription-independent manner. Future experiments will be needed to gain further insight into how the array of p53 isoforms modulates the function of p53 and the different biological activities, which will undoubtedly impact the fields of cancer, embryo development, and aging. The first results on p53/p63/p73 isoforms are promising and stimulating. They may provide an explanation to the difficulties in clinical studies to link p53 mutation status to cancer prognosis and treatment. The deciphering of the p53/p63/p73 isoforms interplay could help improve cancer prognosis and treatment in the near future.
Acknowledgments
The authors are very thankful to Dr. Terry Burke, Department of Mathematics at the University of Dundee, for the combinatorial calculation of p53RE sequences. They also thank Kenneth Fernandes and Mark Saville for reading and helpful discussions.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
This work was supported by Cancer Research UK (M.K., J.C.B.) and Breast Cancer Campaign (J.C.B.).
References
- 1. El-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B. Definition of a consensus binding site for p53. Nat Genet. 1992;1:45-9 [DOI] [PubMed] [Google Scholar]
- 2. Bourdon JC, Deguin-Chambon V, Lelong JC, et al. Further characterisation of the p53 responsive element: identification of new candidate genes for trans-activation by p53. Oncogene. 1997;14:85-94 [DOI] [PubMed] [Google Scholar]
- 3. Lane D, Levine A. p53 research: the past thirty years and the next thirty years. Cold Spring Harb Perspect Biol. 2010;2:a000893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bertheau P, Espie M, Turpin E, et al. TP53 status and response to chemotherapy in breast cancer. Pathobiology. 2008;75:132-9 [DOI] [PubMed] [Google Scholar]
- 5. Murray-Zmijewski F, Slee EA, Lu X. A complex barcode underlies the heterogeneous response of p53 to stress. Nat Rev Mol Cell Biol. 2008;9:702-12 [DOI] [PubMed] [Google Scholar]
- 6. Menendez D, Inga A, Resnick MA. The expanding universe of p53 targets. Nat Rev Cancer. 2009;9:724-37 [DOI] [PubMed] [Google Scholar]
- 7. Kaghad M, Bonnet H, Yang A, et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell. 1997;90:809-19 [DOI] [PubMed] [Google Scholar]
- 8. Yang A, Kaghad M, Wang Y, et al. p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell. 1998;2:305-16 [DOI] [PubMed] [Google Scholar]
- 9. Murray-Zmijewski F, Lane DP, Bourdon JC. p53/p63/p73 isoforms: an orchestra of isoforms to harmonise cell differentiation and response to stress. Cell Death Differ. 2006;13:962-72 [DOI] [PubMed] [Google Scholar]
- 10. Yang A, Walker N, Bronson R, et al. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumors. Nature. 2000;404:99-103 [DOI] [PubMed] [Google Scholar]
- 11. Tomasini R, Tsuchihara K, Wilhelm M, et al. TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Dev. 2008;22:2677-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rosenbluth JM, Pietenpol JA. The jury is in: p73 is a tumor suppressor after all. Genes Dev. 2008;22:2591-5 [DOI] [PubMed] [Google Scholar]
- 13. Tissir F, Ravni A, Achouri Y, Riethmacher D, Meyer G, Goffinet AM. DeltaNp73 regulates neuronal survival in vivo. Proc Natl Acad Sci U S A. 2009;106:16871-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wilhelm MT, Rufini A, Wetzel MK, et al. Isoform-specific p73 knockout mice reveal a novel role for delta Np73 in the DNA damage response pathway. Genes Dev. 2010;24:549-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bourdon JC, Fernandes K, Murray-Zmijewski F, et al. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005;19:2122-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Khoury MP, Bourdon JC. The isoforms of the p53 protein. Cold Spring Harb Perspect Biol. 2010;2:a000927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Marcel V, Perrier S, Aoubala M, et al. Delta160p53 is a novel N-terminal p53 isoform encoded by Delta133p53 transcript. FEBS Lett. 2010;584:4463-8 [DOI] [PubMed] [Google Scholar]
- 18. Chen J, Ruan H, Ng SM, et al. Loss of function of def selectively up-regulates Delta113p53 expression to arrest expansion growth of digestive organs in zebrafish. Genes Dev. 2005;19:2900-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen J, Ng SM, Chang C, et al. p53 isoform delta113p53 is a p53 target gene that antagonizes p53 apoptotic activity via BclxL activation in zebrafish. Genes Dev. 2009;23:278-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Davidson WR, Kari C, Ren Q, Daroczi B, Dicker AP, Rodeck U. Differential regulation of p53 function by the N-terminal DeltaNp53 and Delta113p53 isoforms in zebrafish embryos. BMC Dev Biol. 2010;10:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Aoubala M, Murray-Zmijewski F, Khoury MP, et al. p53 directly transactivates Delta133p53alpha, regulating cell fate outcome in response to DNA damage. Cell Death Differ. 2011;18:248-58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fujita K, Mondal AM, Horikawa I, et al. p53 isoforms Delta133p53 and p53beta are endogenous regulators of replicative cellular senescence. Nat Cell Biol. 2009;11:1135-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Song W, Huo SW, Lu JJ, et al. Expression of p53 isoforms in renal cell carcinoma. Chin Med J (Engl). 2009;122:921-6 [PubMed] [Google Scholar]
- 24. Neubauer A, He M, Schmidt CA, Huhn D, Liu ET. Genetic alterations in the p53 gene in the blast crisis of chronic myelogenous leukemia: analysis by polymerase chain reaction based techniques. Leukemia. 1993;7:593-600 [PubMed] [Google Scholar]
- 25. Chow VT, Quek HH, Tock EP. Alternative splicing of the p53 tumor suppressor gene in the Molt-4 T-lymphoblastic leukemia cell line. Cancer Lett. 1993;73:141-8 [DOI] [PubMed] [Google Scholar]
- 26. Flaman J-M, Waridel F, Estreicher A, et al. The human tumor suppressor gene p53 is alternatively spliced in normal cells. Oncogene. 1996;12:813-8 [PubMed] [Google Scholar]
- 27. Graupner V, Schulze-Osthoff K, Essmann F, Janicke RU. Functional characterization of p53beta and p53gamma, two isoforms of the tumor suppressor p53. Cell Cycle. 2009;8:1238-48 [DOI] [PubMed] [Google Scholar]
- 28. Ghosh A, Stewart D, Matlashewski G. Regulation of human p53 activity and cell localization by alternative splicing. Mol Cell Biol. 2004;24:7987-97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ray PS, Grover R, Das S. Two internal ribosome entry sites mediate the translation of p53 isoforms. EMBO Rep. 2006;7:404-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yang DQ, Halaby MJ, Zhang Y. The identification of an internal ribosomal entry site in the 5′-untranslated region of p53 mRNA provides a novel mechanism for the regulation of its translation following DNA damage. Oncogene. 2006;25:4613-9 [DOI] [PubMed] [Google Scholar]
- 31. Candeias MM, Powell DJ, Roubalova E, et al. Expression of p53 and p53/47 are controlled by alternative mechanisms of messenger RNA translation initiation. Oncogene. 2006;25:6936-47 [DOI] [PubMed] [Google Scholar]
- 32. Yin Y, Stephen CW, Luciani MG, Fahraeus R. p53 stability and activity is regulated by Mdm2-mediated induction of alternative p53 translation products. Nat Cell Biol. 2002;4:462-7 [DOI] [PubMed] [Google Scholar]
- 33. Courtois S, Verhaegh G, North S, et al. Delta N-p53, a natural isoform of p53 lacking the first transactivation domain, counteracts growth suppression by wild-type p53. Oncogene. 2002;21:6722-8 [DOI] [PubMed] [Google Scholar]
- 34. Grover R, Ray PS, Das S. Polypyrimidine tract binding protein regulates IRES-mediated translation of p53 isoforms. Cell Cycle. 2008;7:2189-98 [DOI] [PubMed] [Google Scholar]
- 35. Montanaro L, Calienni M, Bertoni S, et al. Novel dyskerin-mediated mechanism of p53 inactivation through defective mRNA translation. Cancer Res. 2010;70:4767-77 [DOI] [PubMed] [Google Scholar]
- 36. Bellodi C, Kopmar N, Ruggero D. Deregulation of oncogene-induced senescence and p53 translational control in X-linked dyskeratosis congenita. EMBO J. 2010;29:1865-76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Grover R, Sharathchandra A, Ponnuswamy A, Khan D, Das S. Effect of mutations on the p53 IRES RNA structure: implications for de-regulation of the synthesis of p53 isoforms. RNA Biol. 2011;8. [DOI] [PubMed] [Google Scholar]
- 38. Zhu J, Zhou W, Jiang J, Chen X. Identification of a novel p53 functional domain that is necessary for mediating apoptosis. J Biol Chem. 1998;273:13030-6 [DOI] [PubMed] [Google Scholar]
- 39. Marcel V, Hainaut P. p53 isoforms: a conspiracy to kidnap p53 tumor suppressor activity? Cell Mol Life Sci. 2009;66:391-406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bourougaa K, Naski N, Boularan C, et al. Endoplasmic reticulum stress induces G2 cell-cycle arrest via mRNA translation of the p53 isoform p53/47. Mol Cell. 2010;38:78-88 [DOI] [PubMed] [Google Scholar]
- 41. Wolf D, Harris N, Goldfinger N, Rotter V. Isolation of a full-length mouse cDNA clone coding for an immunologically distinct p53 molecule. Mol Cell Biol. 1985;5:127-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Arai N, Nomura D, Yokota K, et al. Immunologically distinct p53 molecules generated by alternative splicing. Mol Cell Biol. 1986;6:3232-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wu Y, Liu Y, Lee L, Miner Z, Kulesz-Martin M. Wild-type alternatively spliced p53: binding to DNA and interaction with the major p53 protein in vitro and in cells. EMBO J. 1994;13:4823-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Han KA, Kulesz-Martin MF. Alternatively spliced p53 RNA in transformed and normal cells of different tissue types. Nucleic Acids Res. 1992;20:1979-81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kulesz-Martin MF, Lisafeld B, Huang H, Kisiel ND, Lee L. Endogenous p53 protein generated from wild-type alternatively spliced p53 RNA in mouse epidermal cells. Mol Cell Biol. 1994;14:1698-708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Will K, Warnecke G, Bergmann S, Deppert W. Species- and tissue-specific expression of the C-terminal alternatively spliced form of the tumor suppressor p53. Nucleic Acids Res. 1995;23:4023-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wu Y, Huang H, Miner Z, Kulesz-Martin M. Activities and response to DNA damage of latent and active sequence-specific DNA binding forms of mouse p53. Proc Natl Acad Sci U S A. 1997;94:8982-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wolkowicz R, Peled A, Elkind NB, Rotter V. Augmented DNA-binding activity of p53 protein encoded by a carboxyl-terminal alternatively spliced mRNA is blocked by p53 protein encoded by the regularly spliced form. Proc Natl Acad Sci U S A. 1995;92:6842-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Miner Z, Kulesz-Martin M. DNA binding specificity of proteins derived from alternatively spliced mouse p53 mRNAs. Nucleic Acids Res. 1997;25:1319-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hupp TR, Meek DW, Midgley CA, Lane DP. Regulation of the specific DNA binding function of p53. Cell. 1992;71:875-86 [DOI] [PubMed] [Google Scholar]
- 51. Hupp TR, Lane DP. Allosteric activation of latent p53 tetramers. Curr Biol. 1994;4:865-75 [DOI] [PubMed] [Google Scholar]
- 52. Hupp TR, Sparks A, Lane DP. Small peptides activate the latent sequence-specific DNA binding function of p53. Cell. 1995;83:237-45 [DOI] [PubMed] [Google Scholar]
- 53. Almog N, Li R, Peled A, et al. The murine C′-terminally alternatively spliced form of p53 induces attenuated apoptosis in myeloid cells. Mol Cell Biol. 1997;17:713-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Almog N, Goldfinger N, Rotter V. p53-dependent apoptosis is regulated by a C-terminally alternatively spliced form of murine p53. Oncogene. 2000;19:3395-403 [DOI] [PubMed] [Google Scholar]
- 55. Almog N, Milyavsky M, Stambolsky P, Falcovitz A, Goldfinger N, Rotter V. The role of the C′ terminus of murine p53 in the p53/mdm-2 regulatory loop. Carcinogenesis. 2001;22:779-85 [DOI] [PubMed] [Google Scholar]
- 56. Tyner SD, Venkatachalam S, Choi J, et al. p53 mutant mice that display early ageing-associated phenotypes. Nature. 2002;415:45-53 [DOI] [PubMed] [Google Scholar]
- 57. Maier B, Gluba W, Bernier B, et al. Modulation of mammalian life span by the short isoform of p53. Genes Dev. 2004;18:306-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pehar M, O’Riordan KJ, Burns-Cusato M, et al. Altered longevity-assurance activity of p53:p44 in the mouse causes memory loss, neurodegeneration and premature death. Aging Cell. 2010;9:174-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Avery-Kiejda KA, Zhang XD, Adams LJ, et al. Small molecular weight variants of p53 are expressed in human melanoma cells and are induced by the DNA-damaging agent cisplatin. Clin Cancer Res. 2008;14:1659-68 [DOI] [PubMed] [Google Scholar]
- 60. Anensen N, Oyan AM, Bourdon JC, Kalland KH, Bruserud O, Gjertsen BT. A distinct p53 protein isoform signature reflects the onset of induction chemotherapy for acute myeloid leukemia. Clin Cancer Res. 2006;12:3985-92 [DOI] [PubMed] [Google Scholar]
- 61. Boldrup L, Bourdon JC, Coates PJ, Sjostrom B, Nylander K. Expression of p53 isoforms in squamous cell carcinoma of the head and neck. Eur J Cancer. 2007;43:617-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Marabese M, Marchini S, Marrazzo E, et al. Expression levels of p53 and p73 isoforms in stage I and stage III ovarian cancer. Eur J Cancer. 2008;44:131-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bourdon JC, Khoury MP, Diot A, et al. p53 mutant breast cancer patients expressing p53gamma have as good a prognosis as wild-type p53 breast cancer patients. Breast Cancer Res. 2011;13:R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hofstetter G, Berger A, Fiegl H, et al. Alternative splicing of p53 and p73: the novel p53 splice variant p53delta is an independent prognostic marker in ovarian cancer. Oncogene. 2010;29:1997-2004 [DOI] [PubMed] [Google Scholar]
- 65. Marcel V, Vijayakumar V, Fernandez-Cuesta L, et al. p53 regulates the transcription of its Delta133p53 isoform through specific response elements contained within the TP53 P2 internal promoter. Oncogene. 2010;29:2691-700 [DOI] [PubMed] [Google Scholar]
- 66. Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations [see comments]. Science. 1994;265:346-55 [DOI] [PubMed] [Google Scholar]
- 67. McKinney K, Mattia M, Gottifredi V, Prives C. p53 linear diffusion along DNA requires its C terminus. Mol Cell. 2004;16:413-24 [DOI] [PubMed] [Google Scholar]
- 68. Juven T, Barak Y, Zauberman A, George DL, Oren M. Wild type p53 can mediate sequence-specific transactivation of an internal promoter within the mdm2 gene. Oncogene. 1993;8:3411-6 [PubMed] [Google Scholar]
- 69. Liang H, Lunec J. Characterisation of a novel p53 down-regulated promoter in intron 3 of the human MDM2 oncogene. Gene. 2005;361:112-8 [DOI] [PubMed] [Google Scholar]
- 70. Budhram-Mahadeo V, Morris PJ, Smith MD, Midgley CA, Boxer LM, Latchman DS. p53 suppresses the activation of the Bcl-2 promoter by the Brn-3a POU family transcription factor. J Biol Chem. 1999;274:15237-44 [DOI] [PubMed] [Google Scholar]
- 71. Bredow S, Juri DE, Cardon K, Tesfaigzi Y. Identification of a novel Bcl-2 promoter region that counteracts in a p53-dependent manner the inhibitory P2 region. Gene. 2007;404:110-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wu Y, Mehew JW, Heckman CA, Arcinas M, Boxer LM. Negative regulation of bcl-2 expression by p53 in hematopoietic cells. Oncogene. 2001;20:240-51 [DOI] [PubMed] [Google Scholar]
- 73. Sbisa E, Catalano D, Grillo G, et al. p53FamTaG: a database resource of human p53, p63 and p73 direct target genes combining in silico prediction and microarray data. BMC Bioinformatics. 2007;8 Suppl 1:S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Noureddine MA, Menendez D, Campbell MR, et al. Probing the functional impact of sequence variation on p53-DNA interactions using a novel microsphere assay for protein-DNA binding with human cell extracts. PLoS Genet. 2009;5:e1000462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bandele OJ, Wang X, Campbell MR, Pittman GS, Bell DA. Human single-nucleotide polymorphisms alter p53 sequence-specific binding at gene regulatory elements. Nucleic Acids Res. 2011;39:178-89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Smeenk L, van Heeringen SJ, Koeppel M, et al. Characterization of genome-wide p53-binding sites upon stress response. Nucleic Acids Res. 2008;36:3639-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Perez CA, Ott J, Mays DJ, Pietenpol JA. p63 consensus DNA-binding site: identification, analysis and application into a p63MH algorithm. Oncogene. 2007;26:7363-70 [DOI] [PubMed] [Google Scholar]
- 78. Harms K, Nozell S, Chen X. The common and distinct target genes of the p53 family transcription factors. Cell Mol Life Sci. 2004;61:822-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Perez CA, Pietenpol JA. Transcriptional programs regulated by p63 in normal epithelium and tumors. Cell Cycle. 2007;6:246-54 [DOI] [PubMed] [Google Scholar]
- 80. Davison TS, Vagner C, Kaghad M, Ayed A, Caput D, Arrowsmith CH. p73 and p63 are homotetramers capable of weak heterotypic interactions with each other but not with p53. J Biol Chem. 1999;274:18709-14 [DOI] [PubMed] [Google Scholar]
- 81. Joerger AC, Rajagopalan S, Natan E, Veprintsev DB, Robinson CV, Fersht AR. Structural evolution of p53, p63, and p73: implication for heterotetramer formation. Proc Natl Acad Sci U S A. 2009;106:17705-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Yang A, Zhu Z, Kapranov P, et al. Relationships between p63 binding, DNA sequence, transcription activity, and biological function in human cells. Mol Cell. 2006;24:593-602 [DOI] [PubMed] [Google Scholar]
- 83. Flores ER, Tsai KY, Crowley D, et al. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature. 2002;416:560-4 [DOI] [PubMed] [Google Scholar]
- 84. Flores ER, Sengupta S, Miller JB, et al. Tumor predisposition in mice mutant for p63 and p73: evidence for broader tumor suppressor functions for the p53 family. Cancer Cell. 2005;7:363-73 [DOI] [PubMed] [Google Scholar]
- 85. Fujitani M, Cancino GI, Dugani CB, et al. TAp73 acts via the bHLH Hey2 to promote long-term maintenance of neural precursors. Curr Biol. 2010;20:2058-65 [DOI] [PubMed] [Google Scholar]
- 86. Guo X, Keyes WM, Papazoglu C, et al. TAp63 induces senescence and suppresses tumorigenesis in vivo. Nat Cell Biol. 2009;11:1451-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Su X, Paris M, Gi YJ, et al. TAp63 prevents premature aging by promoting adult stem cell maintenance. Cell Stem Cell. 2009;5:64-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Su X, Chakravarti D, Cho MS, et al. TAp63 suppresses metastasis through coordinate regulation of Dicer and miRNAs. Nature. 2010;467:986-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Koster MI, Dai D, Marinari B, et al. p63 induces key target genes required for epidermal morphogenesis. Proc Natl Acad Sci U S A. 2007;104:3255-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Koster MI, Marinari B, Payne AS, Kantaputra PN, Costanzo A, Roop DR. DeltaNp63 knockdown mice: a mouse model for AEC syndrome. Am J Med Genet A. 2009;149A:1942-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Lowe SW, Schmitt EM, Smith SW, Osborne BA, Jacks T. p53 is required for radiation-induced apoptosis in mouse thymocytes [see comments]. Nature. 1993;362:847-9 [DOI] [PubMed] [Google Scholar]
- 92. Xue W, Zender L, Miething C, et al. Senescence and tumor clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Smeenk L, van Heeringen SJ, Koeppel M, et al. Role of p53 serine 46 in p53 target gene regulation. PLoS One. 2011;6:e17574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Suh EK, Yang A, Kettenbach A, et al. p63 protects the female germ line during meiotic arrest. Nature. 2006;444:624-8 [DOI] [PubMed] [Google Scholar]
- 95. Deutsch GB, Zielonka EM, Coutandin D, et al. DNA damage in oocytes induces a switch of the quality control factor TAp63alpha from dimer to tetramer. Cell. 2011;144:566-76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Burge S, Teufel DP, Townsley FM, Freund SM, Bycroft M, Fersht AR. Molecular basis of the interactions between the p73 N terminus and p300: effects on transactivation and modulation by phosphorylation. Proc Natl Acad Sci U S A. 2009;106:3142-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Collavin L, Lunardi A, Del Sal G. p53-family proteins and their regulators: hubs and spokes in tumor suppression. Cell Death Differ. 2010;17:901-11 [DOI] [PubMed] [Google Scholar]
- 98. Meek DW, Anderson CW. Posttranslational modification of p53: cooperative integrators of function. Cold Spring Harb Perspect Biol. 2009;1:a000950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Stenger JE, Mayr GA, Mann K, Tegtmeyer P. Formation of stable p53 homotetramers and multiples of tetramers. Mol Carcinog. 1992;5:102-6 [DOI] [PubMed] [Google Scholar]
- 100. Wang P, Reed M, Wang Y, et al. p53 domains: structure, oligomerization, and transformation. Mol Cell Biol. 1994;14:5182-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Jiao Y, Cherny DI, Heim G, Jovin TM, Schaffer TE. Dynamic interactions of p53 with DNA in solution by time-lapse atomic force microscopy. J Mol Biol. 2001;314:233-43 [DOI] [PubMed] [Google Scholar]
- 102. Huang H, Kaku S, Knights C, Park B, Clifford J, Kulesz-Martin M. Repression of transcription and interference with DNA binding of TATA-binding protein by C-terminal alternatively spliced p53. Exp Cell Res. 2002;279:248-259 [DOI] [PubMed] [Google Scholar]