

Abstract
Although the majority of pediatric malignancies express wild-type p53, it is well established that germline TP53 mutations or functional inactivation of this pathway by other means contribute to childhood cancer. Epidemiology studies have revealed the existence of diverse inherited mutant TP53 alleles that display different levels of tumor suppressor activity, which correlate with cancer risk in terms of penetrance, age of onset, and tumor types. In this monograph, the authors describe those childhood cancers associated with functional inactivation of TP53 focusing on adrenocortical carcinoma as a model for tissues that are highly sensitive to loss of p53 activity.
Keywords: p53 mutation, adrenocortical tumors, Arg337His
Introduction
Pediatric malignancies are rare, complex, and heterogeneous disorders. Altogether they represent only about 1% to 2% of all human malignancies.1 If we assume that childhood cancer is a developmental genetic disorder, it is reasonable to postulate that a combination of alterations crippling one or more signaling pathways is sufficient to initiate a cascade of events establishing a malignant clone. As a group, TP53 mutations are among the most common genetic alterations observed in human cancers and occur in about 50% of unselected sporadic tumors.2,3
When TP53 mutations are inherited, carriers have an increased lifetime predisposition to cancer.4 Although the spectrum of tumor types in these individuals is similar to that found in the sporadic counterpart, they tend to develop tumors much earlier in life. The anticipation of tumor development, particularly in certain tissues, suggests that cells with constitutional TP53 mutations are less dependent on naturally occurring events, such as aging or environmental carcinogens (e.g., UV, benzo[a]pyrene) for initiating tumorigenesis. Therefore, it can be postulated that additional constitutional genetic polymorphisms, mutations, and other epigenetic mechanisms cooperate with p53 insufficiency to promote tumor development. If this premise is correct, the study of pediatric tumors associated with TP53 mutations provides an important opportunity to uncover these cooperating events for establishing the cancer phenotype. It is also plausible that these same constitutional genetic alterations play a role in the development of sporadic tumors later in life.
For many years, our clinical and laboratory efforts have focused on improving the outcome of children with a rare malignancy, adrenocortical tumors (ACT), which are usually associated with TP53 mutations. Understanding the changes in incidence over time and differences between different groups (e.g., gender and ethnicity) provides essential clues to the etiology of disease, particularly the balance between genetic factors versus environment in the causation of the disease. We approached this task first by creating a tumor registry to collect demographic, epidemiologic, and clinical information, including family history of cancer, outcome, and long-term follow up data of families with a proband with ACT. Second, we established uniform treatment guidelines for the management of these patients. Finally, we have been conducting extensive cellular and molecular studies of p53 and its associated pathways to learn more about the mechanisms driving adrenocortical tumorigenesis with the goal of developing more effective therapies for these patients.
TP53 Tumor Suppression Function
p53 (encoded by human gene TP53) functions primarily as a tetrameric transcription factor within a signaling pathway that is essential for maintaining normal cell growth and survival. Cell stress caused by DNA damaging agents, deregulation of oncogenes, or other environmental insults, such as hypoxia, can activate p53, resulting in cell cycle arrest, cellular senescence, and/or apoptosis. In addition, p53 can affect glycolytic pathways, DNA repair, angiogenesis, longevity, and aging.5 Upon cellular stress, p53 undergoes posttranslational modifications that stabilize and activate the protein to induce the expression of its downstream target genes.6 These p53-responsive genes are differentially regulated depending on cell context, the extent of damage, and various other yet unidentified parameters.7
Mutational Inactivation of TP53
During tumorigenesis, strong selective pressures prevent the activation of p53.8 About 50% of all human tumors sustain TP53 mutations, whereas the remaining cases acquire other genetic or epigenetic alterations that compromise p53 function.9 The majority of tumor-derived TP53 mutations are single-nucleotide substitutions clustering within the DNA binding domain (DBD), resulting in the expression of a missense protein that is defective in sequence-specific binding, transactivation, and growth inhibition.10 In addition, p53 can be functionally inactivated through the loss of upstream inducers (ATM, p14ARF) or downstream mediators (caspase 9, APAF1) or by overexpressing negative regulators (Mdm2, Mdm4, human papilloma virus E6), for example.11
Approximately 80% of TP53 mutations occur within DBD,12 with the majority occurring at 6 hot spot codons (175, 245, 248, 249, 273, and 282). These specific sites account for about 40% of all TP53 missense mutations and are naturally selected during tumorigenesis due to their deleterious effects on p53’s ability to bind DNA and regulate transcription.9 However, an increasing number of studies highlight the importance of mutations outside of this domain.13-15
Most missense p53 proteins are stable and expressed at high levels in human tumors.8 It has been shown that some p53 mutants exhibit a dominant-negative phenotype by binding the normal p53 monomer (expressed by the wild-type allele), resulting in an inactive heterodimer and tetramer.16,17 Consistent with these findings, human cancers that express mutant p53 while retaining the wild-type TP53 allele have been described.18 Similarly, mice that are heterozygous for a dominant-negative TP53 point mutation develop tumors without loss of the wild-type TP53 allele.19 Partial inactivation of wild-type p53 function by mutant p53 might allow for some selective advantage during tumor progression. However, many tumors that harbor TP53 point mutations also show loss of heterozygosity (LOH)13,14 by eliminating the wild-type allele through either deletion or other mechanisms.
Negative Regulators of p53
p53 tumor suppressor activity is limited primarily by Mdm2, which binds and conceals the N-terminus of p53. Consequently, Mdm2 inhibits p53 transcription function and, through its E3 ubiquitin ligase activity, targets p53 for proteasomal degradation. Notably, Mdm2 forms an elegant negative-feedback loop with p53, as Mdm2 transcription can be directly positively regulated by p53.20,21 The importance of this interaction in regulating p53-dependent tumor suppression is highlighted by the following findings: 1) Studies in animal models have shown that the embryonic lethality of Mdm2 knockout mice can be fully rescued by deleting p53, 2) Mdm2 transgenic mice are tumor prone, 3) MDM2 is frequently amplified and overexpressed in human tumors that retain wild-type TP53 alleles, and 4) Nutlin, an MDM2 inhibitor, can reactivate p53 function in tumors expressing wild-type TP53 alleles.22
A single-nucleotide polymorphism (SNP) within the MDM2 promoter at nucleotide 309T>G (rs2279744) has been shown to enhance MDM2 transcription, resulting in the accumulation of MDM2 protein and consequently the attenuation of p53 function.23 Consistent with these findings, the MDM2 SNP309 G allele is associated with earlier age of onset of tumors among patients with Li-Fraumeni syndrome (LFS).24
p53 inactivation in human cancers can also occur through the amplification and overexpression of MDM4, which shares homology with MDM2.25,26 MDM4 is genetically amplified in a subset of breast, colon, and lung carcinomas; gliomas; and approximately 65% of children with retinoblastoma.27 A general consensus is emerging that in addition to Nutlin and derivative compounds, an MDM4-specific inhibitor will need to be developed to effectively treat retinoblastoma and other tumors that retain expression of wild-type p53.26
Familial Mutant p53 Syndromes
Germline inherited TP53 mutations cause a rare type of familial cancer predisposition known as LFS, in which family members can present with diverse tumor types occurring over a wide age range, including during childhood. This syndrome is characterized by the following criteria: a proband with a sarcoma aged less than 45 years, a first-degree relative younger than 45 years with any cancer, plus an additional first- or second-degree relative in the same lineage with any cancer before age 45 years or a sarcoma at any age.28
Individuals with LFS have a greater risk for cancer at younger ages than those in the general population. It is estimated that 50% of LFS-associated malignancies occur by age 30 years.29 LFS is associated with increased susceptibility to diverse malignancies such as soft tissue sarcomas, osteosarcomas, brain tumors, choroid plexus carcinomas (CPC), and pediatric ACT, as well as multiple primary tumors.30 In particular, women with LFS are at a significantly high risk of premenopausal breast cancer.31 In assessing families for LFS, an unusually young age at diagnosis may be as important a variable as the specific type of malignancy.
More inclusive clinical classification schemes have been described that characterize Li-Fraumeni-like syndrome (LFL) as having features of LFS without fulfilling its strict requirements.32 Additional criteria by Chompret and colleagues33 allow for the possibility of a proband with an LFS-related tumor but a negative family history of cancer.
TP53 is the only gene known to date to be strongly associated with LFS and LFL. Clinical testing involves sequence analysis of the entire TP53 coding region. This method detects about 95% of TP53 mutations, most of which are missense mutations. It is estimated that 70% of individuals with LFS and 8% to 22% of individuals with LFL have detectable TP53 mutations.34,35 The frequency of de novo mutations in LFS ranges between 7% and 20%.30 It is not too surprising that the inherited TP53 mutations associated with LFS are localized to the same hot spot sites found in sporadic tumors. One of the most commonly inherited TP53 mutations is Arg175His. This mutant fails to fold properly, bind DNA, activate transcription, or suppress cell growth. Carriers of this mutation, which is considered to be highly penetrant, are at greater risk for cancer at early ages.11
Pediatric Cancers Associated with TP53 Mutations
Compared with the cancer incidence in adults, pediatric tumors are rare, accounting for only 3.0% of all cases in the United States. However, cancer is the primary cause of disease-related deaths in children.36 Childhood tumors comprise a variety of malignancies with incidences varying worldwide by age, sex, ethnicity, and geography, and these factors can provide important insights into cancer etiology.37
Pediatric ACT is rare and occurs at an estimated incidence of 0.3 per million in individuals younger than 15 years.1 ACT is associated with isolated hemi-hypertrophy or Beckwith-Wiedemann syndrome38 and rarely occurs in children with multiple endocrine neoplasia type 1.39 However, pediatric ACT may also be the first indication of a germline TP53 mutation and LFS within a family.28 Sporadic cases of ACT have also been noted,40 and approximately 80% have atypical germline mutations of TP53 associated with a lower but increased cancer risk in relatives.14,41
Affected children with ACT typically present with symptoms related to excessive production of adrenal cortex steroids, and virilization and Cushing syndrome are the most common endocrine manifestations. Disease stage, which includes tumor size, is the most important predictor of outcome. Patients who have residual disease after incomplete tumor resection or metastatic disease have a dismal prognosis.42
Tumor Registry
We have established an International Pediatric Adrenocortical Tumor Registry (IPACTR) at St. Jude Children’s Research Hospital, which collects clinical information and biological specimens, including blood and tumor. Our data (manuscript in preparation) show that approximately 75% of ACTs are positive for TP53 mutations (8% somatic and 92% germline). In our cohort of 48 ACTs, we detected 23 independent TP53 mutations within and outside the DBD, and of these, only 3 were detected more than once (Arg175His, Arg273Cys, and Arg337His; Figure 1). In our cohort, we also identified a novel germline variant, Arg175Leu.43 Surprisingly, the family is not tumor prone or associated with LFS. In vitro, the Arg175Leu mutant displayed intermediate tumor suppressor activity in the regulation of transcription, colony formation, and apoptosis when compared to wild-type and hot spot mutant Arg175His. These findings suggest that Arg175Leu retains sufficient activity to suppress LFS but not ACT. Therefore, not all TP53 mutations are functionally equivalent, and the biochemical nature of the mutant may significantly influence clinical outcome.11,43
Figure 1.
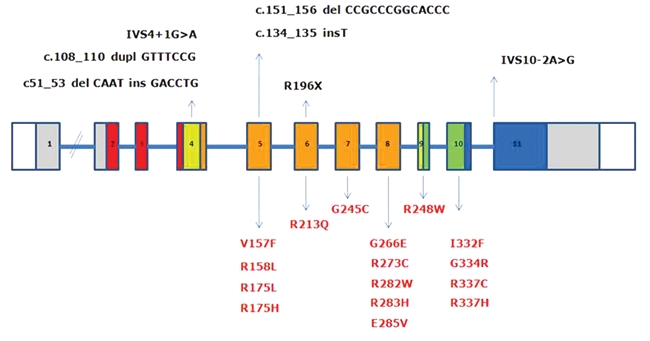
TP53 mutations in childhood adrenocortical tumors. Blood and tumor samples were banked through the St. Jude Children’s Research Hospital International Pediatric Adrenocortical Tumor Registry (institutional review board approved). In a cohort of 48 patients with adrenocortical tumors, 36 TP53 mutations were detected (75%) within and outside the DNA binding domain. Three mutations were detected in multiple cases (Arg175His, Arg273Cys, and Arg337His). Most mutations represent single-nucleotide substitutions (red), although non-sense and complex mutations were also observed (black). The schematic outlines the coding region of TP53 spanning exons 1 to 11 (colors represent functional domains).
Pediatric ACT is frequently associated with LFS families carrying high-penetrance TP53 mutations. However, there is growing evidence that low-penetrance TP53 mutations, such as the one found in southern Brazil, increases the predisposition for childhood ACT but not that of other Li-Fraumeni core component tumors.44 On the basis of these observations, we propose that the adrenal cortex is particularly vulnerable to TP53 loss of function.
According to the National Cancer Institute (Surveillance Epidemiology and End Results [SEER]), 79 cases of pediatric ACT were reported from 17 geographic areas in the United States during a 34-year period (1973-2007) (Table 1).1 Remarkably, the number of cases in the SEER database represents approximately 60% of the cases treated at a single institution in southern Brazil (n = 124; Hospital das Clínicas de Curitiba) during a similar time period (1966-2003) and time span (37 years).45
Table 1.
Cases of Childhood Adrenocortical Carcinoma Registered in the Surveillance Epidemiology and End Results (SEER) Database1
| Cases | ||
|---|---|---|
| Number | % | |
| Sex | ||
| Female | 53 | 67.1 |
| Male | 26 | 32.9 |
| Age at diagnosis, y | ||
| 0-4 | 36 | 46.6 |
| 5-9 | 14 | 17.7 |
| 10-14 | 11 | 14.0 |
| 15-19 | 18 | 22.7 |
| Disease stage | ||
| Local | 37 | 34.2 |
| Regional | 10 | 46.8 |
| Distant metastasis | 27 | 12.7 |
| Unknown | 5 | 6.3 |
The predisposition for childhood ACT in southern Brazil is associated with the germline TP53-Arg337His mutation.13,14 This mutation lies within the p53 tetramerization domain (amino acids 336-353). Specifically, arginine-337 of one subunit forms a salt bridge with aspartate-352 in the corresponding monomer to stabilize the complex. Structural studies have shown that the Arg337His mutant is unstable and does not efficiently dimerize at physiologic pH in vitro 14,46 but was found to display wild-type p53 activity when ectopically expressed in fibroblasts and osteosarcoma cells.14 These observations could explain the lack of association of Arg337His with LFS. In contrast, the substitution of cysteine at codon 337 has a very different biochemical and clinical outcome.47 The Arg337Cys mutant improperly folds irrespective of pH and exhibits only partial biological activities.48 Consistent with this significant loss of functional activity, carriers of a germline Arg337Cys mutation are susceptible to multiple tumor types, including breast carcinoma.47 Therefore, Arg337His in most cases has a markedly less severe impact on tumor predisposition than Arg337Cys, and this difference correlates with the relative degree of activity that can be measured in the laboratory.11 These observations support the concept that single-site mutations, depending on the amino acid substitution, have different consequences on the structure, function, and degree of activity that, in turn, affects tumor susceptibility.11
The TP53-Arg337His mutation is the most common germline mutation reported in the International Agency for Research on Cancer (IARC) database (http://www.iarc.fr/). Although Arg337His was initially observed in pediatric ACT,14 detailed studies of families with this mutation also revealed an association with choroid plexus carcinoma in children49 and breast and stomach tumors in young adults.50 The Arg337His mutation was also observed in 7% of pediatric osteosarcomas in southern Brazil.49,51 This frequency is much higher than that reported for the general population of the region (0.3%) and is greater than that reported for nonselected North American patients with osteosarcoma who had any constitutional TP53 mutation (3%).52 These findings implicate genetic background and cooperating factors that could influence tumor susceptibility in the presence of Arg337His.
Although it is recognized that individuals with certain TP53 mutations (i.e., Arg175His) are at high risk for cancer at early ages and multiple tumors, this is not the case for Arg337His. In fact, many carriers remain asymptomatic.14,53 The incomplete penetrance of Arg337His in pediatric ACT (2%-10% of carriers develop disease) may be related to its inability to act as a dominant negative regulator of wild-type p53. This is consistent with nearly 100% of the ACT cases undergoing LOH by eliminating the wild-type p53 allele.14,49,54
Molecular studies identify the same haplotype for the TP53 locus in individuals carrying the Arg337His mutation, indicating a common origin for this mutation (founder effect).55 The majority of the population in the southern region of Brazil is of European extraction (Portuguese, Italians, Spanish, and Germans) with native Brazilian Indian and African influence.56 The high incidence of ACT in patients from this region is therefore explained by the presence of the germline Arg337His mutation, which was likely inherited from a common founder.55 Nevertheless, Arg337His has been detected in isolated ACT cases outside of Brazil but not yet as a common TP53 mutation in other populations.
Cooperating Factors in Pediatric ACT
Additional factors that have been implicated in ACT include defects in imprinting and overexpression of insulin-like growth factor-2 (IGF-2). Gene expression profiling of sporadic human ACT revealed dysregulation of the 11p15.5 region, and several studies have shown IGF-2 to be the single most up-regulated transcript in 80% to 90% of ACTs.57-59 IGF-2 mainly elicits its cellular effects through the ubiquitously expressed type 1 IGF receptor (IGF-1R). Interestingly, during organ development in the human fetus, the adrenal gland has one of the highest levels of IGF-2 expression, which subsequently declines after birth.60 These observations suggest that activation of the IGF pathway is a common pathological mechanism used by tumor cells during adrenocortical tumorigenesis.
Using comparative genomic hybridization, it has been shown that childhood ACTs are characterized by a high frequency of chromosomal aberrations, especially involving amplification of 9q33-q34.61 This chromosomal region harbors the human steroidogenic factor 1 (SF-1) gene, and an increased SF-1 copy number was detected in 8 of 9 Brazilian pediatric ACTs using fluorescence in situ hybridization, suggesting an association between SF-1 gene amplification and childhood adrenocortical tumorigenesis.62 Consistent with these findings, each case of ACT overexpressed SF-1 protein.63 SF-1 is an orphan member of the nuclear receptor family of transcription factors and plays an important role in endocrine function, including the regulation of steroid hydroxylases, development and function of the adrenal cortex, and male sexual differentiation.64 Furthermore, increased SF-1 dosage promotes cell proliferation and triggers tumorigenesis in mice.65 Based on these observations, SF1 functions as an oncogene that contributes to the development of ACT.
Inhibin-α (Inha) was found to be an important determinant of cortical adrenal tumor development in mice.66 Molecular analysis of Brazilian childhood ACTs identified mutations in INHIBIN-α (INHA), and LOH was observed in 8 of 9 tumors, suggesting INHA functions as a tumor suppressor of ACT in carriers of the Arg337His TP53 mutation.67 Therefore, in keeping with a multihit model for tumorigenesis, the incomplete penetrance of ACT in carriers of the Arg337His mutation may be explained by the apparent requirement for loss of relevant tumor suppressors (e.g., INHA) and activation of cooperating oncogenes (e.g., SF1 and IGF2).
Concluding Remarks
It is becoming clear that most human cancers have acquired defects within the p53 tumor suppressor signaling pathway, either by TP53 mutation or by deregulation of upstream regulators or downstream effectors. Our clinical and molecular studies of pediatric ACT have advanced our understanding of the basic biology of p53 in these tumors. We have uncovered important genotype-phenotype relationships between the biochemical consequence of germline TP53 mutations on p53 function and their association with inherent cancer risk. These findings have important implications for genetic counseling and clinical management. By focusing on a rare childhood cancer, such as ACT, it is possible to learn not only a great deal regarding how p53 functions as a tumor suppressor but also about the disease itself and ultimately how to improve long-term outcome.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
This work was supported in part by grant CA-21765 from the National Institutes of Health (U.S. Department of Health and Human Services), by a Center of Excellence grant from the State of Tennessee, and by the American Lebanese Syrian Associated Charities (ALSAC).
References
- 1. Altekruse SF, Kosary CL, Krapcho M, et al., editors. SEER Cancer Statistics Review, 1975-2007, based on November 2009 SEER data submission, posted to the SEER web site, 2010. Bethesda, MD: National Cancer Institute; http://seer.cancer.gov/csr/1975_2007/ [Google Scholar]
- 2. Strong LC, Williams WR, Tainsky MA. The Li-Fraumeni syndrome: from clinical epidemiology to molecular genetics. Am J Epidemiol. 1992;135:190-9 [DOI] [PubMed] [Google Scholar]
- 3. Birch JM, Blair V, Kelsey AM, et al. Cancer phenotype correlates with constitutional TP53 genotype in families with the Li-Fraumeni syndrome. Oncogene. 1998;17:1061-8 [DOI] [PubMed] [Google Scholar]
- 4. Bennett WP, Hussain SP, Vahakangas KH, Khan MA, Shields PG, Harris CC. Molecular epidemiology of human cancer risk: gene-environment interactions and p53 mutation spectrum in human lung cancer. J Pathol. 1999;187:8-18 [DOI] [PubMed] [Google Scholar]
- 5. Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8:275-83 [DOI] [PubMed] [Google Scholar]
- 6. Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer. 2009;9:701-13 [DOI] [PubMed] [Google Scholar]
- 7. Oren M. Decision making by p53: life, death and cancer. Cell Death Differ. 2003;10:431-42 [DOI] [PubMed] [Google Scholar]
- 8. Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nat Rev Cancer. 2002;2:594-604 [DOI] [PubMed] [Google Scholar]
- 9. Weisz L, Oren M, Rotter V. Transcription regulation by mutant p53. Oncogene. 2007;26:2202-11 [DOI] [PubMed] [Google Scholar]
- 10. Yan W, Chen X. Characterization of functional domains necessary for mutant p53 gain of function. J Biol Chem. 2010;285:14229-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zambetti GP. The p53 mutation “gradient effect” and its clinical implications. J Cell Physiol. 2007;213:370-3 [DOI] [PubMed] [Google Scholar]
- 12. Petitjean A, Mathe E, Kato S, et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat. 2007;28:622-9 [DOI] [PubMed] [Google Scholar]
- 13. Latronico AC, Pinto EM, Domenice S, et al. An inherited mutation outside the highly conserved DNA-binding domain of the p53 tumor suppressor protein in children and adults with sporadic adrenocortical tumors. J Clin Endocrinol Metab. 2001;86:4970-3 [DOI] [PubMed] [Google Scholar]
- 14. Ribeiro RC, Sandrini F, Figueiredo B, et al. An inherited p53 mutation that contributes in a tissue-specific manner to pediatric adrenal cortical carcinoma. Proc Natl Acad Sci U S A. 2001;98:9330-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pinto EM, Ribeiro RC, Kletter GB, et al. Inherited germline TP53 mutation encodes a protein with an aberrant C-terminal motif in a case of pediatric adrenocortical tumor. Fam Cancer. 2011;10:141-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Milner J, Medcalf EA. Cotranslation of activated mutant p53 with wild type drives the wild-type p53 protein into the mutant conformation. Cell. 1991;65:765-74 [DOI] [PubMed] [Google Scholar]
- 17. de Vries A, Flores ER, Miranda B, et al. Targeted point mutations of p53 lead to dominant-negative inhibition of wild-type p53 function. Proc Natl Acad Sci U S A. 2002;99:2948-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Russell-Swetek A, West AN, Mintern JE, et al. Identification of a novel TP53 germline mutation E285V in a rare case of paediatric adrenocortical carcinoma and choroid plexus carcinoma. J Med Genet. 2008;45:603-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu G, McDonnell TJ, Montes de Oca Luna R, et al. High metastatic potential in mice inheriting a targeted p53 missense mutation. Proc Natl Acad Sci U S A. 2000;97:4174-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69:1237-45 [DOI] [PubMed] [Google Scholar]
- 21. Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296-9 [DOI] [PubMed] [Google Scholar]
- 22. Wade M, Wahl GM. Targeting Mdm2 and Mdmx in cancer therapy: better living through medicinal chemistry? Mol Cancer Res. 2009;7:1-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bond GL, Hu W, Bond EE, et al. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell. 2004;119:591-602 [DOI] [PubMed] [Google Scholar]
- 24. Bougeard G, Baert-Desurmont S, Tournier I, et al. Impact of the MDM2 SNP309 and p53 Arg72Pro polymorphism on age of tumour onset in Li-Fraumeni syndrome. J Med Genet. 2006;43:531-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shvarts A, Steegenga WT, Riteco N, et al. MDMX: a novel p53-binding protein with some functional properties of MDM2. EMBO J. 1996;15:5349-57 [PMC free article] [PubMed] [Google Scholar]
- 26. Reed D, Shen Y, Shelat AA, et al. Identification and characterization of the first small molecule inhibitor of MDMX. J Biol Chem. 2010;285:10786-96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Laurie NA, Donovan SL, Shih CS, et al. Inactivation of the p53 pathway in retinoblastoma. Nature. 2006;444:61-6 [DOI] [PubMed] [Google Scholar]
- 28. Li FP, Fraumeni JF, Jr, Mulvihill JJ, et al. A cancer family syndrome in twenty-four kindreds. Cancer Res. 1998;48:5358-62 [PubMed] [Google Scholar]
- 29. Lustbader ED, Williams WR, Bondy ML, Strom S, Strong LC. Segregation analysis of cancer in families of childhood soft-tissue-sarcoma patients. Am J Hum Genet. 1992;51:344-56 [PMC free article] [PubMed] [Google Scholar]
- 30. Gonzalez KD, Noltner KA, Buzin CH, et al. Beyond Li Fraumeni syndrome: clinical characteristics of families with p53 germline mutations. J Clin Oncol. 2009;27:1250-6 [DOI] [PubMed] [Google Scholar]
- 31. Olivier M, Goldgar DE, Sodha N, et al. Li-Fraumeni and related syndromes: correlation between tumor type, family structure, and TP53 genotype. Cancer Res. 2003;63:6643-50 [PubMed] [Google Scholar]
- 32. Birch JM. Familial cancer syndromes and clusters. Br Med Bull. 1994;50:624-39 [DOI] [PubMed] [Google Scholar]
- 33. Chompret A, Abel A, Stoppa-Lyonnet D, et al. Sensitivity and predictive value of criteria for p53 germline mutation screening. J Med Genet. 2001;38:43-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Birch JM, Hartley AL, Tricker KJ, et al. Prevalence and diversity of constitutional mutations in the p53 gene among 21 Li-Fraumeni families. Cancer Res. 1994;54:1298-304 [PubMed] [Google Scholar]
- 35. Varley JM. Germline TP53 mutations and Li-Fraumeni syndrome. Hum Mutat. 2003;21:313-20 [DOI] [PubMed] [Google Scholar]
- 36. Jemal A, Murray T, Samuels A, Ghafoor A, Ward E, Thun MJ. 2003. Cancer statistics, 2003. CA Cancer J Clin. 2003;53:5-26 [DOI] [PubMed] [Google Scholar]
- 37. Munir CS, Nectoux J. International patterns of cancer. In: Schottenfeld D, Fraumeni JF, Jr, editors. Cancer epidemiology and prevention. 2nd ed. New York: Oxford University Press; 1996. p. 141-67 [Google Scholar]
- 38. Teinturier C, Pauchard MS, Brugieres L, Landais P, Chaussain JL, Bougneres PF. Clinical and prognostic aspects of adrenocortical neoplasms in childhood. Med Pediatr Oncol. 1999;32:106-11 [DOI] [PubMed] [Google Scholar]
- 39. Langer P, Cupisti K, Bartsch DK, et al. Adrenal involvement in multiple endocrine neoplasia type 1. World J Surg. 2002;26:891-6 [DOI] [PubMed] [Google Scholar]
- 40. Rosati R, Cerrato F, Doghman M, et al. High frequency of loss of heterozygosity at 11p15 and IGF2 overexpression are not related to clinical outcome in childhood adrenocortical tumors positive for the R337H TP53 mutation. Cancer Genet Cytogenet. 2008;186:19-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wagner J, Portwine C, Rabin K, Leclerc JM, Narod SA, Malkin D. High frequency of germline p53 mutations in childhood adrenocortical cancer. J Natl Cancer Inst. 1994;86:1707-10 [DOI] [PubMed] [Google Scholar]
- 42. Michalkiewicz E, Sandrini R, Figueiredo B, et al. Clinical and outcome characteristics of children with adrenocortical tumors: a report from the International Pediatric Adrenocortical Tumor Registry. J Clin Oncol. 2004;22:838-45 [DOI] [PubMed] [Google Scholar]
- 43. West AN, Ribeiro RC, Jenkins J, et al. Identification of a novel germ line variant hotspot mutant p53-R175L in pediatric adrenal cortical carcinoma. Cancer Res. 2006;66:5056-62 [DOI] [PubMed] [Google Scholar]
- 44. Varley JM, McGown G, Thorncroft M, et al. Are there low-penetrance TP53 alleles? Evidence from childhood adrenocortical tumors. Am J Hum Genet. 1999;65:995-1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pianovski MA, Maluf EM, de Carvalho DS, et al. Mortality rate of adrenocortical tumors in children under 15 years of age in Curitiba, Brazil. Pediatr Blood Cancer. 2006;47:56-60 [DOI] [PubMed] [Google Scholar]
- 46. DiGiammarino EL, Lee AS, Cadwell C, et al. A novel mechanism of tumorigenesis involving pH-dependent destabilization of a mutant p53 tetramer. Nat Struct Biol. 2002;9:12-6 [DOI] [PubMed] [Google Scholar]
- 47. Lomax ME, Barnes DM, Hupp TR, Picksley SM, Camplejohn RS. Characterization of p53 oligomerization domain mutations isolated from Li-Fraumeni and Li-Fraumeni like family members. Oncogene. 1998;17:643-9 [DOI] [PubMed] [Google Scholar]
- 48. Davison TS, Yin P, Nie E, Kay C, Arrowsmith CH. Characterization of the oligomerization defects of two p53 mutants found in families with Li-Fraumeni and Li-Fraumeni-like syndrome. Oncogene. 1998;7:651-6 [DOI] [PubMed] [Google Scholar]
- 49. Seidinger AL, Mastellaro MJ, Fortes FP, et al. Association of the highly prevalent TP53 R337H mutation with pediatric choroid plexus carcinoma and osteosarcoma in southeast Brazil. Cancer. 2010. December 29 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 50. Assumpcao JG, Seidinger AL, Mastellaro MJ, et al. Association of the germline TP53 R337H mutation with breast cancer in southern Brazil. BMC Cancer. 2008;8:357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Oliveira CR, Mendonca BB, Camargo OP, et al. Classical osteoblastoma, atypical osteoblastoma, and osteosarcoma: a comparative study based on clinical, histological, and biological parameters. Clinics (Sao Paulo). 2007;62:167-74 [DOI] [PubMed] [Google Scholar]
- 52. McIntyre JF, Smith-Sorensen B, Friend SH, et al. Germline mutations of the p53 tumor suppressor gene in children with osteosarcoma. J Clin Oncol. 1994;12:925-30 [DOI] [PubMed] [Google Scholar]
- 53. Figueiredo BC, Sandrini R, Zambetti GP, et al. Penetrance of adrenocortical tumours associated with the germline TP53 R337H mutation. J Med Genet. 2006;43:91-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pinto EM, Billerbeck AE, Fragoso MC, Mendonca BB, Latronico AC. Deletion mapping of chromosome 17 in benign and malignant adrenocortical tumors associated with the Arg337His mutation of the p53 tumor suppressor protein. J Clin Endocrinol Metab. 2005;90:2976-81 [DOI] [PubMed] [Google Scholar]
- 55. Pinto EM, Billerbeck AE, Villares MC, Domenice S, Mendonca BB, Latronico AC. Founder effect for the highly prevalent R337H mutation of tumor suppressor p53 in Brazilian patients with adrenocortical tumors. Arq Bras Endocrinol Metabol. 2004;48:647-50 [DOI] [PubMed] [Google Scholar]
- 56. Parra FC, Amado RC, Lambertucci JR, Jr, Rocha J, Antunes CM, Pena SD. Color and genomic ancestry in Brazilians. Proc Natl Acad Sci U S A. 2003;100:177-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Giordano TJ, Thomas DG, Kuick R, et al. Distinct transcriptional profiles of adrenocortical tumors uncovered by DNA microarray analysis. Am J Pathol. 2003;162:521-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Velazquez-Fernandez D, Laurell C, Geli J, et al. Expression profiling of adrenocortical neoplasms suggests a molecular signature of malignancy. Surgery. 2005;138:1087-94 [DOI] [PubMed] [Google Scholar]
- 59. West AN, Neale GA, Pounds S, et al. Gene expression profiling of childhood adrenocortical tumors. Cancer Res. 2007;67:600-8 [DOI] [PubMed] [Google Scholar]
- 60. Han VK, Lund PK, Lee DC, D’Ercole AJ. Expression of somatomedin/insulin-like growth factor messenger ribonucleic acids in the human fetus: identification, characterization, and tissue distribution. J Clin Endocrinol Metab. 1998;66:422-9 [DOI] [PubMed] [Google Scholar]
- 61. Figueiredo BC, Stratakis CA, Sandrini R, et al. Comparative genomic hybridization analysis of adrenocortical tumors of childhood. J Clin Endocrinol Metab. 1999;84:1116-21 [DOI] [PubMed] [Google Scholar]
- 62. Figueiredo BC, Cavalli LR, Pianovski MA, et al. Amplification of the steroidogenic factor 1 gene in childhood adrenocortical tumors. J Clin Endocrinol Metab. 2005;90:615-9 [DOI] [PubMed] [Google Scholar]
- 63. Pianovski MA, Cavalli LR, Figueiredo BC, et al. SF-1 overexpression in childhood adrenocortical tumours. Eur J Cancer. 2006;42:1040-3 [DOI] [PubMed] [Google Scholar]
- 64. Parker KL, Schimmer BP. Steroidogenic factor 1: a key determinant of endocrine development and function. Endocr Rev. 1997;18:361-77 [DOI] [PubMed] [Google Scholar]
- 65. Doghman M, Karpova T, Rodrigues GA, et al. Increased steroidogenic factor-1 dosage triggers adrenocortical cell proliferation and cancer. Mol Endocrinol. 2007;21:2968-87 [DOI] [PubMed] [Google Scholar]
- 66. Matzuk MM, Finegold MJ, Mather JP, Krummen L, Lu H, Bradley A. Development of cancer cachexia-like syndrome and adrenal tumors in inhibin-deficient mice. Proc Natl Acad Sci U S A. 1994;91:8817-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Longui CA, Lemos-Marini SH, Figueiredo B, et al. Inhibin alpha-subunit (INHA) gene and locus changes in paediatric adrenocortical tumours from TP53 R337H mutation heterozygote carriers. J Med Genet. 2004;41:354-9 [DOI] [PMC free article] [PubMed] [Google Scholar]