

Abstract
Numerous observations indicate a strong link between chronic inflammation and cancer. This link is supported by substantial experimental evidence indicating mutual negative regulation of NF-κB, the major regulator of inflammation, and p53, the major tumor suppressor. This antagonistic relationship reflects the opposite principles of the physiological responses driven by these transcription factors, which act as sensors and mediators of intrinsic and extrinsic cell stresses, respectively. Constitutive activation of NF-κB, the underlying cause of chronic inflammation, is a common acquired characteristic of tumors. A variety of experimental methods have been used to demonstrate that constitutive activation of NF-κB reduces the tumor suppressor activity of p53, thereby creating permissive conditions for dominant oncogene-mediated transformation. Loss of p53 activity is also a characteristic of the majority of tumors and results in unleashed inflammatory responses due to loss of p53-mediated NF-κB suppression. On the other hand, in natural or pharmacological situations of enforced p53 activation, NF-κB activity, inflammation, and immune responses are reduced, resulting in different pathologies. It is likely that the chronic inflammation that is commonly acquired in various tissues of older mammals leads to general suppression of p53 function, which would explain the increased risk of cancer observed in aging animals and humans. Although the molecular mechanisms underlying reciprocal negative regulation of p53 and NF-κB remain to be deciphered, this phenomenon has important implications for pharmacological prevention of cancer and aging and for new approaches to control inflammation.
Keywords: cancer, infection, immune response, NF-κB, cytokines, chemokines, apoptosis, senescence, aging
Inflammation and Cancer Development
At the dawn of cancer research, cancer was first assigned to the category of diseases caused by infectious agents. Indeed, the first cancer-related Nobel Prize was awarded for work supporting the theory of helminthes being cancer-causing agents.1 This and a number of other similar theories were eventually either disproved or defined as laboratory observations with no direct relevance to human diseases (e.g., recombinant retroviruses transducing dominant oncogenes). Yet there remain a handful of infectious agents whose link to cancer is well proven and are capable of inducing transformation directly by transducing cells with dominant oncogenes. These include the lymphotropic oncogenic viruses (Epstein-Barr virus, human herpesvirus 8, and human T-lymphotropic virus 1) and human papilloma viruses, which promote transformation of epithelial cells and significantly contribute to head and neck, anal, and cervical cancer development. The role of infectious agents in cancer is much broader than indicated by these few cases, however. There are abundant examples of infectious agents that do not directly cause cell transformation but are nevertheless clearly associated with increased frequency of development of certain types of cancer. These include microbes representing different kingdoms of life such as Helicobacter pylori, which is associated with gastric cancer and MALT-lymphoma2,3; Schistosoma haematobium, which is associated with bladder cancer4; Opisthorchis viverrini, which is associated with cholangiocarcinoma5; hepatitis B and C viruses, which are associated with liver cancer6-8; and many others (for review, see Rook and Dalgleish9). The only unifying theme among this diverse group of pathogens is their ability to cause long-lasting, frequently latent infections accompanied by mild chronic inflammation that may last for years with no or minimal symptoms. This suggests that it is not the microorganisms themselves but rather the chronic inflammation that they cause that is a cancer predisposing factor. Further support for this idea is the fact that some other pathogens that used to cause severe acute human diseases but now, due to medical advances, cause relatively benign chronic infections are associated with cancers originating from the infected tissues. Examples of this include the association of lung cancer with Mycobacterium tuberculosis and Chlamydophila pneumoniae infection,10-12 colorectal cancer with Streptococcus bovis and Staphylococcus infection,13,14 and gallbladder cancer with chronic Salmonella typhimurium infection.13
Another group of facts pointing to inflammation as a cancer predisposing factor are examples of cancer outgrowth from pathologies that involve noninfectious inflammatory conditions such as Barrett’s esophagitis, chronic pancreatitis, inflammatory bowel diseases (ulcerative colitis and Crohn’s disease), age-related inflammation of prostate tissue, and so on.15-18 Finally, many cancer-inducing factors that were originally assumed to cause cancer through physical or chemical damage to tissues (e.g., smoking, asbestos inhalation, etc.) also cause chronic inflammation, and the contribution of such inflammation to induction of cancer in these patients needs to be further evaluated.19,20
Inflammation is a complex condition that involves a spectrum of local and systemic changes in tissues associated with ongoing immune responses. It involves changes in the activity of multiple pathways in virtually all cells within the inflamed area. These changes are caused by interaction of cellular receptors with numerous secreted inflammation-associated humoral factors. Significant changes occur in the cellular content of inflamed tissues since humoral factors produced at the site of inflammation attract various components of the immune system. Invasion of these components into the tissue further contributes to alteration of the microenvironment by adding new types of humoral, as well as cell-cell, interactions. Inflammation is frequently associated with an increased local concentration of reactive oxygen species and with tissue damage caused by infiltrating immunocytes and their products. These events may trigger tissue regeneration processes that add even more complexity to an already extremely complex condition.
What are the tumor-promoting factors within this dynamic mixture of molecular, cellular, and physiological events taking place in inflamed tissues? Is there any common denominator that is responsible for the elevated risk of a carcinogenic process originating from chronic inflammation? After reviewing the bulk of information relevant to inflammation, one can conclude that there is one central component involved in all inflammation-associated conditions, regardless of whether they are caused by an infectious agent or not, and in all signal transduction pathways activated during inflammation. This central component is the transcription factor, NF-κB.21 In fact, NF-κB is involved in all stages of inflammation: It collects information about damage directly from pathogens via PAMP (pathogen-associated molecular pattern) receptors or from damaged cells via signaling of DAMP (damage-associated molecular pattern) produced by dead cells through the same or similar PAMP receptors.22 Following its activation by ligand-activated PAMP receptors, NF-κB induces inflammation further by producing cytokines and chemokines that attract different cells of the innate and adaptive immune systems to the site of damage or pathogen invasion and induce their activation. NF-κB also serves to significantly increase the self-defense of cells at the site of inflammation by inducing expression of anti-apoptotic factors and scavengers of reactive oxygen species. Finally, NF-κB-regulated expression of growth factors and angiogenic factors helps to restore tissue integrity.21
Given the unique role of NF-κB as the central director of inflammation, its functional links to mechanisms controlling genomic and epigenomic stability and transformation are likely to be responsible for the pro-carcinogenic effects of inflammation. Indeed, accumulating information points to strong functional links between NF-κB and the major tumor suppressor, p53, which seem to be connected by reciprocal negative regulation (see below). In this review, we analyze information about the relationship between p53 and NF-κB within the context of inflammation-induced carcinogenesis.
p53 and NF-κB: Opposite Approaches to Physiological Stress Responses
The p53 and NF-κB pathways are the two major cellular stress response pathways. As summarized in Table 1, there are multiple technical similarities in the mechanisms that control these two pathways. Both pathways are stress responsive (i.e., inducible by dangerous circumstances). Under normal conditions, both p53 and NF-κB reside in the cytoplasm in an inactive form due to binding of specific negative regulators. In both cases, initiation of signal transduction under stress conditions occurs via neutralization of the activity of the inhibitory protein. Upon release from these inhibitors, p53 and NF-κB are translocated into the nucleus, where they bind to numerous DNA sites specific to each factor and regulate (up or down) transcription of numerous responsive genes. These p53- and NF-κB-regulated transcriptional programs produce phenotypic alterations that allow the cell to adequately respond to the applied challenge/stress. Activated p53 and NF-κB both induce expression of genes that encode protein antagonists of themselves. This results in negative feedback regulatory loops that provide for efficient shutdown of the responses, making them temporary and oscillating. The spectrum of genes modulated by each transcription factor is highly cell type specific, which leads to tissue-specific differences in stress responses.23 Finally, both the p53 and NF-κB pathways are commonly (almost universally) deregulated in cancer.24,25
Table 1.
Common Properties Shared by the p53 and NF-κB Pathways
| Property | p53 | NF-κB |
|---|---|---|
| Mediation of cell response to stressful conditions | √ | √ |
| Under normal conditions reside in the cytoplasm bound to inhibitory factor | √ | √ |
| Activation occurs via release from inhibitory factor and nuclear translocation | √ | √ |
| Upon activation, modulate transcription (up or down) of multiple target genes | √ | √ |
| Inhibitory proteins are among positively regulated targets | √ | √ |
| Activation of response is short lasting and self-suppressing | √ | √ |
| Commonly deregulated in cancer via mutations or epigenetic changes | √ | √ |
| Excessive activation can result in severe acute pathology | √ | √ |
In sharp contrast to the multiple mechanistic similarities mentioned above, the phenotypic outcomes of p53 and NF-κB activation are strikingly different and, in general, opposing (Table 2). Activation of NF-κB typically promotes cell survival by making cells resistant to apoptosis and stimulating cell growth (although, as for most biological systems, some exceptions have been observed). On the other hand, p53 activation is growth inhibitory and, depending on the cell type and the type and strength of stress, can result in temporary cell cycle arrest, irreversible arrest (senescence), or apoptosis.26 These cardinal differences in the outcomes of p53 and NF-κB activation are determined by the nature of the genes that they modulate as transcription factors. NF-κB activates transcription of positive growth regulators (e.g., G-CSF), various anti-apoptotic factors (e.g., IAPs, BCL2 family members), and secreted attractants of the immune response (cytokines and chemokines). p53, however, induces genes that encode cell cycle checkpoint regulators (e.g., 21), pro-apoptotic factors (e.g., BAX, PUMA, NOXA), and secreted growth inhibitors.27 Consistent with these functional differences, p53 and NF-κB are deregulated in opposite directions in tumors. Although p53 is a tumor suppressor that is commonly inactivated, NF-κB behaves like an oncogene, showing constitutive activation in the majority of cancers.28
Table 2.
Opposite Direction of p53- and NF-κB-Driven Responses
| p53 | NF-κB |
|---|---|
| Sensor and mediator of intrinsic stresses | Sensor and mediator of extrinsic stresses |
| Tumor suppressor frequently inactivated in cancer | Oncogene frequently activated in tumors |
| Growth suppressive, pro-apoptotic | Growth stimulating, anti-apoptotic |
| Long-term inactivation leads to cancer | Long-term activation leads to cancer |
| Anticancer context: target for activation | Anticancer context: target for repression |
| Tissue protection context: target for repression | Tissue protection context: target for activation |
Another interesting similarity between the p53 and NF-κB pathways is that, although both are deregulated in cancer, neither is deregulated to the greatest possible extreme. Instead of complete loss of expression (which is typical for other tumor suppressors, such as Rb, p16, Arf, and PTEN, and would likely be easier to achieve during tumor progression), the p53 gene is still expressed (either in a mutant or the wild-type form) in the majority of cancers.29 The deregulation of p53 in tumors stems from impairment of its transcription regulatory activity, not necessarily loss of expression. This suggests that p53 may provide some benefits to tumors via an activity distinct from its function as a transcription factor.30,31 Similarly, the constitutive activation of NF-κB that is typically observed in tumors never reaches the maximum possible level of activity for this pathway (NF-κB-activating agents can induce the pathway still further). The threshold for the degree of NF-κB activation in tumors likely represents a compromise between the benefits of having apoptosis suppressed and the risk of attraction of an antitumor immune response.
The biological sense of the opposite stress response strategies executed by p53 and NF-κB lies in the basic differences in the types of stress that they respond to. p53 is the major cellular responder for intrinsic stress signals (e.g., DNA damage, oncogene expression, spindle poisoning, telomere shortening) for which a pro-apoptotic, anti-proliferative outcome is the most beneficial to the organism.25 Propagation of intrinsically damaged cells is restricted by the internal cell decision driven by p53. This is a rational strategy for protection of multicellular organisms from the hazards of amplification of potentially dangerous mutant cells originating from tissues consisting of rapidly dividing cells (i.e., the hematopoietic system, gastrointestinal tract, hair follicles). In this regard, p53’s role is analogous to that of the human conscience: an intrinsic mechanism that prevents members of a social society from unethical actions (which, in extreme cases, could lead to suicidal death, analogous to apoptosis in the context of this metaphor). This type of function must be lost in order to allow unethical actions and establishment of an asocial element, whether human or cellular in nature. On the other hand, NF-κB is the major cellular responder for extrinsic stress signals (presence of infectious agents or traces—direct or indirect—of their presence). Altruistic self-restrictive and self-punishing behavior is not a rational strategy when one is under extrinsic attack, which requires mobilization of all internal and external protective mechanisms. It is, therefore, not surprising that NF-κB activation results in a temporary ban on apoptosis and creates conditions (via secretion of bioactive chemokines and cytokines) for attraction and stimulation of the innate (and, subsequently, an adaptive) immune response. Powerful mobilization of pro-survival functions likely underlies the frequent activation of NF-κB observed in tumor cells.
Reciprocal Negative Regulation of p53 and NF-κB
Given the generally opposite nature of the responses initiated by p53 and NF-κB, there is a clear physiological need to coordinately regulate their relative activities to best serve the organism. There is a growing body of evidence indicating that a reciprocal antagonistic relationship exists between the p53 and NF-κB pathways within cells.32-35 Crosstalk between the p53 and NF-κB pathways occurs on multiple levels, and the results of this crosstalk depend on the specific cellular context and stress stimulus.36 Interplay between p53 and NF-κB leading to coactivation of p53 and NF-κB in some cell systems has been reported.37,38 However, a growing number of reports indicate that cells expressing NF-κB shut down p53 activity and p53 responses and, reciprocally, that cells with activated p53 shut down NF-κB transcriptional activity. For example, pro-inflammatory NF-κB-induced cytokines (such as IL-6 and MIF) can suppress p53 transcriptional activity,37,39,40 and drugs suppressing NF-κB cause activation of p53.41 Despite substantial efforts, our understanding of the mechanisms that regulate p53/NF-κB crosstalk remains limited. Below, we will focus on the mutually antagonistic relationship between p53 and NF-κB and possible mechanisms underlying this phenomenon.
It was shown that p53 and NF-κB can repress the activities of each other owing to their competition for limited pools of the transcriptional coactivator proteins p300 and the closely related CREB binding protein (CBP).42,43 Both p53 and NF-κB have been shown to physically interact with CBP and to require such interaction to maximize their activities.44,45
Among documented models explaining NF-κB-mediated negative regulation of p53 is NF-κB-dependent upregulation of the levels of the major p53 inhibitor, Mdm2, the p53 E3 ubiquitin ligase.46-48 Gu et al.49 demonstrated that in the absence of p53, MDM2 directly increased p65 promoter activity. Another relevant study showed that IKKβ, a major component and mediator of the conventional NF-κB pathway, can phosphorylate p53, leading to its ubiquitination and degradation independent of Mdm2.50 Thus, it is possible that IKK, which is constitutively activated by extrinsic mediators of chronic inflammation (cytokines), might cause long-term inhibition of p53 activity. Products of NF-κB-responsive genes can also suppress p53 function by acting against its downstream targets: For example, NF-κB-induced anti-apoptotic factors can neutralize the effects of pro-apoptotic products induced by p53.51 A recent study demonstrated that IKKα-induced CBP phosphorylation switches CBP’s binding preference from p53 to NF-κB, which results in increased NF-κB-mediated and decreased p53-mediated transcription.43,52,53 It is important to note that IKKα was also found to negatively regulate p53 function through an alternative mechanism involving NF-κB subunit p52.54,55
Despite the large volume of information summarized and discussed in several recent reviews,23,36,51,56 the mechanisms underlying attenuation of NF-κB activity by p53 remain largely unclear. There are data indicating that suppression of NF-κB activity by p53 may be realized through protein-protein interactions, particularly via regulation of the activities of components of the NF-κB pathway such as IKKα57 and IKKβ.58 p53 can restrict activation of the IKKβ-NF-κB pathway through suppression of glycolysis as well (Figure 1).56,59,60
Figure 1.
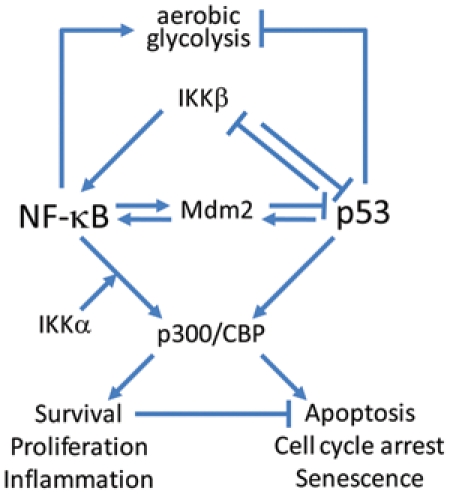
Schematic model summarizing the molecular mechanisms responsible for reciprocal negative regulation of p53 and NF-κB. See explanations in the text.
NF-κB against p53: A Double-Edged Sword
Let us look at the phenomenon of mutual antagonistic regulation between the p53 and NF-κB stress response pathways within the physiological context of the organism (Figure 2). Under what conditions does one pathway dominate over the other? From an organismal standpoint, it would seem most beneficial to give priority to the pathway that deals with the most immediate and more risky stress condition. In this regard, NF-κB responses deal with situations of extreme emergency (extrinsic assault by a variety of infectious agents), whereas p53 responses are aimed at preventing the more delayed, albeit serious, problem of cancer development. Hence, one would expect that under conditions of NF-κB response (e.g., acute infectious disease), it would benefit the organism to impair p53 function in order to reduce unnecessary cell losses that might otherwise occur in tissues prone to p53-dependent apoptosis due to toxicities associated with infection. Experimental data supporting this concept are discussed below in the section devoted to use of NF-κB activators as tissue-protecting agents.
Figure 2.
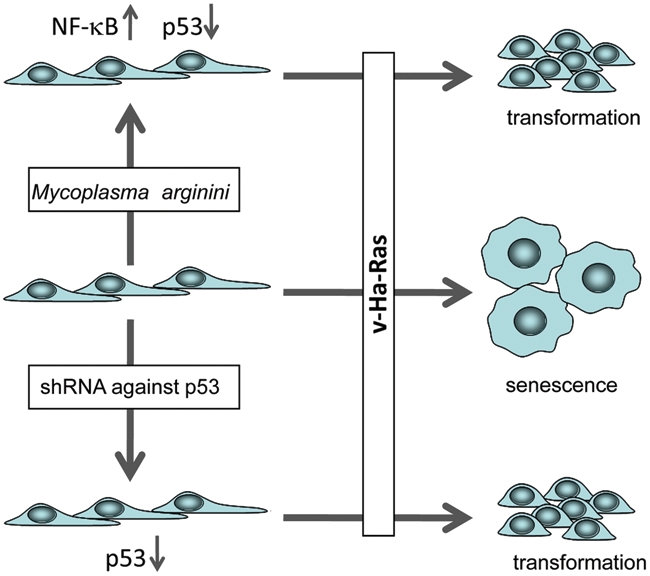
Scheme of the experiments demonstrating the transformation-enabling effect of mycoplasma infection in normal rodent fibroblasts (see text for details).
Despite the association between loss of p53 function and cancer, NF-κB-mediated suppression of p53 is not carcinogenic. Numerous concerns were originally raised about the safety of p53-suppressive drugs; however, it is now clear that short-term, reversible suppression of p53 does not translate into an increased risk of cancer development.61-63 Thus, although long-term (chronic) suppression of p53 is pathogenic and dangerous, short-term (acute) reversible suppression of p53 is benign and actually useful to the organism under some conditions. A similar situation exists for NF-κB (see below).
As described in the introductory section, long-term NF-κB activation, regardless of its underlying cause, is associated with increased risk of cancer development. Since long-term inactivation of p53 creates cancer-prone conditions and NF-κB, as we reviewed above, is a p53 suppressor, it would be natural to link these two facts together and hypothesize that chronic inflammation is tumorigenic due to suppression of p53 by activated NF-κB (Figure 2). The first support for this idea was provided by our finding that the functional inactivation/attenuation of wild-type p53 that is observed in cancers that retain wild-type p53 expression can be maintained by the function of constitutively active NF-κB. In such tumor cells, p53 function can be restored by genetic (ectopic expression of a super-repressor of IκB) or pharmacological inhibition of NF-κB41 (see below). These findings suggest that tumors that acquire activation of NF-κB do not benefit further from p53 mutations since wild-type p53 activity is already “eliminated” through the activity of NF-κB, which creates conditions phenotypically equivalent to genetic p53 deficiency.
If NF-κB promotes malignant transformation via suppression of p53 activity, then it would be expected that the effects of active NF-κB in cell transformation models in vitro would be similar to those caused by p53 suppression. In fact, one of the key tumor suppressor functions of p53 is its Arf-mediated response to the activation of dominant oncogenes resulting in establishment of premature senescence.64 Hence, if NF-κB activation leads to sufficient suppression of p53 activity, one would expect that under conditions of constitutively active NF-κB, cell transformation via oncogenic Ras could occur even in the presence of wild-type p53 expression. We tested this possibility in vitro using mycoplasma infection as a constitutive extrinsic NF-κB-activating agent. We found that various species of mycoplasma or their structural components were able to stimulate NF-κB activation as a result of their interaction with TLR2/6 and TLR2/CD14 receptor complexes. We showed that infection of cells expressing toll-like receptors TLR2/6 with Mycoplasma arginini led to suppression of apoptosis induced by chemotherapeutic agents and reduced activation of p53 and its responsive genes. Infection with M. arginini made rat and mouse embryo fibroblasts susceptible to transformation with oncogenic H-Ras, whereas mycoplasma-free cells underwent irreversible p53-dependent growth arrest (Figure 2). Mycoplasma infection was as effective as shRNA-mediated knockdown of p53 expression in making rodent fibroblasts permissive to Ras-induced transformation. These observations indicate that mycoplasma infection plays the role of a p53-suppressing oncogene that cooperates with Ras in cell transformation and suggest that the carcinogenic and mutagenic effects of mycoplasma might be due to NF-κB-mediated inhibition of p53.65
The observed ability of mycoplasma infection to cooperate with a dominant oncogene in cell transformation is likely to be more than just a laboratory phenomenon. Inspired by our in vitro findings, we recently undertook a clinical study in which we looked at the presence of mycoplasma in the prostates of men suspected of having prostate cancer due to elevated prostate-specific antigen (PSA) levels.66 Mycoplasma hominis was detected in more than 30% of prostate biopsies. Stratification of patients according to diagnosis showed that M. hominis was present at 3 times higher frequency in patients with prostate cancer than in those with benign prostatic hyperplasia. No M. hominis was detected in the prostates of 27 men without detectable prostate disease. In addition, prostate cancer–positive men had higher titers of antibodies against M. hominis, and average PSA levels were higher in M. hominis–positive men. These data, together with previous observations linking mycoplasma infection with cell transformation, genomic instability, and resistance to apoptosis, suggest that M. hominis infection may be involved in the development of prostate cancer and may, therefore, be a potential diagnostic marker and/or target for improved prevention and treatment of this disease.
The effects of live mycoplasma could be partially mimicked in vitro by a lipopeptide component of the mycoplasma surface, the TLR2 agonist R-Pam2. Supplementation of the cell culture medium with R-Pam2 resulted in a substantial delay in the onset of replicative senescence in mouse embryo fibroblasts, which is known to be regulated by p53 (D. Logunov, V. Natarajan, and A. V. Gudkov, unpublished observation).
Finally, we found that genetic elements encoding NF-κB-activating peptides can act as mild p53 suppressors and permit transformation of rodent and human fibroblasts by oncogenic Ras (V. Natarajan, A. Komarov, and A. V. Gudkov, in preparation).
Thus, we have used three different means of mimicking the tumor-specific property of constitutive NF-κB activation—chronic infection (mycoplasma), a pharmacological agent (TLR2 agonist), and genetic modification (NF-κB-activating peptides)—and found that all lead to attenuation of p53 function and establishment of permissive conditions for transformation.
Why Does Inflammation Promote Cancer? NF-κB as a p53-Suppressing Oncogene
In summary, activation of NF-κB functionally leads to “weakening” of p53 activities and phenotypically resembles partial deficiency of p53. Different aspects of p53 function are repressed by NF-κB to different degrees. Analysis of the available data suggests that NF-κB is especially effective in suppressing p53-mediated apoptosis. This is due to the activity of a number of anti-apoptotic factors encoded by NF-κB-responsive genes, which are efficient suppressors of p53-dependent mediators of apoptosis. At the same time, p53-dependent growth arrest, which is largely mediated through induction of the cyclin-dependent kinase inhibitor p21, seems to be less significantly affected by NF-κB activation. This indicates that activation of NF- κB is not fully equivalent to p53 knockdown.
Overall, NF-κB activation and establishment of inflammation create a situation in which p53 can no longer effectively exert its function as an eradicator of transformation-prone cells (Figure 3). The severity of the consequences that follow depends dramatically on the duration of inflammation. For “typical” acute infections, strong NF-κB activity lasts from a few hours to a few weeks. As shown by Gerard Evan’s group, this duration of a p53-suppressed state is not sufficient to cause a detectable increase in spontaneous carcinogenesis in vivo in mice.63 Everyday experience also supports the safety of short-term NF-κB induction in terms of cancer promotion: No association has been found between occurrence of diseases associated with acute inflammation and cancer. Chronic inflammation, however, is a completely different story since the cells in inflamed tissues may exist in an NF-κB-activated/p53-suppressed state for amounts of time sufficient for the acquisition of genetic and epigenetic alterations supporting advanced transformed phenotypes. This makes chronic inflammation a dangerous physiological condition that is functionally equivalent to a p53- suppressing oncogene (Figure 3).
Figure 3.
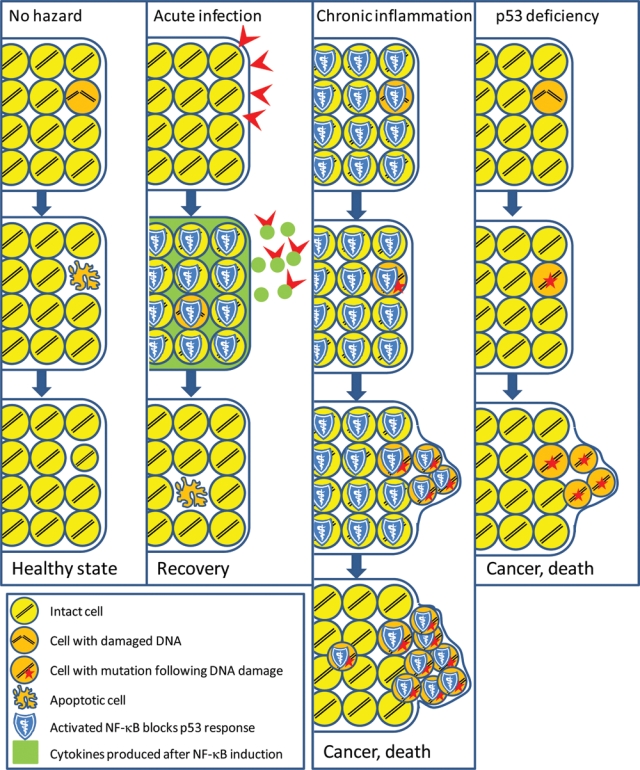
Comparison of the tissue effects of acute and chronic inflammation (scheme is applicable to apoptosis-prone tissues [e.g., hematopoietic tissues]). “No hazard” column: p53-dependent apoptosis helps to clear the cell population of cells that underwent rare spontaneous DNA damage; lost cells are quickly replaced. “Acute infection” column: Attack by infectious agents leads to activation of NF-κB signaling, resulting in short-term (for the time of inflammation) suppression of apoptosis and changes of the microenvironment to alert immune responses (acute inflammation). After eradication of infection, apoptosis is permitted again and eliminates cells that acquired DNA damage during inflammation. “Chronic inflammation” column: Constitutively active NF-κB blocks p53-dependent apoptosis, thereby allowing cells with DNA damage and subsequent mutations to accumulate and form a population with high risk of selection of malignant variants. Under the cover of NF-κB-mediated p53 suppression, tumor progression occurs. This results in advanced tumors, which may no longer depend on constitutive extrinsic activation of NF-κB and continue growing even after conditions of chronic inflammation have been resolved. This situation mimics the ongoing carcinogenic process in p53-deficient organisms (“p53 deficiency” column).
p53 as an Inhibitor of Inflammation
There is a growing body of evidence indicating that p53 is a negative regulator of inflammation. Manifestations of autoimmune diseases, including collagen-induced arthritis67 and experimental autoimmune encephalitis,68 were found to be more severe in p53-deficient mice than in wild-type mice. In addition, inflammatory infiltration of the lung and subsequent disruption of alveolar architecture caused by chronic exposure to the DNA damaging agent bleomycin was markedly increased in p53-null mice and transgenic mice expressing mutant p53 in the lung as compared to wild-type mice.69,70 Accelerated growth of atherosclerotic plaques was observed in p53−/−/apoE−/− mice as compared to p53+/+/apoE−/− mice and in LDL receptor-knockout mice that were lethally irradiated and transplanted with bone marrow from p53−/− mice as compared to the same mice transplanted with p53-wild-type bone marrow.71-74 The observed acceleration in plaque growth was associated with increased invasion of activated macrophages into the plaques. It was also shown that ionizing radiation induces faster and stronger invasion of inflammatory cells and fibroblasts into damaged tissues in p53-null mice than in wild-type mice.75 Specific ablation of the p53 gene in mouse epidermis led to spontaneous development of aggressive squamous cell carcinoma preceded by inflammation.76 It is interesting that mice with a P72R mutation (substitution of proline with arginine) in their p53 gene have a markedly enhanced response to inflammatory challenges, whereas in humans, codon-72 polymorphism is associated with a cancer-prone phenotype.77 Also, mice harboring a hypomorphic mutant p53, R172P, a mutation that abrogates p53-mediated apoptosis while keeping cell cycle control intact, are more sensitive to ultraviolet light–induced skin inflammation than wild-type mice.78
Finally, a significant proportion of tumor-prone p53-null mice (25%) die before tumor development from unresolved inflammation that results in abscesses, gastroenteritis, or myocarditis,16,76 suggesting that the control of innate immune response is deregulated in these mice.79
Consistent with the observations described above, we found that p53 is a general inhibitor of inflammation and that this activity is due to its antagonism of NF-κB (Figure 4).80 This was first suggested by our observation of striking similarities in the global gene expression profiles of human LNCaP prostate cancer cells transduced with a p53-inhibitory genetic element or treated with tumor necrosis factor (TNF). These data suggested that p53 inhibits transcription of TNF-inducible genes, many of which are known to be regulated by NF-κB. In support of this, ectopically expressed p53 was shown to inhibit transcription from NF-κB-dependent promoters. Furthermore, suppression of inflammatory responses by p53 was observed in vivo by comparing wild-type and p53-null mice at the molecular level (inhibition of transcription of genes encoding cytokines and chemokines, reducing accumulation of reactive oxygen species and protein clearance), the organismal level (high levels of metabolic markers of inflammation in tissues of p53-deficient mice), and the cellular level (activation of macrophages and neutrophils and their hypersensitivity to lipopolysaccharide [LPS]) (Figure 4). More severe inflammatory responses and accelerated development of fibrosis in the lung were also observed in p53−/− mice as compared to wild-type mice (E. A. Komarova and A. V. Gudkov, unpublished observations).
Figure 4.
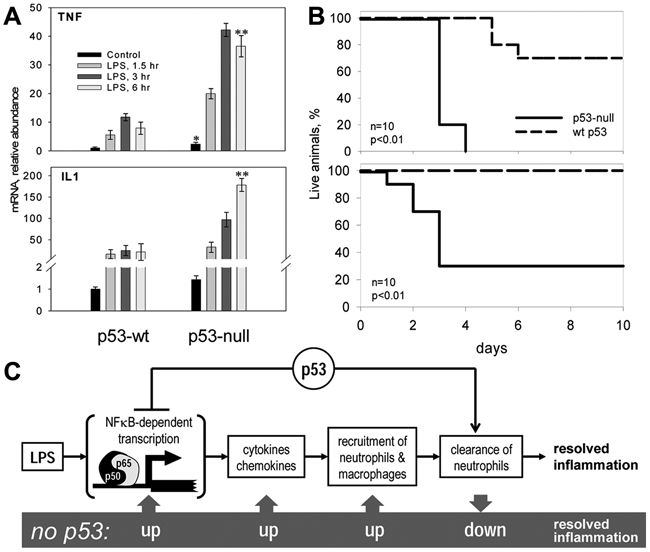
Increased inflammatory response in p53-null mice.68 (A) Dramatic difference in induction of mRNA for inflammatory cytokines by lipopolysaccharide (LPS) treatment (8 mg/kg for 3 hours) between wild-type (wt) and p53-null mice (real-time PCR results). TNF = tumor necrosis factor. **P < 0.01 for p53-null vs. p53 wild-type mice by Student t test. (B) p53-null mice are hypersensitive to LPS-induced septic shock. Survival curves for 2 doses of intraperitoneal-injected LPS: 20 mg/kg (top) and 12 mg/kg (bottom). (C) Scheme of p53-mediated regulation of inflammation. p53 acts at least in two stages of inflammation: as a general inhibitor of NF-κB-dependent transcription and as a positive regulator of neutrophil clearance by macrophages. Lack of p53 results in overreaction to pro-inflammatory stimuli and hypersensitivity of p53-null mice.
Later, similar results were described in a study that used a mouse model of LPS-induced lung injury.81 In this study, neutrophils and macrophages from p53-knockout mice had elevated responses to LPS stimulation as compared to wild-type cells, showing stronger induction of pro-inflammatory cytokines and enhanced NF-κB DNA binding activity. In addition, p53-knockout mice were more susceptible to LPS-induced acute lung injury as compared to wild-type mice.81 Interestingly, p21-null mice have an overactivated inflammatory phenotype that is similar to that of p53-null mice. p21−/− mice displayed increased susceptibility to endotoxic shock, which was associated with increased serum levels of cytokines. Elevated NF-κB activity and secretion of cytokines was also found in LPS-stimulated p21-deficient mouse and human macrophages as compared to similarly treated wild-type cells.82,83 It will be interesting to learn whether the increased inflammatory status of p21−/− mice is p53 dependent.
The observations described above indicate that p53, acting through suppression of NF-κB, plays the role of a general “buffer” of innate immune responses in vivo. This role is consistent with both the tumor suppressor function of p53 (since inflammation is frequently associated with tumorigenesis24) and the constitutive activation of NF-κB that is commonly observed in tumors, the majority of which are p53 deficient.41
Use of p53-NF-κB Mutual Antagonism to Treat Pathologies Associated with Deregulation of the p53 and NF-κB Pathways
Excessive activation or abnormal regulation of p53 or NF-κB can be underlying causes of a variety of pathologies. Acute overactivation of NF-κB, which can result from exposure to powerful bacterial agonists of receptors of innate immunity, injection of pro-inflammatory cytokines, or other situations, can produce severe systemic acute inflammation known as septic shock. This is a common pathology and one of the most frequent causes of human death.84 Acute activation of p53 following systemic genotoxic stress associated with exposure to ionizing radiation or treatment with cytotoxic chemotherapeutic drugs results in massive apoptosis in sensitive tissues, such as the hematopoietic system, gastrointestinal tract, and hair follicles.27 These p53-dependent responses contribute to development of acute radiation syndrome and the side effects of chemotherapy that cause morbidity and treatment-limiting toxicities in millions of cancer patients.27,85 Thus, for both NF-κB and p53, normal physiological responses can lead to lethal consequences if activated to excessive levels. Therefore, availability of p53 and NF-κB inhibitors would likely have a substantial clinical impact on treatment and/or prevention of such pathologies.86-88
Other subsets of diseases stem from chronic deregulation of either the p53 or NF-κB pathways. For example, even partial genetic deficiency in p53 function produces severe cancer-prone conditions (e.g., Li-Fraumeni syndrome in humans).89 As previously mentioned, constitutive activation of NF-κB by extrinsic or intrinsic stimuli is an underlying cause of a broad variety of inflammatory disorders and, in many instances, dramatically increases the risk of cancer development.24,90 On the other hand, insufficient activation of immune responses, which are all mediated through different branches of NF-κB signaling, results in immunodeficiency. Agents capable of restoring or enhancing p53 functions or modulating (positively or negatively) NF-κB activity are likely to be useful for treatment and/or prevention of these pathological conditions.
Our increasing understanding of the reciprocal negative regulation of p53 and NF-κB suggests that pharmacological modulation of one pathway might simultaneously produce an opposite effect in the other pathway. This possibility of simultaneous targeting of the p53 and NF-κB pathways opens up interesting opportunities for new approaches to prevention and treatment of the above-mentioned spectrum of diseases. These approaches are briefly described below.
Inducing Inflammation to Suppress p53-Mediated Acute Pathologies
Similar to the toxicity caused by genotoxic chemotherapeutic agents, the toxicity of ionizing radiation (IR) is associated with massive apoptosis in radiosensitive organs. Our studies have demonstrated that by triggering this apoptosis, p53 is responsible for the development of myelosuppression, alopecia, and rapid apoptosis occurring in several layers of proliferating epithelial cells in radiosensitive divisions of the gastrointestinal tract.91,92 Earlier, we validated small molecule inhibitors of p53 functions as potential radioprotective agents.61,93 Later, after discovery of the p53-suppressive role of NF-κB, we considered using NF-κB activators to protect radiosensitive cells from p53-mediated death.
As a sensor and mediator of numerous extrinsic stresses, NF-κB can be activated by a large number of natural agents, which might be expected to simplify and expedite drug development. Indeed, in our work, we chose to search for pharmacological activators of NF-κB that might be prototype drugs among natural factors produced by the mammalian microflora. This was hypothesized to be a potentially promising source of bioactive yet safe molecules since the microflora has undergone evolutionary adaption to be benign for the mammalian organism. We found that agonists of the Toll-like receptors TLR2 (lipopeptides of mycoplasma) and, especially, TLR5 (flagellin proteins of gram-negative bacteria) act as powerful radioprotectants in mice.94 Subsequently, we generated a pharmacologically optimized polypeptide derived from Salmonella flagellin, CBLB502, and showed that it both protects against and mitigates radiation damage to the hematopoietic and gastrointestinal systems of lethally irradiated mice and rhesus macaques. The dramatically increased postirradiation survival of CBLB502-treated animals was associated with strong suppression of p53-dependent apoptosis of hematopoietic precursors in the bone marrow and gastrointestinal precursors in crypts of the small intestine,94 demonstrating that CBLB502 indeed acted as a suppressor of a p53 function (Figure 5). Remarkably, survival benefit from a single injection of CBLB502 was detectable in monkeys when the injection was given between 24 hours before and 48 hours after irradiation.
Figure 5.
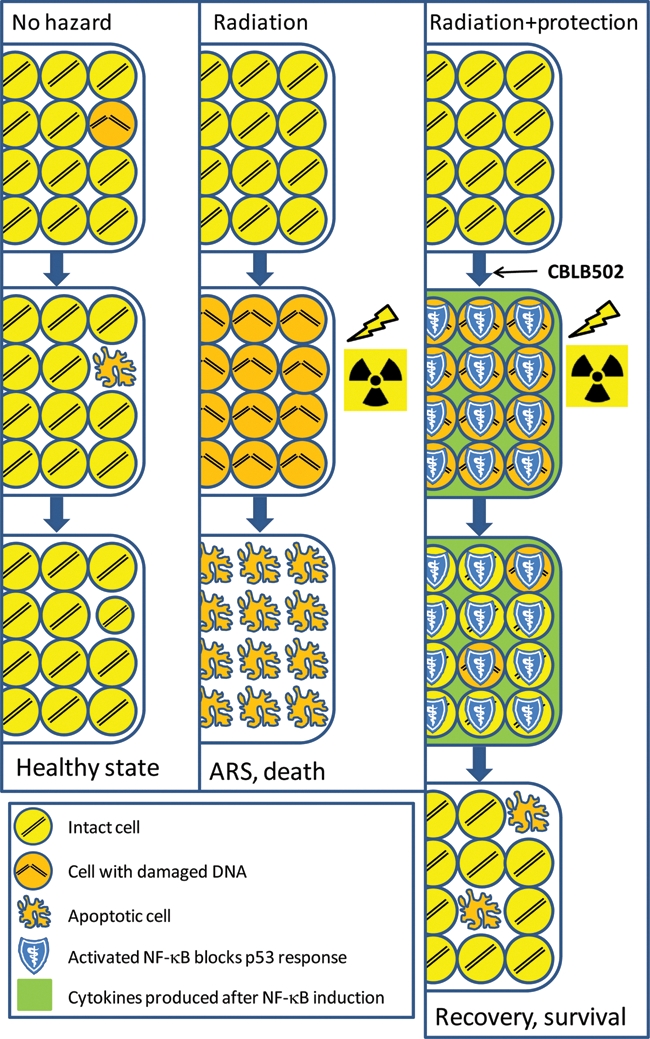
Short-term activation of NF-κB can neutralize tissue damage mediated by p53-dependent apoptosis following systemic genotoxic stress (e.g., ionizing radiation). The scheme is applicable to radiosensitive tissues (e.g., the hematopoietic system or epithelium of the gastrointestinal tract). “No hazard” column: p53-dependent apoptosis helps to clear the cell population of cells that underwent rare spontaneous DNA damage; lost cells are quickly replaced. “Radiation” column: Radiation-induced DNA damage (even technically repairable) is translated into p53-dependent apoptosis, leading to massive cell death in radiosensitive tissues and development of acute radiation syndrome (ARS). “Radiation + protection” column: Administration of an NF-κB-inducing agent (e.g., the TLR5 agonist CBLB502) prior to irradiation leads to transient activation of NF-κB and suppression of apoptosis, thus providing damaged cells an opportunity to repair their DNA during the time of NF-κB activity. Once the effect of NF-κB activation is over, p53 activity is restored and eradicates cells with remaining DNA damage or other apoptosis-triggering conditions (e.g., activation of oncogenic pathways), thus reducing the risk associated with further maintenance and propagation of altered cells.
It is noteworthy that despite its ability to protect normal cells and tissues from radiation-induced death and damage, CBLB502 was found to not reduce the radiosensitivity of tumors in numerous mouse models.94 A similar lack of tumor-protecting activity was demonstrated earlier for the p53 inhibitor pifithrin-α.61,95 These differences in drug activity on tumor versus normal cells are likely due to the intrinsic differences in the activity of the p53 and NF-κB pathways in tumor cells as compared to normal cells.
In addition, a theoretical risk of using CBLB502 to protect against the adverse side effects of cancer treatment is that suppression of apoptosis induced by the systemic genotoxic stress of the treatment might actually promote cancer. However, neither the timing nor frequency of tumor appearance in p53+/− mice was affected by administration of CBLB502 prior to 4 Gy total body irradiation applied to stimulate carcinogenesis. Again, a similar conclusion of no carcinogenic effect was reached for pharmacological inhibition of p53 with pifithrin-α (see above).61,95
Overall, we have found that the radioprotective capacity of NF-κB-activating TLR5 agonists such as CBLB502 exceeds that of p53 inhibitors.94 CBLB502 is currently in the advanced stages of development as a drug for reducing the adverse side effects of cancer radiotherapy and as a radiation antidote to be used in radiation emergencies.
Suppressing Inflammation to Restore p53 Function for Cancer Treatment and Prophylaxis
Discovery of the NF-κB-dependent mechanism of p53 suppression in tumors41 presented an attractive opportunity for simultaneous modulation, in the desired directions, of two major cancer treatment targets, p53 and NF-κB. In fact, inhibition of constitutively active NF-κB by genetic or pharmacological means was found to result in induction of p53-dependent apoptosis in cells in which the p53 pathway is inactive and cannot be activated by “standard” p53-inducing treatments (i.e., DNA damage). Using a renal cell carcinoma (RCC)–derived cell line as a representative of this type of cells, we screened a diverse chemical library for small molecules capable of simultaneously restoring p53-dependent transactivation and suppressing NF-κB activity.41 Among the compounds isolated was an antimalaria drug, quinacrine, which strongly induced p53 function not only in RCC cells but also in other types of cancer cells. Induction of p53 by these compounds was found to depend on NF-κB suppression and does not involve genotoxic stress. A large-scale screening effort was undertaken to isolate more powerful molecules with the same properties as quinacrine. Subsequent hit-to-lead optimization and pharmacological characterization of the resulting compounds led to development of a novel class of small molecules named curaxins. Curaxins are structurally distinct from quinacrine, having carbazole-based structures. However, they are functionally similar to quinacrine in their ability to simultaneously activate p53 and inhibit NF-κB without induction of any detectable genotoxicity. The most powerful of the isolated curaxin molecules exceeds the specific activity of quinacrine by more than 200-fold and demonstrates strong antitumor effects in all mouse tumor models tested to date (more than 10) (A. Gasparian, C. Burkhart, A. Purmal, A. V. Gudkov, and K. V. Gurova, submitted). The lack of genotoxicity and p53-activating ability of these novel NF-κB inhibitors prompted us to consider them not only as potential cancer treatments but also as possible cancer prophylactic agents. Indeed, we found that long-term administration of curaxins in drinking water resulted in a substantial delay in the appearance and reduction in the frequency of tumor development in two cancer-prone mouse models (M. Yakubovskaya, A. V. Gudkov, and K. V. Gurova, in preparation). Thus, exploitation of the phenomenon of NF-κB-mediated p53 suppression has led to a new approach for development of novel cancer preventive agents.
Nongenotoxic Activation of p53 to Suppress Inflammation
Theoretically, the NF-κB-suppressive activity of p53 might be employed as an anti-inflammatory strategy. The best reagent currently available to test the feasibility of this approach is Nutlin-3A, a small molecule inhibitor of the p53-Mdm2 interaction that was isolated by Lyubomir Vassilev.96 Nutlin-3A has shown strong promise as an anticancer agent for the treatment of malignancies retaining wild-type p53 expression.97,98 p53 activation by Nutlin-3A is nongenotoxic and well tolerated by normal tissues.99 Pharmacologically optimized analogues of Nutlin-3A are currently in phase I clinical trials.100
On the basis of our finding that p53 is a suppressor of inflammatory responses, we proposed that pharmacological activation of p53 by Nutlin-3A96 in mice might inhibit inflammatory responses induced by pro-inflammatory stimuli. In fact, Nutlin-3A administration significantly increased the survival of mice following LPS-induced septic shock (E. A. Komarova, L. Vassilev, and A. V. Gudkov, in preparation). These results are consistent with earlier reports by Liu et al.81 and Groskreutz et al.,101 who showed anti-inflammatory effects of Nutlin-3A in models of LPS-induced and respiratory syncytial virus–induced lung injury, respectively. It was shown previously in vitro that Nutlin-3A can down-regulate TNFα- and IL-1-induced activation of NF-κB-dependent reporter gene expression and inhibit the expression of proteins encoded by the NF-κB target genes ICAM-1 and MCP-1, which are known to be critical for cancer cell invasion.102 In our in vivo experiments, mice given Nutlin-3A by gavage showed significantly reduced serum levels of cytokines, including IL-1β, IL-6, TNFα, and GM-CSF (E. A. Komarova, L. Vassilev, and A. V. Gudkov, in preparation).
Aging-Associated Inflammation as a p53-Suppressing Carcinogen
There are numerous indications that aging in mammals commonly involves processes driven by p53 and NF-κB, suggesting that suppression of p53 by inflammation may have implications for aging. Gradual increase in systemic inflammation is one of the universal hallmarks of aging. It includes elevation of serum cytokine levels, infiltration of immunocytes into tissues, and a high frequency of inflammation-associated diseases in elderly subjects,103 all indicative of elevated NF-κB activity. In fact, we recently confirmed this prediction directly by measuring NF-κB activity in tissues of mice of different ages that carried an NF-κB-responsive luciferase reporter construct in their germline. In these experiments, we observed a gradual increase in reporter activity, with a maximal degree of NF-κB-driven transcription being observed at the end of life (I. Gitlin, L. Burdelya, and A. V. Gudkov, unpublished observations). Consistent with this and the reciprocal negative regulation of NF-κB and p53, the activity of p53 in mouse tissues gradually declines with age.104
The source of aging-associated inflammation has not been unequivocally defined. Aging tissues show gradual accumulation of senescent cells characterized by acquisition of the so-called pathological secretion phenotype, which is considered one of the important systemic pathological alterations accompanying, and possibly contributing to, aging.105 NF-κB-driven transcription of cytokines and chemokines is a major component of this phenotype, determining, at least in part, the phenomenon of age-associated chronic inflammation.
At the same time, there is a large body of somewhat controversial evidence linking p53 with regulation of longevity and aging,106,107 including a role in cellular senescence. On one hand, p53, acting largely through positive regulation of the cyclin kinase inhibitor p21, has been shown to mediate replicative senescence in fibroblasts in culture,108 as well as “premature senescence” caused by aberrant oncogene activation.64,109,110 On the other hand, there are cell models in which p53 acts as a suppressor of senescence.26,111 This presumably results from the ability of p53 to negatively regulate mTOR and AKT signaling,112 which mechanistically mimics calorie restriction known to increase longevity. In fact, the role of p53 as a suppressor of senescence can be pharmacologically mimicked by the mTOR inhibitor rapamycin.113 To resolve this apparent controversy, one can hypothesize that p53 contributes to establishment of senescence by inducing initial cell cycle arrest (quiescence) via its genotoxic or oncogenic stress response but delays the decision on the irreversibility of this arrest via its regulation of cell metabolism.26,112,113
The complexity of the relationship between p53 and aging is reflected by a variety of age-related phenotypes observed in mice with deregulated p53 expression or function. For example, the increased p53 gene dose in transgenic “super p53” mice carrying an extra copy of the p53 gene significantly protected them from cancer and did not result in premature aging.114 Similarly, “super Ink4a/Arf” mice carrying an extra transgenic copy of the entire Ink4a/Arf locus have elevated p53 activity and show higher resistance to cancer than wild-type mice and normal aging and life span as well.115 Consistent with this, mice in which Mdm2 expression was genetically reduced had a normal life span and were resistant to tumor development.116,117 However, mice in which overexpression of p53 was accompanied by an imbalance in the normal ratios of different p53 isoforms showed an alarming premature aging phenotype.118 Also, several knockout and transgenic mouse lines that exhibited increased p53 activity had premature aging phenotypes.119,120 In some cases, these aging phenotypes were partially rescued by reduction of the p53 dosage.106,121
We propose a hypothesis (Figure 6) that accommodates most of the above-mentioned facts and largely resolves the existing controversies regarding the dual role of p53 in aging. The center of this model is a gradual accumulation of senescent cells with constitutively active NF-κB, which drives abnormal secretion of pro-inflammatory factors and determines the pathological secretion phenotype. This further amplifies inflammation caused by NF-κB activation in secondary cells. Senescent cells accumulate in tissues through life as a result of a two-step process of cell conversion from proliferation-competent to a quiescent state and then to a senescent state. The first step (quiescence) occurs as a result of p53-mediated growth arrest in response to spontaneous rare spikes in proto-oncogene pathway activation and episodes of oxidative stress causing DNA damage. The second step depends on metabolic stimulation of quiescent cells and requires mTOR/AKT activity; it can be effectively suppressed by the mTOR inhibitor rapamycin and by nongenotoxic activation of p53 (with Nutlin-3A).111,113,122 It is the second step that makes growth arrest irreversible and converts it to senescence with a pathological secretion phenotype. The role of p53 is 2-fold: On one hand, it recruits cells into the quiescent state and, on the other hand, inhibits their transition from quiescence into senescence by suppressing mTOR/AKT.
Figure 6.
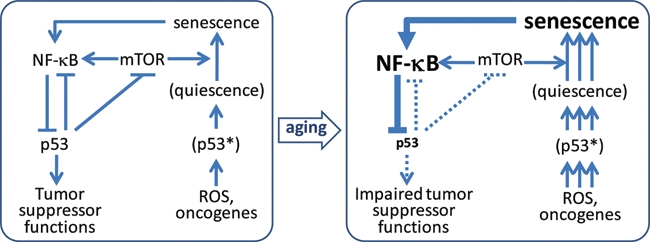
Schematic description of our aging hypothesis, which explains mechanisms of increasing imbalance of NF-κB (inflammation) and p53 activities with age. This imbalance leads to a gradual progressive increase in the severity of inflammation, decline in p53 tumor suppressor functions, and elevation of the risk of development of cancer and other age-related diseases. See explanation in the text. ROS = reactive oxygen species.
With age, the proportion of senescent cells in tissues gradually increases, particularly in connective and other tissues consisting of proliferation-competent cells that cannot be eliminated by apoptosis (as in the hematopoietic system) or constant renewal of cell populations (as in rapidly growing epithelia). With accumulation of senescent cells, the tissue concentration of NF-κB-driven pro-inflammatory cytokines (intrinsic and produced by bystander cells) increases, leading to the establishment of inflammation. Inflammation, in turn, inhibits p53 activities, including its suppression of NF-κB and mTOR. At the same time, the growth arrest–inducing function of p53 (which seems to be less dependent on NF-κB) continues generating quiescent precursors of senescent cells; the number of such cells increases with the increase in inflammation-associated oxidative stress. This creates a positive feedback loop that strengthens the severity of inflammation with time. Together, these events establish the conditions of age-related syndrome: tissue poisoning by the products of senescent cells leading to systemic inflammation, changes in the contents of cell populations in inflamed tissues, reduction in the regenerating capacity of inflamed tissues, and elevated risk of cancer due to the decline in activity of the major tumor suppressor p53.
The proposed hypothesis accommodates and explains many aging-associated phenomena. It combines Judy Campisi’s aging model (gradually increasing tissue poisoning by accumulation of senescent cells with abnormal inflammation-inducing secretion)105 and indications of a general decline in p53 function in aging tissues123 with the phenomenon of p53 suppression by NF-κB and two distinct roles of p53: 1) its role as a suppressor of senescence via inhibition of mTOR/AKT26,112 and 2) its role as an inducer of growth arrest in response to oxidative or oncogenic stresses, which can eventually lead to irreversible senescence.64,109 It provides a mechanistic basis for the observed age-related increase in cancer incidence, linking it to inflammation-mediated p53 suppression. It is consistent with the anti-aging effects of the mTOR inhibitor rapamycin, anti-inflammatory agents, and antioxidants, but clearly shows that these agents may be useful only for prophylaxis, not reversion, of age-related syndrome. This model also predicts that restoration of p53 function by targeting the mechanism of NF-κB-mediated p53 suppression has the potential to slow down self-accelerating age-associated inflammation.
Conclusions
Inflammation presents a major challenge for the tumor suppressor function of p53. Mechanistically, it is exerted through NF-κB-mediated attenuation of the transactivation function of p53. This situation occurs during chronic infections and aging, thereby creating conditions that stimulate cancer development. In tumors, overactivation of NF-κB is acquired through selection of cell clones in which p53 is functionally inactivated. This situation underscores the role of anti-inflammatory therapy as a potential approach for the treatment and prophylaxis of cancer and aging. Technically, this type of therapy could be achieved either by eradicating the source of inflammation (e.g., antimycoplasma treatment, killing of senescent cells) or by using pharmacological inhibitors of NF-κB or other components of the mechanism of NF-κB-mediated suppression of p53.
Acknowledgments
We thank Patricia Stanhope-Baker for valuable help in manuscript preparation and Mikhail Blagosklonny for creative discussions.
Footnotes
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Andrei Gudkov is a shareholder and consultant of Cleveland Biolabs, Incuron, and Tartis. He is principal investigator (PI) on research projects supported by Cleveland Biolabs and Tartis. Katerina Gurova is PI on a contract research project funded by Incuron. Elena Komarova is a consultant of Cleveland Biolabs.
This work was supported by grants CA75179, AI080446, and AI087616 from the National Institutes of Health and Cleveland Biolabs to A.V.G.
References
- 1. Nobel Lectures, Physiology or Medicine 1922-1941. Amsterdam, London, and New York: Elsevier; 1965 [Google Scholar]
- 2. Loffeld RJ, Willems I, Flendrig JA, Arends JW. Helicobacter pylori and gastric carcinoma. Histopathology. 1990;17(6):537-41 [DOI] [PubMed] [Google Scholar]
- 3. Stolte M. Helicobacter pylori gastritis and gastric MALT-lymphoma. Lancet. 1992;339(8795):745-6 [DOI] [PubMed] [Google Scholar]
- 4. Gelfand M, Weinberg RW, Castle WM. Relation between carcinoma of the bladder and infestation with Schistosoma haematobium. Lancet. 1967;1(7502):1249-51 [DOI] [PubMed] [Google Scholar]
- 5. Schwartz DA. Helminths in the induction of cancer: Opisthorchis viverrini, Clonorchis sinensis and cholangiocarcinoma. Trop Geogr Med. 1980;32(2):95-100 [PubMed] [Google Scholar]
- 6. Chang MH. Hepatitis B virus infection. Semin Fetal Neonatal Med. 2007;12(3):160-7 [DOI] [PubMed] [Google Scholar]
- 7. Kiyosawa K, Akahane Y, Nagata A, Furuta S. Hepatocellular carcinoma after non-A, non-B posttransfusion hepatitis. Am J Gastroenterol. 1984;79(10):777-81 [PubMed] [Google Scholar]
- 8. Sherlock S, Fox RA, Niazi SP, Scheuer PJ. Chronic liver disease and primary liver-cell cancer with hepatitis-associated (Australia) antigen in serum. Lancet. 1970;1(7659):1243-7 [DOI] [PubMed] [Google Scholar]
- 9. Rook GA, Dalgleish A. Infection, immunoregulation, and cancer. Immunol Rev. 2011;240(1):141-59 [DOI] [PubMed] [Google Scholar]
- 10. Dacosta NA, Kinare SG. Association of lung carcinoma and tuberculosis. J Postgrad Med. 1991;37(4):185-9 [PubMed] [Google Scholar]
- 11. Engels EA, Shen M, Chapman RS, et al. Tuberculosis and subsequent risk of lung cancer in Xuanwei, China. Int J Cancer. 2009;124(5):1183-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shiels MS, Albanes D, Virtamo J, Engels EA. Increased risk of lung cancer in men with tuberculosis in the alpha-tocopherol, Beta-carotene cancer prevention study. Cancer Epidemiol Biomarkers Prev. 2011;20(4):672-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Boleij A, Muytjens CM, Bukhari SI, et al. Novel clues on the specific association of Streptococcus gallolyticus subsp gallolyticus with colorectal cancer. J Infect Dis. 2011;203(8):1101-9 [DOI] [PubMed] [Google Scholar]
- 14. De Flora S, Bonanni P. The prevention of infection-associated cancers. Carcinogenesis. 2011. March 24 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Raimondi S, Lowenfels AB, Morselli-Labate AM, Maisonneuve P, Pezzilli R. Pancreatic cancer in chronic pancreatitis: aetiology, incidence, and early detection. Best Pract Res Clin Gastroenterol. 2010;24(3):349-58 [DOI] [PubMed] [Google Scholar]
- 16. Sandhu JS. Prostate cancer and chronic prostatitis. Curr Urol Rep. 2008;9(4):328-32 [DOI] [PubMed] [Google Scholar]
- 17. Tamura H, Schulman SA. Barrett-type esophagus associated with squamous carcinoma. Chest. 1971;59(3):330-3 [DOI] [PubMed] [Google Scholar]
- 18. Terzic J, Grivennikov S, Karin E, Karin M. Inflammation and colon cancer. Gastroenterology. 2010;138(6):2101-14, e5 [DOI] [PubMed] [Google Scholar]
- 19. Adcock IM, Caramori G, Barnes PJ. Chronic obstructive pulmonary disease and lung cancer: new molecular insights. Respiration. 2011;81(4):265-84 [DOI] [PubMed] [Google Scholar]
- 20. Chapman EA, Thomas PS, Yates DH. Breath analysis in asbestos-related disorders: a review of the literature and potential future applications. J Breath Res. 2010;4(3):034001. [DOI] [PubMed] [Google Scholar]
- 21. Karin M. NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb Perspect Biol. 2009;1(5):a000141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hajishengallis G, Lambris JD. Microbial manipulation of receptor crosstalk in innate immunity. Nat Rev Immunol. 2011;11(3):187-200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lane D, Levine A. p53 Research: the past thirty years and the next thirty years. Cold Spring Harb Perspect Biol. 2010;2(12):a000893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441(7092): 431-6 [DOI] [PubMed] [Google Scholar]
- 25. Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. 2009;9(10):749-58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Feng Z, Hu W, Rajagopal G, Levine AJ. The tumor suppressor p53: cancer and aging. Cell Cycle. 2008;7(7):842-7 [DOI] [PubMed] [Google Scholar]
- 27. Gudkov AV, Komarova EA. Pathologies associated with the p53 response. Cold Spring Harb Perspect Biol. 2010;2(7):a001180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lu T, Burdelya LG, Swiatkowski SM, et al. Secreted transforming growth factor beta2 activates NF-kappaB, blocks apoptosis, and is essential for the survival of some tumor cells. Proc Natl Acad Sci U S A. 2004;101(18):7112-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. IARC p53 Database. Available at: www.IARC.fr/p53/index.html.
- 30. Goh AM, Coffill CR, Lane DP. The role of mutant p53 in human cancer. J Pathol. 2011;223(2):116-26 [DOI] [PubMed] [Google Scholar]
- 31. Goldstein I, Marcel V, Olivier M, Oren M, Rotter V, Hainaut P. Understanding wild-type and mutant p53 activities in human cancer: new landmarks on the way to targeted therapies. Cancer Gene Ther. 2011;18(1):2-11 [DOI] [PubMed] [Google Scholar]
- 32. Lee TL, Yang XP, Yan B, et al. A novel nuclear factor-kappaB gene signature is differentially expressed in head and neck squamous cell carcinomas in association with TP53 status. Clin Cancer Res. 2007;13(19):5680-91 [DOI] [PubMed] [Google Scholar]
- 33. Scian MJ, Stagliano KE, Anderson MA, et al. Tumor-derived p53 mutants induce NF-kappaB2 gene expression. Mol Cell Biol. 2005;25(22):10097-110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Szoltysek K, Pietranek K, Kalinowska-Herok M, Pietrowska M, Kimmel M, Widlak P. TNFalpha-induced activation of NFkappaB protects against UV-induced apoptosis specifically in p53-proficient cells. Acta Biochim Pol. 2008;55(4):741-8 [PubMed] [Google Scholar]
- 35. Weisz L, Damalas A, Liontos M, et al. Mutant p53 enhances nuclear factor kappaB activation by tumor necrosis factor alpha in cancer cells. Cancer Res. 2007;67(6):2396-401 [DOI] [PubMed] [Google Scholar]
- 36. Schneider G, Kramer OH. NFkappaB/p53 crosstalk-a promising new therapeutic target. Biochim Biophys Acta. 2011;1815(1):90-103 [DOI] [PubMed] [Google Scholar]
- 37. O’Prey J, Crighton D, Martin AG, Vousden KH, Fearnhead HO, Ryan KM. p53-mediated induction of Noxa and p53AIP1 requires NFkappaB. Cell Cycle. 2010;9(5):947-52 [DOI] [PubMed] [Google Scholar]
- 38. Ryan KM, Ernst MK, Rice NR, Vousden KH. Role of NF-kappaB in p53-mediated programmed cell death. Nature. 2000;404(6780):892-7 [DOI] [PubMed] [Google Scholar]
- 39. Hudson JD, Shoaibi MA, Maestro R, Carnero A, Hannon GJ, Beach DH. A proinflammatory cytokine inhibits p53 tumor suppressor activity. J Exp Med. 1999;190(10):1375-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yonish-Rouach E, Resnitzky D, Lotem J, Sachs L, Kimchi A, Oren M. Wild-type p53 induces apoptosis of myeloid leukaemic cells that is inhibited by interleukin-6. Nature. 1991;352(6333):345-7 [DOI] [PubMed] [Google Scholar]
- 41. Gurova KV, Hill JE, Guo C, et al. Small molecules that reactivate p53 in renal cell carcinoma reveal a NF-kappaB-dependent mechanism of p53 suppression in tumors. Proc Natl Acad Sci U S A. 2005;102(48):17448-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ikeda A, Sun X, Li Y, et al. p300/CBP-dependent and -independent transcriptional interference between NF-kappaB RelA and p53. Biochem Biophys Res Commun. 2000;272(2):375-9 [DOI] [PubMed] [Google Scholar]
- 43. Webster GA, Perkins ND. Transcriptional cross talk between NF-kappaB and p53. Mol Cell Biol. 1999;19(5):3485-95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Avantaggiati ML, Ogryzko V, Gardner K, Giordano A, Levine AS, Kelly K. Recruitment of p300/CBP in p53-dependent signal pathways. Cell. 1997;89(7):1175-84 [DOI] [PubMed] [Google Scholar]
- 45. Gerritsen ME, Williams AJ, Neish AS, Moore S, Shi Y, Collins T. CREB-binding protein/p300 are transcriptional coactivators of p65. Proc Natl Acad Sci U S A. 1997;94(7):2927-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Egan LJ, Eckmann L, Greten FR, et al. IkappaB-kinasebeta-dependent NF-kappaB activation provides radioprotection to the intestinal epithelium. Proc Natl Acad Sci U S A. 2004;101(8):2452-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kashatus D, Cogswell P, Baldwin AS. Expression of the Bcl-3 proto-oncogene suppresses p53 activation. Genes Dev. 2006;20(2):225-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tergaonkar V, Pando M, Vafa O, Wahl G, Verma I. p53 stabilization is decreased upon NFkappaB activation: a role for NFkappaB in acquisition of resistance to chemotherapy. Cancer Cell. 2002;1(5):493-503 [DOI] [PubMed] [Google Scholar]
- 49. Gu L, Findley HW, Zhou M. MDM2 induces NF-kappaB/p65 expression transcriptionally through Sp1-binding sites: a novel, p53-independent role of MDM2 in doxorubicin resistance in acute lymphoblastic leukemia. Blood. 2002;99(9):3367-75 [DOI] [PubMed] [Google Scholar]
- 50. Xia Y, Padre RC, De Mendoza TH, Bottero V, Tergaonkar VB, Verma IM. Phosphorylation of p53 by IkappaB kinase 2 promotes its degradation by beta-TrCP. Proc Natl Acad Sci U S A. 2009;106(8):2629-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol. 2007;8(1):49-62 [DOI] [PubMed] [Google Scholar]
- 52. Huang WC, Ju TK, Hung MC, Chen CC. Phosphorylation of CBP by IKKalpha promotes cell growth by switching the binding preference of CBP from p53 to NF-kappaB. Mol Cell. 2007;26(1):75-87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tergaonkar V, Perkins ND. p53 and NF-kappaB crosstalk: IKKalpha tips the balance. Mol Cell. 2007;26(2):158-9 [DOI] [PubMed] [Google Scholar]
- 54. Schumm K, Rocha S, Caamano J, Perkins ND. Regulation of p53 tumour suppressor target gene expression by the p52 NF-kappaB subunit. Embo J. 2006;25(20):4820-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shen HM, Tergaonkar V. NFkappaB signaling in carcinogenesis and as a potential molecular target for cancer therapy. Apoptosis. 2009;14(4):348-63 [DOI] [PubMed] [Google Scholar]
- 56. Ak P, Levine AJ. p53 and NF-kappaB: different strategies for responding to stress lead to a functional antagonism. Faseb J. 2010;24(10):3643-52 [DOI] [PubMed] [Google Scholar]
- 57. Gu L, Zhu N, Findley HW, Woods WG, Zhou M. Identification and characterization of the IKKalpha promoter: positive and negative regulation by ETS-1 and p53, respectively. J Biol Chem. 2004;279(50):52141-9 [DOI] [PubMed] [Google Scholar]
- 58. Kawauchi K, Araki K, Tobiume K, Tanaka N. Activated p53 induces NF-kappaB DNA binding but suppresses its transcriptional activation. Biochem Biophys Res Commun. 2008;372(1):137-41 [DOI] [PubMed] [Google Scholar]
- 59. Kawauchi K, Araki K, Tobiume K, Tanaka N. p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat Cell Biol. 2008;10(5):611-8 [DOI] [PubMed] [Google Scholar]
- 60. Kawauchi K, Araki K, Tobiume K, Tanaka N. Loss of p53 enhances catalytic activity of IKKbeta through O-linked beta-N-acetyl glucosamine modification. Proc Natl Acad Sci U S A. 2009;106(9):3431-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Komarov PG, Komarova EA, Kondratov RV, et al. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science. 1999;285(5434):1733-7 [DOI] [PubMed] [Google Scholar]
- 62. Christophorou MA, Martin-Zanca D, Soucek L, et al. Temporal dissection of p53 function in vitro and in vivo. Nat Genet. 2005;37(7):718-26 [DOI] [PubMed] [Google Scholar]
- 63. Christophorou MA, Ringshausen I, Finch AJ, Swigart LB, Evan GI. The pathological response to DNA damage does not contribute to p53-mediated tumour suppression. Nature. 2006;443(7108):214-7 [DOI] [PubMed] [Google Scholar]
- 64. Evan GI, d’Adda di Fagagna F. Cellular senescence: hot or what? Curr Opin Genet Dev. 2009;19(1):25-31 [DOI] [PubMed] [Google Scholar]
- 65. Logunov DY, Scheblyakov DV, Zubkova OV, et al. Mycoplasma infection suppresses p53, activates NF-kappaB and cooperates with oncogenic Ras in rodent fibroblast transformation. Oncogene. 2008;27(33):4521-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Barykova YA, Logunov DY, Shmarov MM, et al. Association of Mycoplasma hominis infection with prostate cancer. Oncotarget. 2011. April 4 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yamanishi Y, Boyle DL, Pinkoski MJ, et al. Regulation of joint destruction and inflammation by p53 in collagen- induced arthritis. Am J Pathol. 2002;160(1):123-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Okuda Y, Okuda M, Bernard CC. Regulatory role of p53 in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2003;135(1-2):29-37 [DOI] [PubMed] [Google Scholar]
- 69. Davis DW, Weidner DA, Holian A, McConkey DJ. Nitric oxide-dependent activation of p53 suppresses bleomycin-induced apoptosis in the lung. J Exp Med. 2000;192(6):857-69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ghosh S, Mendoza T, Ortiz LA, et al. Bleomycin sensitivity of mice expressing dominant-negative p53 in the lung epithelium. Am J Respir Crit Care Med. 2002;166(6):890-7 [DOI] [PubMed] [Google Scholar]
- 71. Merched AJ, Williams E, Chan L. Macrophage-specific p53 expression plays a crucial role in atherosclerosis development and plaque remodeling. Arterioscler Thromb Vasc Biol. 2003;23(9):1608-14 [DOI] [PubMed] [Google Scholar]
- 72. Guevara NV, Kim HS, Antonova EI, Chan L. The absence of p53 accelerates atherosclerosis by increasing cell proliferation in vivo. Nat Med. 1999;5(3):335-9 [DOI] [PubMed] [Google Scholar]
- 73. van Vlijmen BJ, Gerritsen G, Franken AL, et al. Macrophage p53 deficiency leads to enhanced atherosclerosis in APOE*3-Leiden transgenic mice. Circ Res. 2001;88(8):780-6 [DOI] [PubMed] [Google Scholar]
- 74. von der Thusen JH, van Vlijmen BJ, Hoeben RC, et al. Induction of atherosclerotic plaque rupture in apolipoprotein E-/- mice after adenovirus-mediated transfer of p53. Circulation. 2002;105(17):2064-70 [DOI] [PubMed] [Google Scholar]
- 75. Komarova EA, Kondratov RV, Wang K, et al. Dual effect of p53 on radiation sensitivity in vivo: p53 promotes hematopoietic injury, but protects from gastro-intestinal syndrome in mice. Oncogene. 2004;23(19):3265-71 [DOI] [PubMed] [Google Scholar]
- 76. Martinez-Cruz AB, Santos M, Garcia-Escudero R, et al. Spontaneous tumor formation in Trp53-deficient epidermis mediated by chromosomal instability and inflammation. Anticancer Res. 2009;29(8):3035-42 [PubMed] [Google Scholar]
- 77. Frank AK, Leu JI, Zhou Y, et al. The codon 72 polymorphism of p53 regulates interaction with NF-{kappa}B and transactivation of genes involved in immunity and inflammation. Mol Cell Biol. 2011;31(6):1201-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Tavana O, Benjamin CL, Puebla-Osorio N, et al. Absence of p53-dependent apoptosis leads to UV radiation hypersensitivity, enhanced immunosuppression and cellular senescence. Cell Cycle. 2010;9(16):3328-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Donehower LA, Harvey M, Slagle BL, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356(6366):215-21 [DOI] [PubMed] [Google Scholar]
- 80. Komarova EA, Krivokrysenko V, Wang K, et al. p53 is a suppressor of inflammatory response in mice. Faseb J. 2005. April 5 [DOI] [PubMed] [Google Scholar]
- 81. Liu G, Park YJ, Tsuruta Y, Lorne E, Abraham E. p53 Attenuates lipopolysaccharide-induced NF-kappaB activation and acute lung injury. J Immunol. 2009;182(8):5063-71 [DOI] [PubMed] [Google Scholar]
- 82. Scatizzi JC, Mavers M, Hutcheson J, et al. The CDK domain of p21 is a suppressor of IL-1beta-mediated inflammation in activated macrophages. Eur J Immunol. 2009;39(3):820-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Trakala M, Arias CF, Garcia MI, et al. Regulation of macrophage activation and septic shock susceptibility via p21(WAF1/CIP1). Eur J Immunol. 2009;39(3):810-9 [DOI] [PubMed] [Google Scholar]
- 84. Gustot T. Multiple organ failure in sepsis: prognosis and role of systemic inflammatory response. Curr Opin Crit Care. 2011;17(2):153-9 [DOI] [PubMed] [Google Scholar]
- 85. Gudkov AV, Komarova EA. Prospective therapeutic applications of p53 inhibitors. Biochem Biophys Res Commun. 2005;331(3):726-36 [DOI] [PubMed] [Google Scholar]
- 86. Coupienne I, Bontems S, Dewaele M, et al. NF-kappaB inhibition improves the sensitivity of human glioblastoma cells to 5-aminolevulinic acid-based photodynamic therapy. Biochem Pharmacol. 2011;81(5):606-16 [DOI] [PubMed] [Google Scholar]
- 87. Dey A, Tergaonkar V, Lane DP. Double-edged swords as cancer therapeutics: simultaneously targeting p53 and NF-kappaB pathways. Nat Rev Drug Discov. 2008;7(12):1031-40 [DOI] [PubMed] [Google Scholar]
- 88. Suzuki J, Ogawa M, Muto S, et al. Novel IkB kinase inhibitors for treatment of nuclear factor-kB-related diseases. Expert Opin Investig Drugs. 2011;20(3):395-405 [DOI] [PubMed] [Google Scholar]
- 89. Palmero EI, Achatz MI, Ashton-Prolla P, Olivier M, Hainaut P. Tumor protein 53 mutations and inherited cancer: beyond Li-Fraumeni syndrome. Curr Opin Oncol. 2010;22(1):64-9 [DOI] [PubMed] [Google Scholar]
- 90. Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2(4):301-10 [DOI] [PubMed] [Google Scholar]
- 91. Komarova EA, Gudkov AV. Could p53 be a target for therapeutic suppression? SeminCancer Biol. 1998;8(5):389-400 [DOI] [PubMed] [Google Scholar]
- 92. Komarova EA, Gudkov AV. Chemoprotection from p53-dependent apoptosis: potential clinical applications of the p53 inhibitors. Biochem Pharmacol. 2001;62(6):657-67 [DOI] [PubMed] [Google Scholar]
- 93. Strom E, Sathe S, Komarov PG, et al. Small-molecule inhibitor of p53 binding to mitochondria protects mice from gamma radiation. Nat Chem Biol. 2006;2(9):474-9 [DOI] [PubMed] [Google Scholar]
- 94. Burdelya LG, Krivokrysenko VI, Tallant TC, et al. An agonist of toll-like receptor 5 has radioprotective activity in mouse and primate models. Science. 2008;320(5873):226-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Leonova KI, Shneyder J, Antoch MP, et al. A small molecule inhibitor of p53 stimulates amplification of hematopoietic stem cells but does not promote tumor development in mice. Cell Cycle. 2010;9(7):1434-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Vassilev LT, Vu BT, Graves B, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303(5659):844-8 [DOI] [PubMed] [Google Scholar]
- 97. Vassilev LT. p53 Activation by small molecules: application in oncology. J Med Chem. 2005;48(14):4491-9 [DOI] [PubMed] [Google Scholar]
- 98. Saddler C, Ouillette P, Kujawski L, et al. Comprehensive biomarker and genomic analysis identifies p53 status as the major determinant of response to MDM2 inhibitors in chronic lymphocytic leukemia. Blood. 2008;111(3):1584-93 [DOI] [PubMed] [Google Scholar]
- 99. Shangary S, Wang S. Targeting the MDM2-p53 interaction for cancer therapy. Clin Cancer Res. 2008;14(17):5318-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Shangary S, Wang S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: a novel approach for cancer therapy. Annu Rev Pharmacol Toxicol. 2009;49:223-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Groskreutz DJ, Monick MM, Yarovinsky TO, et al. Respiratory syncytial virus decreases p53 protein to prolong survival of airway epithelial cells. J Immunol. 2007;179(5):2741-7 [DOI] [PubMed] [Google Scholar]
- 102. Dey A, Wong ET, Bist P, Tergaonkar V, Lane DP. Nutlin-3 inhibits the NFkappaB pathway in a p53-dependent manner: implications in lung cancer therapy. Cell Cycle. 2007;6(17):2178-85 [DOI] [PubMed] [Google Scholar]
- 103. Schiffrin EJ, Morley JE, Donnet-Hughes A, Guigoz Y. The inflammatory status of the elderly: the intestinal contribution. Mutat Res. 2010;690(1-2):50-6 [DOI] [PubMed] [Google Scholar]
- 104. Donehower LA. Using mice to examine p53 functions in cancer, aging, and longevity. Cold Spring Harb Perspect Biol. 2009;1(6):a001081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Coppe JP, Patil CK, Rodier F, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6(12):2853-68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Donehower LA. Does p53 affect organismal aging? J Cell Physiol. 2002;192(1):23-33 [DOI] [PubMed] [Google Scholar]
- 107. Campisi J. Cancer and aging: yin, yang, and p53. Sci Aging Knowledge Environ. 2002;2002(1):pe1. [DOI] [PubMed] [Google Scholar]
- 108. Gottlieb TM, Oren M. p53 in growth control and neoplasia. Biochim Biophys Acta. 1996;1287(2-3):77-102 [DOI] [PubMed] [Google Scholar]
- 109. Levine AJ, Hu W, Feng Z. The P53 pathway: what questions remain to be explored? Cell Death Differ. 2006;13(6):1027-36 [DOI] [PubMed] [Google Scholar]
- 110. Krizhanovsky V, Xue W, Zender L, Yon M, Hernando E, Lowe SW. Implications of cellular senescence in tissue damage response, tumor suppression, and stem cell biology. Cold Spring Harb Symp Quant Biol. 2008;73:513-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Demidenko ZN, Korotchkina LG, Gudkov AV, Blagosklonny MV. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci U S A. 2010;107(21):9660-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Feng Z, Levine AJ. The regulation of energy metabolism and the IGF-1/mTOR pathways by the p53 protein. Trends Cell Biol. 2010;20(7):427-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Korotchkina LG, Leontieva OV, Bukreeva EI, Demidenko ZN, Gudkov AV, Blagosklonny MV. The choice between p53-induced senescence and quiescence is determined in part by the mTOR pathway. Aging (Albany NY). 2010;2(6): 344-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Garcia-Cao I, Garcia-Cao M, Martin-Caballero J, et al. “Super p53” mice exhibit enhanced DNA damage response, are tumor resistant and age normally. Embo J. 2002;21(22):6225-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Matheu A, Pantoja C, Efeyan A, et al. Increased gene dosage of Ink4a/Arf results in cancer resistance and normal aging. Genes Dev. 2004;18(22):2736-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Mendrysa SM, McElwee MK, Michalowski J, O’Leary KA, Young KM, Perry ME. mdm2 Is critical for inhibition of p53 during lymphopoiesis and the response to ionizing irradiation. Mol Cell Biol. 2003;23(2):462-72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Mendrysa SM, O’Leary KA, McElwee MK, et al. Tumor suppression and normal aging in mice with constitutively high p53 activity. Genes Dev. 2006;20(1):16-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Maier B, Gluba W, Bernier B, et al. Modulation of mammalian life span by the short isoform of p53. Genes Dev. 2004;18(3):306-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Hinkal GW, Gatza CE, Parikh N, Donehower LA. Altered senescence, apoptosis, and DNA damage response in a mutant p53 model of accelerated aging. Mech Ageing Dev. 2009;130(4):262-71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Matheu A, Maraver A, Serrano M. The Arf/p53 pathway in cancer and aging. Cancer Res. 2008;68(15):6031-4 [DOI] [PubMed] [Google Scholar]
- 121. Purdie CA, Harrison DJ, Peter A, et al. Tumour incidence, spectrum and ploidy in mice with a large deletion in the p53 gene. Oncogene. 1994;9(2):603-9 [PubMed] [Google Scholar]
- 122. Leontieva OV, Blagosklonny MV. DNA damaging agents and p53 do not cause senescence in quiescent cells, while consecutive re-activation of mTOR is associated with conversion to senescence. Aging (Albany NY). 2010;2(12):924-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Feng Z, Hu W, Teresky AK, Hernando E, Cordon-Cardo C, Levine AJ. Declining p53 function in the aging process: a possible mechanism for the increased tumor incidence in older populations. Proc Natl Acad Sci U S A. 2007;104(42):16633-8 [DOI] [PMC free article] [PubMed] [Google Scholar]