

Abstract
p53 is well recognized as a potent tumor suppressor. In its classic role, p53 responds to genotoxic insults by inducing cell cycle exit or programmed cell death to limit the propagation of cells with corrupted genomes. p53 is also implicated in a variety of other cellular processes in which its involvement is less well understood including self-renewal, differentiation, and reprogramming. These activities represent an emerging area of intense interest for cancer biologists, as they provide potential mechanistic links between p53 loss and the stem cell–like cellular plasticity that has been suggested to contribute to tumor cell heterogeneity and to drive tumor progression. Despite accumulating evidence linking p53 loss to stem-like phenotypes in cancer, it is not yet understood how p53 contributes to acquisition of “stemness” at the molecular level. Whether and how stem-like cells confer survival advantages to propagate the tumor also remain to be resolved. Furthermore, although it seems reasonable that the combination of p53 deficiency and the stem-like state could contribute to the genesis of cancers that are refractory to treatment, direct linkages and mechanistic underpinnings remain under investigation. Here, we discuss recent findings supporting the connection between p53 loss and the emergence of tumor cells bearing functional and molecular similarities to stem cells. We address several potential molecular and cellular mechanisms that may contribute to this link, and we discuss implications of these findings for the way we think about cancer progression.
Keywords: p53, stem cell, cancer, dedifferentiation, plasticity, reprogramming
Introduction
p53 (the product of the human TP53 and mouse TRP53 genes) is best known and most extensively studied as a pivotal signaling node that converts diverse upstream stress signals into downstream responses including cell cycle arrest, senescence, DNA repair, and programmed cell death. These activities limit the contribution of corrupted genomes to tissue and have earned p53 the august designation “guardian of the genome”.1 However, the tumor-suppressive effects of p53 likely extend beyond its ability to limit the perpetuation of cells undergoing genome destabilization, given the myriad cellular processes reported to both elicit and respond to p53 activation. Indeed, among tens of thousands of publications on p53, one finds it implicated in diverse aspects of cellular biology including metabolism, meiosis, cell migration, differentiation, embryo implantation, response to teratogens, autophagy, and reprogramming, among others.2-5 While in some cases these “nonclassic” p53 functions may be integrated with classic DNA damage signaling,6 it is possible that they can also act independently to impact tumor susceptibility, tumor progression, and cellular sensitivity to therapy.7 Importantly, elucidation of these relatively understudied p53 functions and their molecular mechanisms has the potential to uncover additional targets for pharmacological intervention in cancer.
An emerging role for p53 in regulating cellular differentiation, self-renewal, and plasticity has generated intense interest, particularly among cancer researchers. Enforced differentiation is a powerful tumor-suppressive mechanism because normal development and differentiation are antithetical to the abnormal development and incomplete differentiation that hallmark cancer. p53 has been implicated as an enforcer of differentiation by virtue of its ability to limit the cardinal stem cell characteristic of self-renewal in several systems.8-11 This, together with the demonstration by Yamanaka et al. that differentiated cells can be reprogrammed to a dedifferentiated state,12 and the demonstration that p53 is a potent reprogramming barrier13-19 have caused a resurgence of interest in the idea that loss of differentiation20 may be linked to p53 pathway disruption in tumors. Recent studies provide additional evidence for the link between p53 and the emergence of dedifferentiated, stem-like phenotypes.9,21
The implications of these findings are far reaching and will cause us to rethink the role that p53 inactivation plays in tumor pathophysiology and the relationship between stem cells and cancer more generally. They call into question existing models for the genesis of cells that perpetuate tumors because plasticity would permit phenotype to change during cancer progression. They also challenge the idea that a static, uniform cancer stem cell pool may be selectively targeted to eradicate the disease. Clearly, optimal therapies will be those that eliminate all malignant cancer cells including both the putative cancer stem cell compartment and cells able to reacquire stem-like characteristics. A better understanding is now needed of the molecular and cellular mechanisms underlying the developmental plasticity and stem-like characteristics that surface when p53 function is corrupted or lost. These mechanisms could well represent targets for the mitigation of critical tumor cell behaviors including chemoresistence, dormancy, and metastasis.
Here, we critically examine the literature to uncover relationships between p53 and the normal or neoplastic stem cell state. Our objectives are to raise questions about the commonality of stem-like cells in cancer and to re-examine the “cancer stem cell” concept in light of the possibility that the large number of cancers in which p53 is inactivated may be susceptible to tumor cell reprogramming “on the fly” to generate multifarious stem-like cells.
p53: Finely Tuned Tumor Suppressor
p53: Tumor Suppressor
First identified as a target for the transforming viral oncogene, large T,22,23 p53 was subsequently found to be mutated in a wide variety of human and murine cancers.24-28 Germline transmission of one mutant allele in humans (e.g., Li-Fraumeni syndrome29) or in engineered mice leads to broad-spectrum tumor formation with high penetrance.30,31 p53-null mice exhibit even more rapid onset of primitive lymphoma and other tumors including various sarcomas and stem-like germ cell teratomas.30-32 Tumors in other tissues including epithelial cancers can be elicited if principal lesions that would otherwise occur are bypassed using conditional alleles, transplant settings, alternative strains, or in the presence of cooperating lesions.33-36 p53 is also found to be mutated in over 50% of sporadic human cancers,37 and it is thought to be functionally inactivated through disruption of other pathway members in a significant proportion of the remainder (Fig. 1).38 The broad spectrum of cancers harboring mutated or inactivated p53 or p53 pathway components indicates that it is a general, rather than tissue-specific, tumor suppressor.
Figure 1.
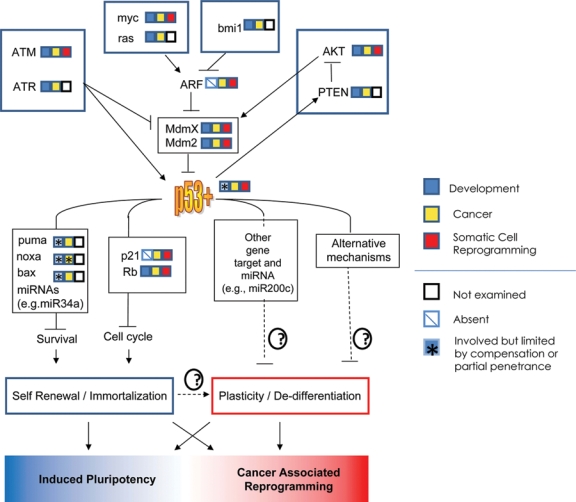
The p53 pathway in development, cancer, and reprogramming. A select set of p53 upstream regulators and downstream effectors are shown in the context of p53-dependent plasticity and self-renewal. The involvement of pathway members in development, cancer, or reprogramming is indicated by their adjacent color-coded squares. Whether induced plasticity leads to induced pluripotent stem cells or stem-like cancer cells may depend on the extent or reversibility of oncogene activation, p53 inactivation, or microenvironmental factor.
In somatic cells, an intact p53 pathway responds to a variety of stimuli ranging from nucleotide depletion6 to oncogene overexpression39 to DNA clastogenic treatments.40,41 p53 activation is accomplished by its rapid nuclear accumulation and subsequent transcriptional activation of numerous genes involved in cell cycle arrest (e.g., p21) apoptosis (e.g., Puma) and other pathways. Additionally, over the past few years, p53 has been shown to directly activate an increasing number of miRNAs able to broadly modulate the function of diverse cellular processes (Fig. 1).42-44 The responses that p53 activation generates are highly stress and tissue specific. For example, in thymocytes, gamma irradiation elicits a robust p53-dependent apoptotic response,41 while the same stimulus in mouse embryo and human fibroblasts induces prolonged cell cycle arrests resembling senescence.40,45 Either response can provide a potent cell-autonomous, tumor-suppressive mechanism. Recently, p53 has been shown to exert profound cell-nonautonomous effects on the stroma and immune system that can be either growth stimulatory or antagonistic.46,47 The cellular responses of apoptosis and cell cycle arrest or senescence have been the principal focus of most p53 tumor suppressor studies to date and provide the most common in vitro readout for chemotherapeutic screens and tumor therapy studies. It has become clear that the nature of the p53 response depends on many factors including the cell type, nature, duration, and intensity of the stress and the status of other prosurvival and cell cycle modulators,7,48,49 and the effects of paracrine factors secreted by the stroma.47,50
p53 Regulation in Brief
p53 function is orchestrated in normal cells by a number of upstream signaling mediators responding to damage, metabolic, proliferative, or other stresses. Most of these pathway components have well-established roles as oncogenes or tumor suppressors (Fig. 1). Stress signals converge principally on 2 related, interacting proteins, Mdm2 and Mdmx, that appear to function cooperatively to promote p53 degradation and thereby inhibit p53 activity.48,49 The levels of p53, Mdm2, and Mdmx are tightly regulated in a coordinated network to provide a sensitive rheostat for stress sensing and response.51 Disruption of this network leads to profound developmental defects, enhanced tumor suppression, or significant protection against oncogenesis. For example, Mdm2 (or Mdmx) knockout in mice leads to early embryonic lethality that can be rescued by p53 ablation.52-54 Conditional knockout of either Mdm2 or Mdmx leads to cell death or reduced proliferation in a variety of different adult tissues and tissue stem/progenitor cells.49,55 Conversely, overexpression of Mdm2 or Mdmx is observed in a variety of cancers in which they overwhelm the stress sensing system by constitutively inactivating p53.56
The polycomb complex component Bmi1 is another p53 pathway regulator with profound consequences in both development and cancer. Bmi1 regulates chromatin to promote stem cell self-renewal.57,58 Its deletion in mice results in hematopoietic and neuronal stem cell loss59 and affects proliferation and differentiation of the mammary epithelium.60 In lymphoma, Bmi1 overexpression can result in silencing of the Ink4a/ARF locus61-63 that encodes p14/ARF (p19/ARF in mice). ARF promotes p53 activation by antagonizing Mdm2-mediated p53 degradation in response to oncogene overexpression.64 Thus, Bmi1 overexpression, or p14/ARF loss of function by epigenetic and other mechanisms, attenuates p53 activity by increasing Mdm2’s ability to antagonize p53.65,66 These mechanisms of p53 inhibition are also observed in bladder, skin, prostate, breast, ovarian, and colorectal cancers and in mantle cell lymphoma.67 Taken in the aggregate, genes that either regulate p53, or that are regulated by p53, comprise networks intimately tied to cellular physiology (e.g., DNA damage sensing and repair, metabolism, epigenetic regulation, etc.) (Fig. 1). In this light, it is not surprising that components of the p53 regulatory pathway can contribute to both tumorigenesis and developmental dysfunction when disrupted (see below).
Emerging Roles for p53 in Stem Cells and Differentiation
p53 in Adult Stem Cells
Recent studies suggest roles for p53 in adult stem cells, but such studies are complicated by several factors. Adult tissue stem cells are very rare, and it is very difficult to identify them unambiguously and to then isolate and purify them.68,69 Indeed, almost all stem cell isolates actually comprise cell mixtures. (For this reason, most stem cell preparations should be referred to as “stem/progenitor populations,” but for simplicity, we will refer to them as “stem cells” below.) Adult stem cells have also proven difficult to maintain in culture without limiting their capacity for self-renewal or inducing differentiation. However, recent improvements offer hope in this area.70,71 In spite of these challenges, studies on adult stem cells, and implications for the functional relevance of p53, continue to appear with increasing frequency.
Small changes in p53 activity profoundly affect hematopoietic stem cell (HSC) abundance and function. HSCs can be quantified by limiting dilution transplantation analyses and by flow cytometry using a variety of surface markers that enable their isolation to a purity of up to 50%.72-74 Impressively, transplantation of as few as one purified HSC rescues the lethality caused by myeloablation.75 Competitive transplantation experiments provide a powerful means of determining the relative abilities of HSCs encoding particular mutations to compete with wild-type HSCs for engraftment of lethally irradiated hosts.72,73 Subsequent to engraftment, the size of the stem cell–containing population defined by flow cytometry can be measured to indicate stem cell expansion/self-renewal. Using such methods, HSCs in genetically engineered models reveal that dysfunctions in p53, ATM, Rb, and p21, among others, significantly affect HSC/progenitor abundance and function at baseline and in response to DNA damage or transplantation-associated stresses.76-80 For example, even the subtle change in baseline and inducible p53 activity caused by deletion of a single Mdm2 allele dramatically reduces the ability of the mutant to compete with wild-type cells during transplantation.81,82 Interestingly, inactivation of the Ink4a/ARF locus along with p53 enables conversion of multipotent progenitors into engrafting stem cells.83 This suggests that p53 function may be required to stabilize the differentiated phenotype of such cells or that its loss enables reprogramming at high efficiency in this cell type.
p53 has also been implicated in regulating mammary stem cell number in vitro and in vivo. Here, stem cells comprise a far lower fraction of cell populations obtained by flow cytometry using cell surface markers, although use of the membrane dye, PKH26, to enrich for quiescent or slowly dividing putative mammary stem cells has been suggested to yield impressively pure populations.84 Mammary stem cells are typically quantified in vitro through generation of mammospheres from single initiating cells and in vivo by limiting dilution transplantation assays measuring the capacity of the cells to fill a de-epithelialized fat pad with a functional mammary gland.84,85 In comparison to wild-type littermates, p53 knockout mice appear to contain a higher concentration of cells able to form mammospheres in culture or to repopulate the gland in vivo.84,85 Furthermore, whereas wild-type sphere-forming cells decline upon serial replating of dissociated mammospheres, the p53-null sphere-forming cells can be expanded.84 Conversely, stem cell depletion accompanies the serial transplantation of mammary cells from mice carrying an overactive p53 gene,86 suggesting that the level of p53 activity dictates the percentage of graftable stem cells in the tissue. However, such interpretations must be moderated because in vitro propagation of mouse cells can activate p53, so the positive growth effects seen in cells lacking p53 may result from the absence of p53-mediated growth arrest responses. Similarly, transplantation may induce stresses that activate p53, as observed in the hematopoietic system. Finally, the identity of the cell(s) that initiate mammosphere formation remains obscure, as does their relationship to the stem cells that underlie gland formation in a transplantation assay.
p53 has also been implicated in regulating stem cell/progenitor compartment dynamics in the epidermis,87 prostate,88 and central nervous system.11,89-91 While these studies generally point to p53 loss in increasing the stem cell pool and p53 activation in blocking stem cell self-renewal, this remains to be demonstrated in a physiological setting using methods able to unambiguously distinguish stem cells from other cells.
Thus, while the existing data make tantalizing links between p53 and stem cell abundance and function, the important role of p53 in the response to diverse stresses complicates unambiguous conclusions. The existence of a substantial number of other cell types in the stem-enriched populations also precludes attribution of differences in inferred stem cell number to the stem cells themselves. In order to make such conclusions, uncovering methods and markers to identify, localize, and quantify stem cells more specifically remains an important challenge to the field.
p53 in Embryonic Stem Cells
Stem cell genomes must be rigorously “guarded” from embryogenesis through adulthood because such cells expand periodically to enable tissue repair and replacement. Thus, faithful genome duplication over a lifetime is required to minimize accumulation of oncogenic lesions during such expansions. Inadequate genomic stability control would be especially deleterious in embryonic stem cells (ESCs), as they are the progenitors of all adult organ systems. However, there are conflicting reports regarding p53 activity in ESCs. We and others found that wild-type mouse embryonic stem cells (mESCs) proliferate rapidly, with cell cycles mainly consisting of S and M phases without the obvious gap phases (G1 and G2) in which p53 normally acts in differentiated cells.92,93 Although mESCs express high levels of p53 protein,93,94 it appears to be preferentially cytoplasmic.92,95,96 Surprisingly, p53 activation by certain types of DNA damage, subsequent nuclear accumulation of p53 protein, and transactivation of specific target genes appear to be inefficient in mESCs.93,95,97,98 On the other hand, mESCs possess robust repair mechanisms that may compensate for p53 to preserve genomic stability and potentially dampen damage responses that would cause cell cycle slowing.97 While high levels of damage induce ESC death or differentiation,10,93,94,99-102 these responses do not appear to be strictly p53 dependent.93,103 In human ESCs (hESCs), p53 has been implicated in damage-induced cell death, but its role inducing apoptosis was again independent of its classic nuclear accumulation and transactivation activity, involving instead a noncanonical function at the mitochondria.104
Mechanisms that inhibit the high levels of mESC p53 from being activated may be required to permit rapid ESC cycling in the presence of strong proliferation signals. For instance, downregulation of cdk2, which is highly expressed in stem cells, lengthened G1 and allowed p53-dependent activation of p21.105-107 Thus, a brief G1 may make it difficult to activate p53, but mechanisms that slow the cycle by lengthening G1 may enable p53 activation. Conversely, the prolonged G1 of most differentiated cells might limit reversion to rapid ESC cycling kinetics by affording an ample opportunity for p53 to act.
Two independent molecular mechanisms may counter the high level of p53 protein in ESCs. p53 deacetylation by SIRT1 prevents its nuclear accumulation at high reactive oxygen species (ROS) levels.96 Thus, increasing ROS level in mESC cultures growing at ambient (~21%) oxygen by removal of reducing reagents caused mESCs lacking SIRT1 to differentiate.96 As ROS levels are affected by metabolism, proliferation, tissue culture stress, oxygen tension, oncogene overexpression,108-112 and many forms of DNA damage,113 effective mechanisms for modulating cellular responses to ROS are critical. The second p53 inhibitory mechanism involves the truncated p53 isoform Δ40-p53 (also referred to as ΔN-p53 or p47 in humans and p44 in the mouse) produced from an internal translational start site in the p53 gene. Δ40-p53 is profoundly defective for p53 transactivation function.114 It is highly expressed in ESCs, and mutant ESCs generated by homologous recombination to lack the internal start site, but that still produce full-length p53, exhibit loss of self-renewal, increased differentiation, and establishment of an obvious G0/G1 population.115 These data are consistent with Δ40-p53 antagonizing wild-type p53 function in ESCs. Interestingly, Δ40-p53 expression was recently identified as a common tumor-associated lesion in a chemically induced bladder cancer model.116 The identification of other p53 isoforms, including several splice variants, adds new opportunities to fine tune p53 activity in specific cell types.117 Whether these other isoforms contribute to development, stem cell function, and genomic stability control remains to be investigated.
While the above functional and mechanistic studies provide evidence of p53 attenuation in ESCs, other studies indicate that p53 is active in mouse and human ESC cultures.10,94,100,101,118 These analyses used reporter constructs to detect active p53 and functional studies to examine p53-mediated responses.94,119 p53 activation was observed in ESC populations following treatment with actinomycin D120 or the Mdm2:p53 interaction disruptor Nutlin 3a.121 Importantly, the effect was reversible by p53 knockdown using shRNA.119 Notably, the reporter analyses revealed high basal expression that was augmented in differentiating cultures in the absence of additional stress.119 As high baseline activity attributed to basal p53 activity in undifferentiated cultures was not appreciably reduced by p53 knockdown, it is likely that p53-independent factors contribute. Earlier functional studies reporting activation of endogenous Mdm2, cyclin G, and Bax in response to DNA damage or induced differentiation with retinoic acid94,100 revealed a correlation between p53 activation and induction of programmed cell death that was reduced or absent in p53 knockout mESC cultures.94,100 Interestingly, time course experiments analyzing p53 localization and target gene activation showed transient p53 activation in a subset of cells.95 The controversy over p53 pathway activation in ESCs extends to hESCs that, although different in important ways from mESCs,122 also have an accelerated cell cycle with abrogated G1 checkpoints.123 Different studies report that hESCs possess or lack a stress-activated p21 response,124,125 undergo p53-dependent apoptosis,126 or execute p53/p21-independent checkpoints in response to genotoxic stresses such as ultraviolet radiation.127 However, persistent p53 activation using continuous Nutlin exposure induces differentiation of mouse and human ESCs.101,128 One interpretation of such results is that ESCs have an altered stress response threshold for p53 activation, and Nutlin is able to consistently meet the activation criteria for these cells.
We offer the following suggestions to resolve the apparent discrepancies in the literature concerning p53 functionality in ESCs. Given the role played by p53 in stress sensing, differences in culture conditions, growth factors,129 and oxygen tension or the level, duration, and type of DNA-damaging insults are likely contributors to differential p53 activation. However, another variable is suggested by the recent identification of 2 distinct ESC/iPS cell states distinguishable based on their miRNA expression profile and by their expression of p53 pathway genes.130 These 2 pluripotent states are interconvertible by expression of appropriate miRNAs or over the course of in vitro differentiation.130 Another potentially related possibility is the existence of distinct pluripotent populations derived from cells with temporally distinct developmental origins.131,132 In this regard, hESCs that are more similar to mouse epiblast-derived stem cells (mEpiSCs) can be reprogrammed to a metastable mESC-like state.133 Given such data, it is reasonable to consider ESC cultures being inherently heterogeneous, with one cell type lacking canonical p53 stress responses but being interconvertible with a second type able to undergo p53 activation (Fig. 2). The latter could lead to differentiation or apoptosis. Thus, the different results reported in the literature might relate to the relative contributions of each cell type to the culture.
Figure 2.
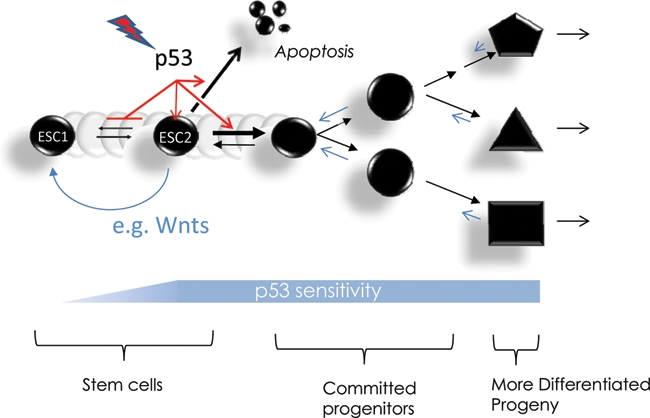
Impact of stem cell state on p53 activation and differentiation. In this model, stem cell populations can be heterogeneous, with some being more responsive to p53 activation. Various stresses can activate p53 to induce differentiation or apoptosis in the sensitive stem, while insensitive cells with dampened p53 responses survive and may proliferate in response to paracrine signals such as Wnt. This model is compatible with recent reports of multiple pluripotent stem cell states in embryonic stem cell cultures130,227 and that multipotent progenitors in the hematopoietic compartment act as stem cells if p53 activation is abrogated in conjunction with Ink4a/ARF deletion.83
Impact of p53 on Development and Differentiation
As p53 responds to many conditions that impact cell growth, and it appears to contribute to stem cell self-renewal and differentiation capacity, one might expect p53 pathway function to also impact development. However, p53-null mice appear superficially normal. This may be due in part to the presence of 2 related proteins, p63 and p73, evolutionary antecedents of p53134 that play key roles in epithelial stem cell self-renewal and neural cell differentiation, respectively.135-137 As these p53 family members can regulate some of the same genes that p53 regulates as they have related DNA-binding domains, it is possible that they can at least partially complement developmental aberrations engendered by p53 deficiency.
p63 also plays a key role in maintaining the integrity of the female germline.138 Note, however, that p53 also contributes to maintaining genomic fidelity during embryogenesis, as indicated by the profound increase in aberrant embryos in p53-null mice exposed to teratogens.5,139-141 In light of p53’s likely descent from a more ancient relative, its response to DNA damage may be viewed as an extension of the role of the ancestral proteins in ensuring germline genomic integrity.142,143 The roles of somatic genome guardian and mediator of responses to diverse stresses that impinge on the soma and that could impact genome stability have mainly been relegated to p53 in mammals, although p73 can serve as a backup when p53 is compromised.144
A closer evaluation of the literature does reveal that p53 plays modulatory, but significant, roles in development. It is commonly observed that intercrosses of p53 heterozygotes generate a lower than Mendelian yield of p53 offspring, and at least 25% of all p53-null mice die from nontumor-related maladies.31,139,141,145 Furthermore, fertility of p53-null mice is compromised by defects in spermatogenesis and embryo implantation,146-148 and their numbers are further reduced by developmental defects in utero 141 such as elevated incidence of exencephaly in females.147,149 Broad effects of p53 deficiency on development might be expected based on its activation in multiple tissues during normal fetal development.150-152 This may explain the eye and tooth defects and polydactyly occasionally observed in p53-deficient mice.149 p53 has also been implicated in regulating normal differentiation of muscle, keratinocytes, neurons, thyroid, and macrophages.153,154 It thus remains possible that closer examination of these and other tissues may uncover additional differentiation abnormalities associated with p53 deficiency. It is also possible that manifestations of p53 loss may only be evident in combination with alterations in other genes and then only in specific tissues. For instance, through mechanisms associated with the retinoblastoma gene (Rb), p53 controls neuron cell cycle exit,155,156 promotes myogenic differentiation in vivo,157 and may inhibit differentiation of adipocytes.158,159
Subtle, nonlethal p53-dependent developmental defects may also impact long-term health. For instance, as cancer is a developmental aberration that manifests over decades in humans, the understated defects caused by p53 malfunction may have profound effects when integrated over time and many cell divisions. Reduced fidelity in lineage commitment may be accompanied by the favorable epigenetic landscapes and expanded stem and/or progenitor cell pools for subsequent transformation. Tumor progression could be further accelerated by loss of p53-mediated genomic stability control. Consistent with this, increased stem/progenitor pools in mammary, neural, and hematopoietic tissues have been reported in p53-null mice.84,85,90,160
Mechanisms by which p53 Modulates Differentiation
The mechanisms by which p53 affects cellular differentiation remain to be enumerated. While numerous possibilities have been suggested,10,161-163 we will focus on 3 to exemplify the range of possibilities.
The first mechanism involves regulation of self-renewal through the Wnt pathway. p53 has been reported to impact on expression of Wnt genes.101,164,165 As noted above, ESCs are heterogeneous, and some of the cells appear more prone to differentiation than others. DNA damage can activate p53 to induce differentiation of a subset of cells within ESC cultures.93,95,101 ESC heterogeneity in response to damage might, therefore, partially derive from the induction of secreted Wnt proteins101 that act in a paracrine fashion to support the self-renewal of nearby undamaged stem cells, enabling them to remain stem like. p53 also induces the E3 ligase SIAH, which reduces β-catenin levels, leading to reduced Wnt pathway signaling164,165 within a cell, which should either lead to a lower probability of cell cycle entry or increased probability of differentiation. Together, these p53-mediated effects on the Wnt pathway enable it to regulate both cell-autonomous and cell-nonautonomous effects. This may provide an elegant mechanism for p53 activation to enable less damaged stem cells to undergo self-renewing divisions to re-establish homeostasis within the stem cell pool under potentially genotoxic conditions.
A second mechanism involves the regulation of symmetric versus asymmetric division. According to this mechanism of differentiation control, symmetric stem cell division generates 2 identical daughter stem cells and thus enables expansion of the stem cell pool. By contrast, asymmetric division produces one stem cell and one proliferative progenitor. Expansion of the latter enables development to proceed but reduces net stem cell concentration within the organ. Recently, p53 loss in the mammary gland was shown to increase the probability of symmetric divisions occurring in an in vitro cell culture system using enriched adult mammary stem cells.84 Asymmetric division was quantified by measuring the distribution of Numb,84,166 a protein linked to asymmetric stem cell division in other systems.167,168 Interestingly, Numb directly interacts with Mdm2 and p53 and has been suggested to activate p53 by preventing Mdm2-mediated ubiquitination.169 While Numb provides a potential direct mechanistic link between p53 and propensity to differentiate via increasing asymmetric cell divisions, the activation and colocalization of p53 with Numb were not analyzed.
A third mechanism involves p53’s ability to limit stem cell self-renewal by inhibiting its ability to cycle rapidly through activation of CDK inhibitors such as p21.170,171 Rapid cell cycles are characteristic of ESCs and are required as an early step in the reprogramming of differentiated cells to iPS cells (see below). Inactivating p53 appears to enable oncogenic lesions to induce such rapid cycles. Conversely, activating p53 induces the lengthened cell cycles that not only typify differentiated cells but may also allow the molecular machinery that implements differentiation programs to function. Overexpression of cyclin/cdk antagonists such as p15, p16, and p21 imposes longer cell cycles and limits reprogramming. Conversely, inactivating Rb, a critical cell cycle regulator downstream of p53, by deleting it, by reducing p21 expression (which enables Rb inactivation through increased activity of G1 cyclin–CDKs), or by overexpressing cyclin D–cdk4 all enable more efficient reprogramming and conversion to S/M cell cycles. As reprogramming represents acquisition of a less differentiated state from differentiated cells, the results suggest that factors such as p53 may impact on the balance between differentiation and stemness by regulating G1 progression.
Beyond Genome Guardian: p53 Deficiency and Stem-Like States
p53 Loss Is Associated with Primitive Tumors, While Activation Induces Differentiation
Although p53 mutation and pathway inactivation are found in the majority of tumors, they appear to be especially concentrated among tumors showing plasticity and loss of differentiation characteristics. p53 loss was almost exclusively associated with poorly differentiated thyroid cancers.172 In breast cancer, p53 mutations are most frequently found within the poorly differentiated basal-like, metaplastic, and medullary types.21,173-175 Collateral mutations of p53 and Pten are the most common tumor suppressor aberrations in glioblastomas, which are poorly differentiated, developmentally plastic brain tumors derived from the neuronal stem/progenitor cells. Here, p53 was observed to limit neurosphere self-renewal.176 A similar association between p53 loss and loss of differentiation characteristics has been observed in lung cancer,177 and recent work in a murine lung cancer model indicates that p53 reactivation suppresses malignant adenocarcinoma progression without affecting less aggressive adenomas.178 Although the mechanism of this suppression remains uncertain, one interpretation is that p53 loss permits tumor plasticity and malignancy in a reversible manner. Likewise, in the classic model of tumor progression from neoplasia to colorectal carcinoma, p53 mutation demarcates tumors progressing from adenomas to less differentiated and more aggressive carcinomas.27 Recently, p53 loss was reported to specifically endow progenitors in AML with self-renewal capacity and correlated with a block to progenitor differentiation.9 Interestingly, undifferentiated stem cell tumors, such as embryonal carcinomas (ECs), infrequently contain mutated p53 (e.g., NCCIT, S2179) but usually express wild-type p53 (such as the F9 line analyzed by Levine et al.180) that is presumably functionally inactivated by one of the many mechanisms summarized in previous sections. The association between p53 loss and the compromised enforcement of differentiation characteristics has also been observed in other settings including mouse models of chemically induced skin carcinogenesis181 and in an Rb model of mammary cancer.182
p53 activation promotes differentiation in various cancers. Evidence from the hematopoietic system suggests that p53 enforces differentiation and prevents ectopic self-renewal at various stages of lineage commitment.183-185 AML blasts could be induced to mature by p53 activation mediated by Nutlin.186 This may also carry over to solid malignancies, as p53 promotes the differentiation of pancreatic carcinoma187 and osteosarcoma cells188 in vivo.
p53 Loss and Cellular Reprogramming
Selection for p53 functional inactivation during cancer progression has typically been attributed to survival benefits accruing due to reduced apoptosis, cell cycle arrest, and increased opportunities for cellular evolution afforded by genomic instability. However, in light of the above discussion, it is also possible that p53 loss might destabilize the differentiated state and enable reversion to a more stem-like state. Recent in vitro model systems, and analyses of tumors in vivo, support this additional role for p53 in the control of developmental plasticity.
Noting the relatively undifferentiated phenotype of carcinomas and some papillomas from p53-null and heterozygous mice relative to corresponding tumors from wild-type mice, Kemp et al. speculated nearly 2 decades ago that p53 loss might drive loss of differentiation and increase malignancy.181 Similarly, over a decade ago, Bond et al. suggested that the apparent “dedifferentiation” accompanying malignant progression in thyroid cancer “. . . may play a causal rather than a passive role in this critical switch in tumour behaviour.”189 Prescient as these interpretations may have been, it seems they were largely forgotten by the mainstream of cancer research. Rather, an alternative model prevailed in which tumors adhere to essentially irreversible hierarchies of cellular differentiation parallel to those proposed for normal tissues. Early experiments reconstituting complex differentiated tumors following transplantation of stem cells from teratomas appeared to confirm this model.190 However, teratomas may be a very peculiar case, as they often encode wild-type p53 that may help enforce differentiation of most daughter cells, and they can be formed from untransformed ESCs.191 Nevertheless, and despite examples from amphibians and other organisms,192 there has been little compelling experimental evidence to directly support the possibility of cellular dedifferentiation in mammalian cells until recently.
This all changed in 2006 when Takahashi and Yamanaka demonstrated that the enforced expression of 4 transcription factors, (Oct4, Sox2, Klf4, and c-Myc) could induce mouse fibroblasts to adopt pluripotent cell fates resembling stem cells.193 These and subsequent studies demonstrated that all cell types can generate induced pluripotent stem (iPS) cells with the appropriate reprogramming gene set. This provided the missing evidence that most, if not all, somatic mammalian cells possess dedifferentiation potential.194 However, the frequency of reprogramming appeared to be extremely low, raising the possibility of cellular barriers against induced reprogramming.
Several genes in the original Yamanaka iPS cocktail, such as c-Myc, generate oncogenic stresses that activate the p53 pathway to induce cell cycle arrest or death.195 Consequently, Myc expression, along with general tissue culture stresses, would be expected to activate p53 during iPS cell generation to reduce reprogramming frequency or rate. Several groups tested the role of the p53 pathway in iPS cell formation and found that inhibiting the p53 pathway dramatically increases the apparent efficiency of iPS cell generation (Fig. 1).14-19 Reducing expression of genes contributing to cell cycle arrest or apoptosis also increased reprogramming. Importantly, a mutation in Mdmx that reduces p53 activity only 2-fold at baseline also dramatically increased reprogramming efficiency.14 These results have several important implications. First, subtle changes in p53 activity are all that is needed to increase the probability of reprogramming. Thus, even subtle elevations in oncogene signaling that are insufficient to activate p53, but sufficient to enable cell cycle progression, might increase reprogramming efficiency. Second, reprogramming is limited by a variety of p53-induced protective pathways, including but not limited to those involved in cell cycle arrest, senescence, and apoptosis. Third, in the absence of p53, fewer factors were needed for reprogramming. Finally, through its ability to inhibit cell cycle progression, p53 provides a potent barrier to the acquisition of the epigenetic changes that underlie the dedifferentiation involved in iPS cell formation.
Understanding the mechanisms by which p53 limits reprogramming is complicated by the various methods used for introduction of the reprogramming factors as well as by the expression levels of these factors. However, generation of mice encoding an inducible set of the reprogramming factors provides a powerful, controllable system for analyzing reprogramming kinetics. Such an analysis indicated that p53 inhibition enhances iPS cell generation probabilistically through cell cycle acceleration,196 although the data left open the possible involvement of cell cycle–independent contributions. Another analysis employing single-cell time-lapse photography of iPS cell formation revealed that a very early step in reprogramming involved oncogene-induced establishment of the very rapid cell cycles that typify ESCs. Here, the effects of p53 loss were cell cycle related, immediate, and restricted to the early phase of the reprogramming process.197 The authors suggested that early cell cycle changes in a subset of fibroblasts are followed by a multistep sequential process that resets the epigenetic architecture of the cell to resemble that of a stem cell, although not perfectly.198
p53 Loss, Cellular Dedifferentiation, and Tumorigenesis
The success of induced pluripotency protocols reveals an inherent reversibility of the steps of cellular differentiation. Given the prominent role of p53 loss in this induced pluripotency, the frequent occurrence of p53 mutations in cancer, and the common occurrence of oncogenic lesions that activate c-Myc or that might phenocopy the effects of other reprogramming factors, it is reasonable to ask whether functional loss of p53 during cancer progression correlates with acquisition of a stem-like state. Recently, several groups have assessed the relationship between cancer and stem cells using comparative gene expression profiling. These comparative studies were facilitated by the archiving of a large number of experimental and disease-associated microarray data sets into publicly accessible databases. Collecting differentially expressed gene lists from a broad set of published studies, Assou et al. derived an ESC expression signature comprised of several hundred genes that are consistently upregulated in ESC culture.199 These signatures included the common reprogramming factors Lin28, Oct3/4, Sox2, and Nanog and many other genes reproducibly identified in ESCs. Subsequently, enrichment analyses using this ESC signature along with other signatures indicative of the ESC state found them to correspond to high-grade tumors in diverse tissues.200 When we applied these signatures to breast and lung tumor data sets in which p53 mutational status was determined by DNA sequencing, we identified a robust correlation between the ESC signatures and p53 mutation.21 The correlation could be extended to functional inactivation of p53 inferred from the broader transcriptional profile or deregulation of upstream p53 pathway genes and to rare tumor subtypes known to bear frequent p53 mutation. An iPS cell signature based on archived iPS cell microarray data using the method of Assou et al. was similarly correlated to p53 status, suggesting significant similarities between p53-mutated cancers and cells that have undergone intentional reprogramming in vitro.
Concurrent work analyzing bladder carcinoma suggested that an ESC-like tumor signature can be driven to a considerable extent by an oncogene such as c-Myc, but these effects appear to be tissue and context dependent, as the signature they employed did not significantly contribute to the stem-like profiles we observed in breast cancers.201 On the other hand, forced c-Myc expression in mammary epithelial cells in vitro has been reported to confer a stem-like phenotype.202 Additionally, given the ability of c-Myc to activate p53, it seems likely that p53 must have been functionally inactivated to enable the strong proliferative signature these authors reported to correlate with tumorigenicity. Interestingly, recent evidence shows that chemically induced tumors in the bladder do harbor the aforementioned dominant-negative embryonic ΔN-p53 isoform.116
The above studies did not identify the classic pluripotency genes Oct3/4, Sox2, and Nanog as significant or consistent contributors to stem-like tumor profiles,21,200,201 suggesting that cancer-associated stem cell states are not identical to ESC states. In this regard, it may be telling that the identification of stem-like states using expression data has thus far required the use of multigene signatures collected from diverse stem cell or iPS cell studies.21,200-203 These signatures are comprised of genes that are frequently, but by no means universally, identified as differentially expressed in stem cells relative to differentiated cells. In fact, Assou et al. found that among 20 studies, only one gene, Oct3/4, was identified in all studies.199 This lack of consistency in the stem cell expression profile may reflect natural variability in the cell types present in the stem cell cultures or the use of different culture conditions. Additionally, other technical and statistical differences likely contribute. Nevertheless, the genes comprising these signatures are inherently crossvalidated in that they are found frequently among independent stem cell studies. Furthermore, the statistical tools to evaluate gene expression similarities do not require that all genes of the signature be equally present in the tumor but rather only that a significant subset is present. Importantly, this subset could vary from one stem-like tumor to another and could indicate diverse and potentially flexible mechanisms for achieving core stem cell functional properties under divergent in vivo pressures.
Tumor Heterogeneity and Cellular Plasticity
A Mixed Stochastic/Cancer Stem Cell Hypothesis
The mechanisms by which cancers initiate and expand into masses exhibiting cellular and genetic heterogeneity at diagnosis have long been debated. Two predominant models have emerged to explain phenotypic and functional heterogeneity in tumors (see recent review215). The first, called the “stochastic” or “clonal evolution” model, is based on classic theories of selection of mutants most fit to survive in particular environments. In this model, stochastic mutations in an appropriate cell type will be selected if they afford that cell a survival or proliferative advantage. This cell then grows, and subsequent mutations in its descendants ultimately yield a tumor with numerous mutations, only some of which were selected for the advantages they provide, while the others are “passengers.” Recent deep sequencing analyses reveal that different types of tumors differ significantly with regard to their mutation load,204,205 and they further reveal the existence of multiple clones within each tumor mass.206 In this model, tumor heterogeneity derives from the constellation of mutations introduced and the phenotypes they engender.
A second model is based on the principles of stem cell biology. In this model, cancers represent an aberration of the molecular and cellular mechanisms that govern development of the corresponding tissue. In normal organs, the final structure derives from a developmental hierarchy in which the undifferentiated stem cell resides at the top, followed by proliferative but relatively undifferentiated descendants, and then (generally) nondividing, terminally differentiated functionally committed cells (Fig. 3). Cellular heterogeneity in tumors is envisioned to result from a “cancer stem cell” that undergoes aberrant differentiation to form the disorganized mass recognized pathologically. Consistent with such a model, some tumors have mixtures of very immature-appearing cells mixed with more differentiated cells,20,190 and specific cells within the tumor can regenerate tumors with the cellular heterogeneity of the original mass in xenografts.207 From this, many researchers have inferred a parent-progeny relationship between tumor-derived cells that initiate xenografts and those that do not208,209 and have defined “cancer stem cell markers” capable of isolating the xenograft-initiating population.207,209
Figure 3.
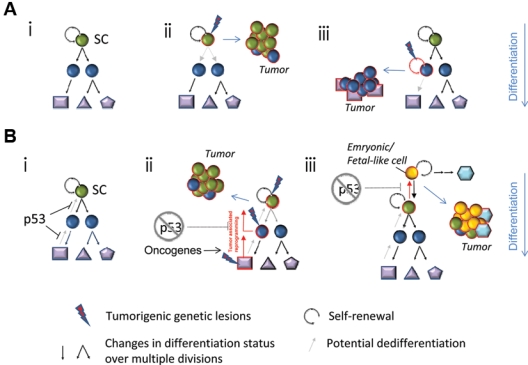
Models of stem cell state acquisition in cancer. (A) A model of the classic differentiation hierarchy initiated by a self-renewing stem cell (A-i). Tumorigenic transformation occurs within a self-renewing stem cell (A-ii). Mutations engender self-renewal competence in a progenitor cell (A-iii). (B) In normal tissues, p53 function limits the possibility of reprogramming to a stem-like state (B-i). Cancer-associated reprogramming following p53 inactivation would permit the evolution of a stem-like cancer from stem or non–stem cell antecedents (B-ii). It is also possible that reprogramming could drive cells toward even more primitive embryonic stem cell states, as occurs in the reprogramming of differentiated cells to induced pluripotent cells (B-iii). SC = stem cell.
Cancer stem cells in the hierarchical model may arise from mutations in a multitude of target cells. For example, transforming mutations may occur in normal tissue stem cells, which would confer obvious advantages deriving from the presumed capacity of such cells to self-renew (Fig. 3A-ii).210 Alternatively, a more limited progenitor may acquire mutations and/or epigenetic changes conferring the ectopic capacity for perpetual self-renewal (Fig. 3A-iii).210 Several reports now document genetic lesions conferring self-renewal to progenitors,211,212 including p53 loss.9,211,212 Importantly, in the epidermis and small intestine, the target cell within which such lesions occur impacts disease phenotype and outcome.213,214 In such cases, aberrant differentiation of the mutated stem-like cell generates the nontumorigenic cells that can constitute the bulk of the tumor mass. However, the mutations that initiate the disease are insufficient for tumor progression, implying that mutation accumulation according to the stochastic model must also occur to generate aggressive tumor clones.215
Reprogramming the Cancer Stem Cell Hypothesis
Neither of the traditional models incorporates the possibility of tumor-associated cellular reprogramming and plasticity associated with loss of p53 function. Given the impact of p53 inactivation on cellular dedifferentiation in the presence of appropriate oncogenic lesions, and the commonality of p53 defects in all human cancers, it is reasonable to consider induction of developmental plasticity as an important correlate of tumor progression. In breast cancer, where p53 mutations are inferred to occur late173,216 and are associated with stem-like states in basal-like cancers,21 we infer that cells that acquire this property have survival advantages that enable their accumulation. Acquisition of developmental plasticity by p53 inactivation may explain why targeting Brca1 deficiency to either basal or luminal cells in the mammary gland generated tumors that could not be distinguished by gene expression but did show morphological differences.217,218
We suggest that a more accurate description of the complex events occurring during tumor progression requires incorporating the potential for cellular reprogramming with the stochastic and cancer stem cell models. Here, cells with stem-like properties may be formed at any time during cancer progression, so long as p53 (or other factors that phenocopy its function) is disabled and appropriate oncogenic lesions that can drive proliferation and enable epigenetic reprogramming to a stem-like state are present (Fig. 3B-ii and 3B-iii). This model could help explain why it has been difficult to apply hierarchical models to some tumors.219 In fact, plasticity has been demonstrated with regard to numerous putative cancer stem cell markers,219-222 and it remains to be determined if there are markers that enable general identification of “stemness” in a given tumor. However, one interesting candidate is CD44, a putative cancer stem cell marker frequently reported to segregate with the capacity for xenograft initiation (e.g., in estrogen receptor–negative breast cancer222). CD44 is repressed by p53 directly and through p53-mediated induction of miRNA34a.223,224 Thus, upregulated CD44 expression may be a surrogate marker for p53 inactivation and associated plasticity.
A corollary of this composite model is that tumor-associated reprogramming may generate aberrant stem-like states. For example, reprogramming p53- deficient, oncogene-expressing cells could generate phenocopies resembling fetal stem cells or iPS cells (Fig. 3B-iii). Additionally, the aberrant microenvironments created within tumors might influence the type of stem-like cells that arise and may explain why the expression signatures of such cells in tumors are similar to, but distinctly different from, other adult or ESCs. Depending on the probability of reprogramming within tumors, it is conceivable that multiple independently derived and molecularly distinct stem-like clones could evolve (Fig. 4). This might explain the appearance of multiple clonal lineages within tumors identified by single-cell sequencing.206 Finally, it is not inconceivable that unique stem-like states could be created within tumors as a consequence of the influence of microenvironments on cells with p53 deficiency and oncogenic changes. Such states might explain the ability of some tumor cells to transdifferentiate into functional vascular-endothelial cells that resist antiangiogenic therapy,225 to exhibit remarkable plasticity regarding chemoresistance,226 and to migrate and metastasize.
Figure 4.
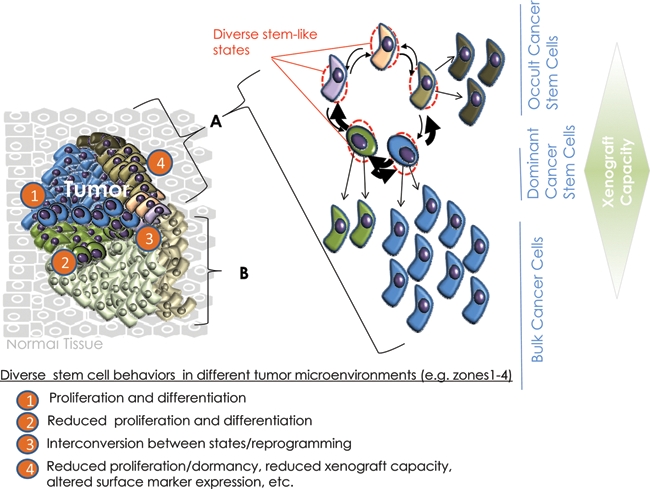
Induced plasticity in cancer and the potential for multiple cancer stem-like cells to coexist. A tumor mass (left) may contain multiple related but genetically distinct and independently propagating clones (A and B).208 Each clone may be sustained by a genetically and epigenetically unstable pool of stem-like cells (right), whose behavior (e.g., proliferation v. dormancy) can be influenced by the level and nature of oncogenic stimuli and the dissimilar local microenvironment in which they are situated. While some stem cells may initiate or perpetuate clonal growth in response to local microenvironments, other stem-like cells may remain indolent until appropriate signals are received. The resulting heterogeneity may be manifested as diverse stem-like states that vary in terms of their proliferative, biomarker, and chemosensitivity profiles as well as their ability to xenograft but not in their net tumorigenicity. Furthermore, disaggregation of the tumor mass for analysis obscures the local heterogeneity initially present. Together, inherent plasticity as well as additional acquired genetic traits may endow these cells with differential capacities for interconversion to more aggressive stem-like states during tumor evolution or recurrence.
Going Forward
Despite differences resulting from their specific genetic lesions and microenvironmental contexts, stem-like cancer cells are unlikely to arise by “inventing” completely novel biology. Rather, it is more likely that their genesis reflects the corruption of a reactivated normal stem cell repertoire. Thus, to gain greater insight into the mechanisms underlying acquisition of stem-like states in cancer, we are compelled to investigate the spectrum of both normal and neoplastic stem cell states. Achieving this goal imposes significant challenges. The uncontrolled stress effects associated with in vitro sphere and in vivo transplantation assays can activate p53, which may prevent normal stem cell function. The heterogeneity of all stem cell–enriched populations prevents one from assigning a definitive gene signature to the actual stem cell. To overcome these challenges, the field will need to develop methods that minimally perturb the natural tissue or tumor setting, potentially by combining in vivo lineage tracing with the robust but physiologically less relevant stem cell assays that are currently most often employed. Solving the problem of population heterogeneity will require that we devise genome-wide, single-cell analyses and then reintegrate the information obtained into systems level understanding of the stem cell state.
Finally, given the link between p53 and dedifferentiation and the implications this association has for cancer stem cell models, it may be useful to focus not only on the processes underlying “stemness” but also the plasticity that allows stemness to emerge and diversify in the tumorigenic context. In this regard, perhaps targeting plasticity, rather than selected “stem-like cells,” will afford more robust strategies for tumor management. Furthermore, the apparent commonality of metastable genomes and differentiation states in cancer suggests that developing therapies that enable conversion of cancers to chronic but manageable diseases is a commendable goal in addition to searching for “cures.” Dissecting the role played by p53 in preventing dedifferentiation may provide valuable inroads into such therapeutic strategies.
Acknowledgments
The authors thank Drs. Peter Gray, Jennie Green, and Mark Wade for their reading of the article and critical discussions.
Footnotes
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
This work was supported by the National Cancer Institute [grant number CA61449 (G.M.W.)]; the Susan G. Komen for the Cure, Breast Cancer Research Foundation (G.M.W.); the G. Harold & Leila Y. Mathers Foundation (G.M.W.); a Ruth L. Kirschstein National Research Service Award [grant number T32 CA009523 (B.T.S.)]; and the Department of Defense [grant number BC095773P1 (B.T.S.)].
References
- 1. Lane DP. Cancer: p53, guardian of the genome. Nature. 1992;358:15-6 [DOI] [PubMed] [Google Scholar]
- 2. Maddocks O, Vousden K. Metabolic regulation by p53. J Mol Med. 2011;89:237-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Muller PAJ, Vousden KH, Norman JC. p53 and its mutants in tumor cell migration and invasion. J Cell Biol. 2011;192:209-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8:275-83 [DOI] [PubMed] [Google Scholar]
- 5. Hosako H, Little SA, Barrier M, Mirkes PE. Teratogen-induced activation of p53 in early postimplantation mouse embryos. Toxicol Sci. 2007;95:257-69 [DOI] [PubMed] [Google Scholar]
- 6. Linke SP, Clarkin KC, Di Leonardo A, Tsou A, Wahl GM. A reversible, p53-dependent G0/G1 cell cycle arrest induced by ribonucleotide depletion in the absence of detectable DNA damage. Genes Dev. 1996;10:934-47 [DOI] [PubMed] [Google Scholar]
- 7. Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137:413-31 [DOI] [PubMed] [Google Scholar]
- 8. Molchadsky A, Rivlin N, Brosh R, Rotter V, Sarig R. p53 is balancing development, differentiation and de-differentiation to assure cancer prevention. Carcinogenesis. 2010;31:1501-8 [DOI] [PubMed] [Google Scholar]
- 9. Zhao Z, Zuber J, Diaz-Flores E, et al. p53 loss promotes acute myeloid leukemia by enabling aberrant self-renewal. Genes Dev. 2010;24:1389-402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lin T, Chao C, Saito Si, et al. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat Cell Biol. 2005;7:165-71 [DOI] [PubMed] [Google Scholar]
- 11. Meletis K, Wirta V, Hede S-M, Nister M, Lundeberg J, Frisen J. p53 suppresses the self-renewal of adult neural stem cells. Development. 2006;133:363-9 [DOI] [PubMed] [Google Scholar]
- 12. Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861-72 [DOI] [PubMed] [Google Scholar]
- 13. Zhao Y, Yin X, Qin H, et al. Two supporting factors greatly improve the efficiency of human iPSC generation. Cell Stem Cell. 2008;3:475-9 [DOI] [PubMed] [Google Scholar]
- 14. Kawamura T, Suzuki J, Wang YV, et al. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature. 2009;460:1140-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hong H, Takahashi K, Ichisaka T, et al. Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature. 2009;460:1132-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li H, Collado M, Villasante A, et al. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature. 2009;460:1136-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Marion RM, Strati K, Li H, et al. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature. 2009;460:1149-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Utikal J, Polo JM, Stadtfeld M, et al. Immortalization eliminates a roadblock during cellular reprogramming into iPS cells. Nature. 2009;460:1145-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sarig R, Rivlin N, Brosh R, et al. Mutant p53 facilitates somatic cell reprogramming and augments the malignant potential of reprogrammed cells. J Exp Med. 2009;207:2127-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pierce GB, Speers WC. Tumors as caricatures of the process of tissue renewal: prospects for therapy by directing differentiation. Cancer Res. 1988;48:1996-2004 [PubMed] [Google Scholar]
- 21. Mizuno H, Spike BT, Wahl GM, Levine AJ. Inactivation of p53 in breast cancers correlates with stem cell transcriptional signatures. Proc Natl Acad Sci U S A. 2010;107:22745-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Linzer DIH, Levine AJ. Characterization of a 54K Dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell. 1979;17:43-52 [DOI] [PubMed] [Google Scholar]
- 23. Lane D, Crawford L. T antigen is bound to a host protein in SV40-transformed cells. Nature. 1979;278:261-3 [DOI] [PubMed] [Google Scholar]
- 24. Ben David Y, Prideaux VR, Chow V, Benchimol S, Bernstein A. Inactivation of the p53 oncogene by internal deletion or retroviral integration in erythroleukemic cell lines induced by Friend leukemia virus. Oncogene. 1988;3:179-85 [PubMed] [Google Scholar]
- 25. Mowat M, Cheng A, Kimura N, Bernstein A, Benchimol S. Rearrangements of the cellular p53 gene in erythroleukaemic cells transformed by Friend virus. Nature. 1985;314:633-6 [DOI] [PubMed] [Google Scholar]
- 26. Wolf D, Rotter V. Major deletions in the gene encoding the p53 tumor antigen cause lack of p53 expression in HL-60 cells. Proc Natl Acad Sci U S A. 1985;82:790-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Baker SJ, Fearon ER, Nigro JM, et al. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science. 1989;244:217-21 [DOI] [PubMed] [Google Scholar]
- 28. Wolf D, Rotter V. Inactivation of p53 gene expression by an insertion of Moloney murine leukemia virus-like DNA sequences. Mol Cell Biol. 1984;4:1402-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Malkin D, Li FP, Strong LC, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250:1233-8 [DOI] [PubMed] [Google Scholar]
- 30. Jacks T, Remington L, Williams BO, et al. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1-7 [DOI] [PubMed] [Google Scholar]
- 31. Donehower LA, Harvey M, Slagle BL, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215-21 [DOI] [PubMed] [Google Scholar]
- 32. Michalak EM, Vandenberg CJ, Delbridge ARD, et al. Apoptosis-promoted tumorigenesis: γ-irradiation-induced thymic lymphomagenesis requires Puma-driven leukocyte death. Genes Dev. 2010;24:1608-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kuperwasser C, Hurlbut GD, Kittrell FS, et al. Development of spontaneous mammary tumors in BALB/c p53 heterozygous mice: a model for Li-Fraumeni syndrome. Am J Pathol. 2000;157:2151-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Artandi SE, Chang S, Lee S-L, et al. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature. 2000;406:641-5 [DOI] [PubMed] [Google Scholar]
- 35. Jonkers J, Berns A. Conditional mouse models of sporadic cancer. Nat Rev Cancer. 2002;2:251-65 [DOI] [PubMed] [Google Scholar]
- 36. Liu X, Holstege H, van der Gulden H, et al. Somatic loss of BRCA1 and p53 in mice induces mammary tumors with features of human BRCA1-mutated basal-like breast cancer. Proc Natl Acad Sci U S A. 2007;104:12111-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2:a001008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Soussi T, Wiman KG. Shaping genetic alterations in human cancer: the p53 mutation paradigm. Cancer Cell. 2007;12:303-12 [DOI] [PubMed] [Google Scholar]
- 39. Lowe SW, Jacks T, Housman DE, Ruley HE. Abrogation of oncogene-associated apoptosis allows transformation of p53-deficient cells. Proc Natl Acad Sci U S A. 1994;91:2026-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 1991;51:6304-11 [PubMed] [Google Scholar]
- 41. Lowe SW, Ruley HE, Jacks T, Housman DE. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell. 1993;74:957-67 [DOI] [PubMed] [Google Scholar]
- 42. He L, He X, Lim LP, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Suzuki HI, Yamagata K, Sugimoto K, Iwamoto T, Kato S, Miyazono K. Modulation of microRNA processing by p53. Nature. 2009;460:529-33 [DOI] [PubMed] [Google Scholar]
- 44. Toledo F, Bardot B. Cancer: three birds with one stone. Nature. 2009;460:466-7 [DOI] [PubMed] [Google Scholar]
- 45. Di Leonardo A, Linke SP, Clarkin K, Wahl GM. DNA damage triggers a prolonged p53-dependent G1 arrest and long-term induction of Cip1 in normal human fibroblasts. Genes Dev. 1994;8:2540-51 [DOI] [PubMed] [Google Scholar]
- 46. Xue W, Zender L, Miething C, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. DuPage M, Cheung AF, Mazumdar C, et al. Endogenous T cell responses to antigens expressed in lung adenocarcinomas delay malignant tumor progression. Cancer Cell. 2011;19:72-85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zilfou JT, Lowe SW. Tumor suppressive functions of p53. Cold Spring Harb Perspect Biol. 2009;1:a001883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wade M, Wang YV, Wahl GM. The p53 orchestra: Mdm2 and Mdmx set the tone. Trends Cell Biol. 2010;20:299-309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hill R, Song Y, Cardiff RD, Van Dyke T. Selective evolution of stromal mesenchyme with p53 loss in response to epithelial tumorigenesis. Cell. 2005;123:1001-11 [DOI] [PubMed] [Google Scholar]
- 51. Wang YV, Wade M, Wahl GM. Guarding the guardian: Mdmx plays important roles in setting p53 basal activity and determining biological responses in vivo. Cell Cycle. 2009;8:3443-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jackson MW, Berberich SJ. MdmX protects p53 from Mdm2-mediated degradation. Mol Cell Biol. 2000;20:1001-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Montes de Oca Luna R, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature. 1995;378:203-6 [DOI] [PubMed] [Google Scholar]
- 54. Jones SN, Roe AE, Donehower LA, Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature. 1995;378:206-8 [DOI] [PubMed] [Google Scholar]
- 55. Gannon HS, Donehower LA, Lyle S, Jones SN. Mdm2-p53 signaling regulates epidermal stem cell senescence and premature aging phenotypes in mouse skin. Dev Biol. In press [DOI] [PMC free article] [PubMed]
- 56. Marine JC, Francoz S, Maetens M, Wahl G, Toledo F, Lozano G. Keeping p53 in check: essential and synergistic functions of Mdm2 and Mdm4. Cell Death Differ. 2006;13:927-34 [DOI] [PubMed] [Google Scholar]
- 57. Alkema M, Wiegant J, Raap AK, Bems A, van Lohuizen M. Characterization and chromosomal localization of the human proto-oncogene BMI-1. Hum Mol Genet. 1993;2:1597-603 [DOI] [PubMed] [Google Scholar]
- 58. Molofsky AV, Pardal R, Iwashita T, Park I-K, Clarke MF, Morrison SJ. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature. 2003;425:962-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Park IK, Qian D, Kiel M, et al. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003;423:302-5 [DOI] [PubMed] [Google Scholar]
- 60. Pietersen AM, Evers B, Prasad AA, et al. Bmi1 regulates stem cells and proliferation and differentiation of committed cells in mammary epithelium. Curr Biol. 2008;18:1094-9 [DOI] [PubMed] [Google Scholar]
- 61. Haupt Y, Alexander WS, Barri G, Peter Klinken S, Adams JM. Novel zinc finger gene implicated as myc collaborator by retrovirally accelerated lymphomagenesis in E mu-myc transgenic mice. Cell. 1991;65:753-63 [DOI] [PubMed] [Google Scholar]
- 62. van Lohuizen M, Verbeek S, Scheljen B, Wientjens E, van der Guidon H, Berns A. Identification of cooperating oncogenes in Eµ-myc transgenic mice by provirus tagging. Cell. 1991;65:737-52 [DOI] [PubMed] [Google Scholar]
- 63. Jacobs JJ, Scheijen B, Voncken JW, Kieboom K, Berns A, van Lohuizen M. Bmi-1 collaborates with c-Myc in tumorigenesis by inhibiting c-Myc-induced apoptosis via INK4a/ARF. Genes Dev. 1999;13:2678-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lowe SW, Sherr CJ. Tumor suppression by Ink4a-Arf: progress and puzzles. Curr Opin Genet Dev. 2003;13:77-83 [DOI] [PubMed] [Google Scholar]
- 65. Pomerantz J, Schreiber-Agus N, Liegeois NJ, et al. The Ink4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2’s inhibition of p53. Cell. 1998;92:713-23 [DOI] [PubMed] [Google Scholar]
- 66. Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell. 1998;92:725-34 [DOI] [PubMed] [Google Scholar]
- 67. Shakhova O, Leung C, Marino S. Bmi1 in development and tumorigenesis of the central nervous system. J Mol Med. 2005;83:596-600 [DOI] [PubMed] [Google Scholar]
- 69. Stingl J, Eirew P, Ricketson I, et al. Purification and unique properties of mammary epithelial stem cells. Nature. 2006;439:993-7 [DOI] [PubMed] [Google Scholar]
- 69. Shackleton M, Vaillant F, Simpson KJ, et al. Generation of a functional mammary gland from a single stem cell. Nature. 2006;439:84-8 [DOI] [PubMed] [Google Scholar]
- 70. Sato T, Vries RG, Snippert HJ, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262-5 [DOI] [PubMed] [Google Scholar]
- 71. Zeng YA, Nusse R. Wnt proteins are self-renewal factors for mammary stem cells and promote their long-term expansion in culture. Cell Stem Cell. 2010;6:568-77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Yilmaz OH, Kiel MJ, Morrison SJ. SLAM family markers are conserved among hematopoietic stem cells from old and reconstituted mice and markedly increase their purity. Blood. 2006;107:924-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Morrison SJ, Uchida N, Weissman IL. The biology of hematopoietic stem cells. Annu Rev Cell Dev Biol. 1995;11:35-71 [DOI] [PubMed] [Google Scholar]
- 74. Morrison SJ, Wandycz AM, Akashi K, Globerson A, Weissman IL. The aging of hematopoietic stem cells. Nat Med. 1996;2:1011-6 [DOI] [PubMed] [Google Scholar]
- 75. Krause DS, Theise ND, Collector MI, et al. Multi-organ, multi-lineage engraftment by a single bone marrow-derived stem cell. Cell. 2001;105:369-77 [DOI] [PubMed] [Google Scholar]
- 76. Liu Y, Elf SE, Miyata Y, et al. p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell. 2009;4:37-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ito K, Hirao A, Arai F, et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature. 2004;431:997-1002 [DOI] [PubMed] [Google Scholar]
- 78. Spike BT, Dirlam A, Dibling BC, et al. The Rb tumor suppressor is required for stress erythropoiesis. EMBO J. 2004;23:4319-29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Cheng T, Rodrigues N, Shen H, et al. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000;287:1804-8 [DOI] [PubMed] [Google Scholar]
- 80. van Os R, Kamminga LM, Ausema A, et al. A limited role for p21Cip1/Waf1 in maintaining normal hematopoietic stem cell functioning. Stem Cells. 2007;25:836-43 [DOI] [PubMed] [Google Scholar]
- 81. Bondar T, Medzhitov R. p53-mediated hematopoietic stem and progenitor cell competition. Cell Stem Cell. 2010;6:309-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Marusyk A, DeGregori J. Declining cellular fitness with age promotes cancer initiation by selecting for adaptive oncogenic mutations. Biochim Biophys Acta. 2008;1785:1-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Akala OO, Park IK, Qian D, Pihalja M, Becker MW, Clarke MF. Long-term haematopoietic reconstitution by Trp53−/−p16Ink4a−/−p19Arf−/− multipotent progenitors. Nature. 2008;453:228-32 [DOI] [PubMed] [Google Scholar]
- 84. Cicalese A, Bonizzi G, Pasi CE, et al. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell. 2009;138:1083-95 [DOI] [PubMed] [Google Scholar]
- 85. Tao L, Roberts AL, Dunphy KA, Bigelow C, Yan H, Jerry DJ. Repression of mammary stem/progenitor cells by p53 is mediated by Notch and separable from apoptotic activity. Stem Cells. 2011;29:119-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Gatza CE, Dumble M, Kittrell F, et al. Altered mammary gland development in the p53+/m mouse, a model of accelerated aging. Dev Biol. 2008;313:130-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Flores I, Blasco MA. A p53-dependent response limits epidermal stem cell functionality and organismal size in mice with short telomeres. PLoS ONE. 2009;4:e4934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Zhou Z, Flesken-Nikitin A, Nikitin AY. Prostate cancer associated with p53 and Rb deficiency arises from the stem/progenitor cell-enriched proximal region of prostatic ducts. Cancer Res. 2007;67:5683-90 [DOI] [PubMed] [Google Scholar]
- 89. Gil-Perotin S, Marin-Husstege M, Li J, et al. Loss of p53 induces changes in the behavior of subventricular zone cells: implication for the genesis of glial tumors. J Neurosci. 2006;26:1107-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Armesilla-Diaz A, Bragado P, Del Valle I, et al. p53 regulates the self-renewal and differentiation of neural precursors. Neuroscience. 2009;158:1378-89 [DOI] [PubMed] [Google Scholar]
- 91. Zheng H, Ying H, Yan H, et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature. 2008;455:1129-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Savatier P, Lapillonne H, van Grunsven LA, Rudkin BB, Samarut J. Withdrawal of differentiation inhibitory activity/leukemia inhibitory factor up-regulates D-type cyclins and cyclin-dependent kinase inhibitors in mouse embryonic stem cells. Oncogene. 1996;12:309-22 [PubMed] [Google Scholar]
- 93. Aladjem MI, Spike BT, Rodewald LW, et al. ES cells do not activate p53-dependent stress responses and undergo p53-independent apoptosis in response to DNA damage. Curr Biol. 1998;8:145-55 [DOI] [PubMed] [Google Scholar]
- 94. Sabapathy K, Klemm M, Jaenisch R, Wagner EF. Regulation of ES cell differentiation by functional and conformational modulation of p53. EMBO J. 1997;16:6217-29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Solozobova V, Rolletschek A, Blattner C. Nuclear accumulation and activation of p53 in embryonic stem cells after DNA damage. BMC Cell Biol. 2009;10:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Han MK, Song EK, Guo Y, Ou X, Mantel C, Broxmeyer HE. SIRT1 regulates apoptosis and Nanog expression in mouse embryonic stem cells by controlling p53 subcellular localization. Cell Stem Cell. 2008;2:241-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Chuykin IA, Lianguzova MS, Pospelova TV, Pospelov VA. Activation of DNA damage response signaling in mouse embryonic stem cells. Cell Cycle. 2008;7:2922-8 [DOI] [PubMed] [Google Scholar]
- 98. Hong Y, Stambrook PJ. Restoration of an absent G1 arrest and protection from apoptosis in embryonic stem cells after ionizing radiation. Proc Natl Acad Sci U S A. 2004;101:14443-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Hong Y, Cervantes RB, Tichy E, Tischfield JA, Stambrook PJ. Protecting genomic integrity in somatic cells and embryonic stem cells. Mutat Res. 2007;614:48-55 [DOI] [PubMed] [Google Scholar]
- 100. de Vries A, Flores ER, Miranda B, et al. Targeted point mutations of p53 lead to dominant-negative inhibition of wild-type p53 function. Proc Natl Acad Sci U S A. 2002;99:2948-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Lee KH, Li M, Michalowski AM, et al. A genomewide study identifies the Wnt signaling pathway as a major target of p53 in murine embryonic stem cells. Proc Natl Acad Sci U S A. 2010;107:69-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Fluckiger AC, Marcy G, Marchand M, et al. Cell cycle features of primate embryonic stem cells. Stem Cells. 2006;24:547-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Prost S, Bellamy CO, Clarke AR, Wyllie AH, Harrison DJ. p53-independent DNA repair and cell cycle arrest in embryonic stem cells. FEBS Lett. 1998;425:499-504 [DOI] [PubMed] [Google Scholar]
- 104. Qin H, Yu T, Qing T, et al. Regulation of apoptosis and differentiation by p53 in human embryonic stem cells. J Biol Chem. 2007;282:5842-52 [DOI] [PubMed] [Google Scholar]
- 105. Stead E, White J, Faast R, et al. Pluripotent cell division cycles are driven by ectopic Cdk2, cyclin A/E and E2F activities. Oncogene. 2002;21:8320-33 [DOI] [PubMed] [Google Scholar]
- 106. Koledova Z, Kafkova LR, Calabkova L, Krystof V, Dolezel P, Divoky V. Cdk2 inhibition prolongs G1 phase progression in mouse embryonic stem cells. Stem Cells Dev. 2010;19:181-94 [DOI] [PubMed] [Google Scholar]
- 107. Neganova I, Lako M. G1 to S phase cell cycle transition in somatic and embryonic stem cells. J Anat. 2008;213:30-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Lombard DB, Chua KF, Mostoslavsky R, Franco S, Gostissa M, Alt FW. DNA repair, genome stability, and aging. Cell. 2005;120:497-512 [DOI] [PubMed] [Google Scholar]
- 109. Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483-95 [DOI] [PubMed] [Google Scholar]
- 110. Vafa O, Wade M, Kern S, et al. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol Cell. 2002;9:1031-44 [DOI] [PubMed] [Google Scholar]
- 111. Macleod KF. The RB tumor suppressor: a gatekeeper to hormone independence in prostate cancer? J Clin Invest. 2010;120:4179-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Dean JB, Mulkey DK, Henderson RA, 3rd, Potter SJ, Putnam RW. Hyperoxia, reactive oxygen species, and hyperventilation: oxygen sensitivity of brain stem neurons. J Appl Physiol. 2004;96:784-91 [DOI] [PubMed] [Google Scholar]
- 113. Kurz EU, Douglas P, Lees-Miller SP. Doxorubicin activates ATM-dependent phosphorylation of multiple downstream targets in part through the generation of reactive oxygen species. J Biol Chem. 2004;279:53272-81 [DOI] [PubMed] [Google Scholar]
- 114. Courtois S, Verhaegh G, North S, et al. DeltaN-p53, a natural isoform of p53 lacking the first transactivation domain, counteracts growth suppression by wild-type p53. Oncogene. 2002;21:6722-8 [DOI] [PubMed] [Google Scholar]
- 115. Ungewitter E, Scrable H. DeltaN40-p53 controls the switch from pluripotency to differentiation by regulating IGF signaling in ESCs. Genes Dev. 2010;24:2408-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Melis JP, Hoogervorst EM, van Oostrom CT, et al. Genotoxic exposure: novel cause of selection for a functional DeltaN-p53 isoform. Oncogene. 2011;14:1764-72 [DOI] [PubMed] [Google Scholar]
- 117. Bourdon JC, Fernandes K, Murray-Zmijewski F, et al. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005;19:2122-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Xu D, Wilson TJ, Chan D, et al. Ets1 is required for p53 transcriptional activity in UV-induced apoptosis in embryonic stem cells. EMBO J. 2002;21:4081-93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Menendez S, Goh AM, Camus S, et al. MDM4 downregulates p53 transcriptional activity and response to stress during differentiation. Cell Cycle. 2011;10:1100-8 [DOI] [PubMed] [Google Scholar]
- 120. Choong ML, Yang H, Lee MA, Lane DP. Specific activation of the p53 pathway by low dose actinomycin D: a new route to p53 based cyclotherapy. Cell Cycle. 2009;8:2810-8 [DOI] [PubMed] [Google Scholar]
- 121. Vassilev LT, Vu BT, Graves B, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844-8 [DOI] [PubMed] [Google Scholar]
- 122. Ginis I, Luo Y, Miura T, et al. Differences between human and mouse embryonic stem cells. Dev Biol. 2004;269:360-80 [DOI] [PubMed] [Google Scholar]
- 123. Neganova I, Vilella F, Atkinson SP, et al. An important role for CDK2 in G1 to S checkpoint activation and DNA damage response in human embryonic stem cells. Stem Cells. 2011;29:651-9 [DOI] [PubMed] [Google Scholar]
- 124. Becker KA, Ghule PN, Therrien JA, et al. Self-renewal of human embryonic stem cells is supported by a shortened G1 cell cycle phase. J Cell Physiol. 2006;209:883-93 [DOI] [PubMed] [Google Scholar]
- 125. Filion TM, Qiao M, Ghule PN, et al. Survival responses of human embryonic stem cells to DNA damage. J Cell Physiol. 2009;220:586-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Wilson KD, Sun N, Huang M, et al. Effects of ionizing radiation on self-renewal and pluripotency of human embryonic stem cells. Cancer Res. 2010;70:5539-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Barta T, Vinarsky V, Holubcova Z, et al. Human embryonic stem cells are capable of executing G1/S checkpoint activation. Stem Cells. 2011;28:1143-52 [DOI] [PubMed] [Google Scholar]
- 128. Maimets T, Neganova I, Armstrong L, Lako M. Activation of p53 by nutlin leads to rapid differentiation of human embryonic stem cells. Oncogene. 2008;27:5277-87 [DOI] [PubMed] [Google Scholar]
- 129. Chou YF, Chen HH, Eijpe M, et al. The growth factor environment defines distinct pluripotent ground states in novel blastocyst-derived stem cells. Cell. 2008;135:449-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Neveu P, Kye MJ, Qi S, et al. MicroRNA profiling reveals two distinct p53-related human pluripotent stem cell states. Cell Stem Cell. 2011;7:671-81 [DOI] [PubMed] [Google Scholar]
- 131. Brons IG, Smithers LE, Trotter MW, et al. Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature. 2007;448:191-5 [DOI] [PubMed] [Google Scholar]
- 132. Tesar PJ, Chenoweth JG, Brook FA, et al. New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature. 2007;448:196-9 [DOI] [PubMed] [Google Scholar]
- 133. Buecker C, Chen H-H, Polo JM, et al. A murine ESC-like state facilitates transgenesis and homologous recombination in human pluripotent stem cells. Cell Stem Cell. 2010;6:535-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Yang A, Kaghad M, Caput D, McKeon F. On the shoulders of giants: p63, p73 and the rise of p53. Trends Genet. 2002;18:90-5 [DOI] [PubMed] [Google Scholar]
- 135. Senoo M, Pinto F, Crum CP, McKeon F. p63 is essential for the proliferative potential of stem cells in stratified epithelia. Cell. 2007;129:523-36 [DOI] [PubMed] [Google Scholar]
- 136. Moll UM, Slade N. p63 and p73: roles in development and tumor formation. Mol Cancer Res. 2004;2:371-86 [PubMed] [Google Scholar]
- 137. Rocco JW, Leong C-O, Kuperwasser N, DeYoung MP, Ellisen LW. p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis. Cancer Cell. 2006;9:45-56 [DOI] [PubMed] [Google Scholar]
- 138. Suh EK, Yang A, Kettenbach A, et al. p63 protects the female germ line during meiotic arrest. Nature. 2006;444:624-8 [DOI] [PubMed] [Google Scholar]
- 139. Aubrecht J, Secretan MB, Bishop AJ, Schiestl RH. Involvement of p53 in X-ray induced intrachromosomal recombination in mice. Carcinogenesis. 1999;20:2229-36 [DOI] [PubMed] [Google Scholar]
- 140. Baatout S, Jacquet P, Michaux A, et al. Developmental abnormalities induced by X-irradiation in p53 deficient mice. In Vivo. 2002;16:215-21 [PubMed] [Google Scholar]
- 141. Armstrong JF, Kaufman MH, Harrison DJ, Clarke AR. High-frequency developmental abnormalities in p53-deficient mice. Curr Biol. 1995;5:931-6 [DOI] [PubMed] [Google Scholar]
- 142. Belyi VA, Ak P, Markert E, et al. The origins and evolution of the p53 family of genes. Cold Spring Harb Perspect Biol. 2010;2:a001198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Crum CP, McKeon FD. p63 in epithelial survival, germ cell surveillance, and neoplasia. Annu Rev Pathol. 2010;5:349-71 [DOI] [PubMed] [Google Scholar]
- 144. Talos F, Nemajerova A, Flores ER, Petrenko O, Moll UM. p73 suppresses polyploidy and aneuploidy in the absence of functional p53. Mol Cell. 2007;27:647-59 [DOI] [PubMed] [Google Scholar]
- 145. Jacks T, Remington L, Williams BO, et al. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1-7 [DOI] [PubMed] [Google Scholar]
- 146. Hu W, Feng Z, Teresky AK, Levine AJ. p53 regulates maternal reproduction through LIF. Nature. 2007;450:721-4 [DOI] [PubMed] [Google Scholar]
- 147. Rotter V, Schwartz D, Almon E, et al. Mice with reduced levels of p53 protein exhibit the testicular giant-cell degenerative syndrome. Proc Natl Acad Sci U S A. 1993;90:9075-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Beumer TL, Roepers-Gajadien HL, Gademan IS, et al. The role of the tumor suppressor p53 in spermatogenesis. Cell Death Differ. 1998;5:669-77 [DOI] [PubMed] [Google Scholar]
- 149. Sah VP, Attardi LD, Mulligan GJ, Williams BO, Bronson RT, Jacks T. A subset of p53- deficient embryos exhibit exencephaly. Nat Genet. 1995;10:175-80 [DOI] [PubMed] [Google Scholar]
- 150. MacCallum DE, Hupp TR, Midgley CA, et al. The p53 response to ionising radiation in adult and developing murine tissues. Oncogene. 1996;13:2575-87 [PubMed] [Google Scholar]
- 151. Gottlieb E, Haffner R, King A, et al. Transgenic mouse model for studying the transcriptional activity of the p53 protein: age- and tissue-dependent changes in radiation-induced activation during embryogenesis. EMBO J. 1997;16:1381-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Komarova EA, Chernov MV, Franks R, et al. Transgenic mice with p53-responsive lacZ: p53 activity varies dramatically during normal development and determines radiation and drug sensitivity in vivo. EMBO J. 1997;16:1391-400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Almog N, Rotter V. Involvement of p53 in cell differentiation and development. Biochim Biophys Acta. 1997;1333:F1-27 [DOI] [PubMed] [Google Scholar]
- 154. Matas D, Milyavsky M, Shats I, Nissim L, Goldfinger N, Rotter V. p53 is a regulator of macrophage differentiation. Cell Death Differ. 2004;11:458-67 [DOI] [PubMed] [Google Scholar]
- 155. Tsukada T, Tomooka Y, Takai S, et al. Enhanced proliferative potential in culture of cells from p53-deficient mice. Oncogene. 1993;8:3313-22 [PubMed] [Google Scholar]
- 156. Macleod KF, Hu Y, Jacks T. Loss of Rb activates both p53-dependent and independent cell death pathways in the developing mouse nervous system. EMBO J. 1996;15:6178-88 [PMC free article] [PubMed] [Google Scholar]
- 157. Porrello A, Cerone MA, Coen S, et al. p53 regulates myogenesis by triggering the differentiation activity of pRb. J Cell Biol. 2000;151:1295-304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Molchadsky A, Shats I, Goldfinger N, et al. p53 plays a role in mesenchymal differentiation programs, in a cell fate dependent manner. PLoS ONE. 2008;3:e3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Hallenborg P, Feddersen S, Madsen L, Kristiansen K. The tumor suppressors pRB and p53 as regulators of adipocyte differentiation and function. Expert Opin Ther Targets. 2009;13:235-46 [DOI] [PubMed] [Google Scholar]
- 160. TeKippe M, Harrison DE, Chen J. Expansion of hematopoietic stem cell phenotype and activity in Trp53-null mice. Exp Hematol. 2003;31:521-7 [DOI] [PubMed] [Google Scholar]
- 161. Zhao T, Xu Y. p53 and stem cells: new developments and new concerns. Trends Cell Biol. 2010;20:170-5 [DOI] [PubMed] [Google Scholar]
- 162. Mora-Castilla S, Tejedo JR, Hmadcha A, et al. Nitric oxide repression of Nanog promotes mouse embryonic stem cell differentiation. Cell Death Differ. 2010;17:1025-33 [DOI] [PubMed] [Google Scholar]
- 163. Ostrakhovitch EA, Semenikhin OA. p53- mediated regulation of neuronal differentiation via regulation of dual oxidase maturation factor 1. Neurosci Lett. 2011;494:80-5 [DOI] [PubMed] [Google Scholar]
- 164. Fiucci G, Beaucourt S, Duflaut D, et al. Siah-1b is a direct transcriptional target of p53: identification of the functional p53 responsive element in the siah-1b promoter. Proc Natl Acad Sci U S A. 2004;101:3510-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Iwai A, Marusawa H, Matsuzawa S, et al. Siah-1L, a novel transcript variant belonging to the human Siah family of proteins, regulates beta-catenin activity in a p53-dependent manner. Oncogene. 2004;23:7593-600 [DOI] [PubMed] [Google Scholar]
- 166. Pece S, Tosoni D, Confalonieri S, et al. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell. 2010;140:62-73 [DOI] [PubMed] [Google Scholar]
- 167. Knoblich JA, Jan LY, Jan YN. Asymmetric segregation of Numb and Prospero during cell division. Nature. 1995;377:624-7 [DOI] [PubMed] [Google Scholar]
- 168. Wu M, Kwon HY, Rattis F, et al. Imaging hematopoietic precursor division in real time. Cell Stem Cell. 2007;1:541-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Colaluca IN, Tosoni D, Nuciforo P, et al. NUMB controls p53 tumour suppressor activity. Nature. 2008;451:76-80 [DOI] [PubMed] [Google Scholar]
- 170. Pereira L, Yi F, Merrill BJ. Repression of Nanog gene transcription by Tcf3 limits embryonic stem cell self-renewal. Mol Cell Biol. 2006;26:7479-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Ruiz S, Panopoulos AD, Herrerías A, et al. A high proliferation rate is required for cell reprogramming and maintenance of human embryonic stem cell identity. Curr Biol. 2011;21:45-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Fagin JA, Matsuo K, Karmakar A, Chen DL, Tang SH, Koeffler HP. High prevalence of mutations of the p53 gene in poorly differentiated human thyroid carcinomas. J Clin Invest. 1993;91:179-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Miller LD, Smeds J, George J, et al. An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc Natl Acad Sci U S A. 2005;102:13550-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. de Cremoux P, Salomon AV, Liva S, et al. p53 mutation as a genetic trait of typical medullary breast carcinoma. J Natl Cancer Inst. 1999;91:641-3 [DOI] [PubMed] [Google Scholar]
- 175. Kochhar R, Howard EM, Umbreit JN, Lau SK. Metaplastic breast carcinoma with squamous differentiation: molecular and clinical analysis of six cases. Breast J. 2005;11:367-9 [DOI] [PubMed] [Google Scholar]
- 176. Zheng H, Ying H, Yan H, et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature. 2008;455:1129-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Junttila MR, Karnezis AN, Garcia D, et al. Selective activation of p53-mediated tumour suppression in high-grade tumours. Nature. 2010;468:567-71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Feldser DM, Kostova KK, Winslow MM, et al. Stage-specific sensitivity to p53 restoration during lung cancer progression. Nature. 2010;468:572-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Burger H, Nooter K, Boersma AWM, Kortland CJ, Stoter G. Lack of correlation between cisplatin-induced apoptosis, p53 status and expression of Bcl-2 family proteins in testicular germ cell tumour cell lines. Int J Cancer. 1997;73:592-9 [DOI] [PubMed] [Google Scholar]
- 180. Oren M, Reich NC, Levine AJ. Regulation of the cellular p53 tumor antigen in teratocarcinoma cells and their differentiated progeny. Mol Cell Biol. 1982;2:443-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Kemp CJ, Donehower LA, Bradley A, Balmain A. Reduction of p53 gene dosage does not increase initiation or promotion but enhances malignant progression of chemically induced skin tumors. Cell. 1993;74:813-22 [DOI] [PubMed] [Google Scholar]
- 182. Jiang Z, Deng T, Jones R, et al. Rb deletion in mouse mammary progenitors induces luminal-B or basal-like/EMT tumor subtypes depending on p53 status. J Clin Invest. 2010;120:3296-309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Chylicki K, Ehinger M, Svedberg H, Gullberg U. Characterization of the molecular mechanisms for p53-mediated differentiation. Cell Growth Differ. 2000;11:561-71 [PubMed] [Google Scholar]
- 184. Johnson P, Chung S, Benchimol S. Growth suppression of Friend virus-transformed erythroleukemia cells by p53 protein is accompanied by hemoglobin production and is sensitive to erythropoietin. Mol Cell Biol. 1993;13:1456-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Feinstein E, Gale RP, Reed J, Canaani E. Expression of the normal p53 gene induces differentiation of K562 cells. Oncogene. 1992;7:1853-7 [PubMed] [Google Scholar]
- 186. Secchiero P, Zerbinati C, Melloni E, et al. The MDM-2 antagonist nutlin-3 promotes the maturation of acute myeloid leukemic blasts. Neoplasia. 2007;9:853-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Lang D, Miknyoczki SJ, Huang L, Ruggeri BA. Stable reintroduction of wild-type P53 (MTmp53ts) causes the induction of apoptosis and neuroendocrine-like differentiation in human ductal pancreatic carcinoma cells. Oncogene. 1998;16:1593-602 [DOI] [PubMed] [Google Scholar]
- 188. Radinsky R, Fidler IJ, Price JE, et al. Terminal differentiation and apoptosis in experimental lung metastases of human osteogenic sarcoma cells by wild type p53. Oncogene. 1994;9:1877-83 [PubMed] [Google Scholar]
- 189. Bond JA, Oddweig Ness G, Rowson J, Ivan M, White D, Wynford-Thomas D. Spontaneous de-differentiation correlates with extended lifespan in transformed thyroid epithelial cells: an epigenetic mechanism of tumour progression? Int J Cancer. 1996;67:563-72 [DOI] [PubMed] [Google Scholar]
- 190. Kleinsmith LJ, Pierce GB., Jr. Multipotentiality of single embryonal carcinoma cells. Cancer Res. 1964;24:1544-51 [PubMed] [Google Scholar]
- 191. Martin GR. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc Natl Acad Sci U S A. 1981;78:7634-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Casimir CM, Gates PB, Patient RK, Brockes JP. Evidence for dedifferentiation and metaplasia in amphibian limb regeneration from inheritance of DNA methylation. Development. 1988;104:657-68 [DOI] [PubMed] [Google Scholar]
- 193. Takahashi K, Okita K, Nakagawa M, Yamanaka S. Induction of pluripotent stem cells from fibroblast cultures. Nat Protoc. 2007;2:3081-9 [DOI] [PubMed] [Google Scholar]
- 194. Tapia N, Scholer HR. p53 connects tumorigenesis and reprogramming to pluripotency. J Exp Med. 2010;207:2045-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Pelengaris S, Khan M, Evan G. c-MYC: more than just a matter of life and death. Nat Rev Cancer. 2002;2:764-76 [DOI] [PubMed] [Google Scholar]
- 196. Hanna J, Saha K, Pando B, et al. Direct cell reprogramming is a stochastic process amenable to acceleration. Nature. 2009;462:595-601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Smith ZD, Nachman I, Regev A, Meissner A. Dynamic single-cell imaging of direct reprogramming reveals an early specifying event. Nat Biotechnol. 2010;28:521-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Lister R, Pelizzola M, Kida YS, et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011;471:68-73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Assou S, Le Carrour T, Tondeur S, et al. A meta-analysis of human embryonic stem cells transcriptome integrated into a web-based expression atlas. Stem Cells. 2007;25:961-73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Ben-Porath I, Thomson MW, Carey VJ, et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008;40:499-507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Kim J, Woo AJ, Chu J, et al. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell. 2010;143:313-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Wong DJ, Liu H, Ridky TW, Cassarino D, Segal E, Chang HY. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell. 2008;2:333-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Muller FJ, Schuldt BM, Williams R, et al. A bioinformatic assay for pluripotency in human cells. Nat Methods. 2011;8:315-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Chapman MA, Lawrence MS, Keats JJ, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471:467-72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Berger MF, Lawrence MS, Demichelis F, et al. The genomic complexity of primary human prostate cancer. Nature. 2011;470:214-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Navin N, Kendall J, Troge J, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472:90-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730-7 [DOI] [PubMed] [Google Scholar]
- 208. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105-11 [DOI] [PubMed] [Google Scholar]
- 209. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Passegue E, Jamieson CH, Ailles LE, Weissman IL. Normal and leukemic hematopoiesis: are leukemias a stem cell disorder or a reacquisition of stem cell characteristics? Proc Natl Acad Sci U S A. 2003;100 Suppl 1:11842-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Henry MD, Triplett AA, Oh KB, Smith GH, Wagner KU. Parity-induced mammary epithelial cells facilitate tumorigenesis in MMTV-neu transgenic mice. Oncogene. 2004;23:6980-5 [DOI] [PubMed] [Google Scholar]
- 212. Bouras T, Pal B, Vaillant F, et al. Notch signaling regulates mammary stem cell function and luminal cell-fate commitment. Cell Stem Cell. 2008;3:429-41 [DOI] [PubMed] [Google Scholar]
- 213. Barker N, Ridgway RA, van Es JH, et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009;457:608-11 [DOI] [PubMed] [Google Scholar]
- 214. Brown K, Strathdee D, Bryson S, Lambie W, Balmain A. The malignant capacity of skin tumours induced by expression of a mutant H-ras transgene depends on the cell type targeted. Curr Biol. 1998;8:516-24 [DOI] [PubMed] [Google Scholar]
- 215. Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med. 2011;17:313-9 [DOI] [PubMed] [Google Scholar]
- 216. Langerod A, Zhao H, Borgan O, et al. TP53 mutation status and gene expression profiles are powerful prognostic markers of breast cancer. Breast Cancer Res. 2007;9:R30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Molyneux G, Geyer FC, Magnay FA, et al. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell. 2010;7:403-17 [DOI] [PubMed] [Google Scholar]
- 218. Molyneux G, Smalley M. The cell of origin of BRCA1 mutation-associated breast cancer: a cautionary tale of gene expression profiling. J Mammary Gland Biol Neoplasia. 2011;16:51-5 [DOI] [PubMed] [Google Scholar]
- 219. Quintana E, Shackleton M, Foster HR, et al. Phenotypic heterogeneity among tumorigenic melanoma cells from patients that is reversible and not hierarchically organized. Cancer Cell. 2008;18:510-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Roesch A, Fukunaga-Kalabis M, Schmidt EC, et al. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell. 2010;141:583-94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Quintana E, Shackleton M, Foster HR, et al. Phenotypic heterogeneity among tumorigenic melanoma cells from patients that is reversible and not hierarchically organized. Cancer Cell. 2010;18:510-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Meyer MJ, Fleming JM, Lin AF, Hussnain SA, Ginsburg E, Vonderhaar BK. CD44posCD49fhiCD133/2hi defines xenograft-initiating cells in estrogen receptor-negative breast cancer. Cancer Res. 2010;70:4624-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Godar S, Ince TA, Bell GW, et al. Growth-inhibitory and tumor- suppressive functions of p53 depend on its repression of CD44 expression. Cell. 2008;134:62-73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Liu C, Kelnar K, Liu B, et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat Med. 2011;17:211-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Soda Y, Marumoto T, Friedmann-Morvinski D, et al. Transdifferentiation of glioblastoma cells into vascular endothelial cells. Proc Natl Acad Sci U S A. 2011;108:4274-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Sharma SV, Lee DY, Li B, et al. A chromatin- mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141:69-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Hough SR, Laslett AL, Grimmond SB, Kolle G, Pera MF. A continuum of cell states spans pluripotency and lineage commitment in human embryonic stem cells. PLoS ONE. 2009;4:e7708. [DOI] [PMC free article] [PubMed] [Google Scholar]