

Abstract
Macrophage migration inhibitory factor is a leaderless protein that is secreted from cells by a specialized, non-classical export pathway. The release of MIF nevertheless is regulated and its production in response to different inflammatory, mitogenic, and hormonal stimuli plays an important role in diverse physiologic and pathologic processes. We report herein the identification of the Golgi complex-associated protein, p115, as an intracellular binding partner for MIF. MIF interacts with p115 in the cytoplasm and the stimulated secretion of MIF results in the accumulation of both proteins in supernatants, which is consistent with MIF release from cells in conjunction with p115. The depletion of p115 from monocytes/macrophages decreases the release of MIF but not other cytokines following inflammatory stimulation or intracellular bacterial infection. Notably, the small molecule MIF inhibitor, 4-iodo-6-phenylpyrimidine, inhibits MIF secretion by targeting the interaction between MIF and p115. These data reveal p115 to be a critical intermediary component in the regulated secretion of MIF from monocytes/macrophages.
Keywords: cytokines, inflammation, lipopolysaccharide, monocytes/macrophages
Introduction
Macrophage migration inhibitory factor (MIF) is a 12.5 kDa cytokine and an important regulator of innate immunity that is produced in response to pro-inflammatory, mitogenic, or hormonal stimuli. MIF plays a pivotal, upstream role in the host anti-microbial responses, the development of autoimmune inflammatory diseases, and in tumor progression (1). Activating stimuli induce the rapid release of MIF from pre-formed, cytoplasmic pools; this is followed by an upregulation in MIF mRNA expression and the replenishment of intracellular protein content (2, 3). Notably, MIF lacks a signal sequence and is secreted by an unconventional route for protein export. MIF undergoes no Golgi complex-dependent post-translational modifications, and inhibitors of the ER/Golgi transport do not affect LPS-stimulated MIF secretion from monocytes/macrophages (4, 5).
In contrast to the “classical” pathway for secretion, in which a hydrophobic leader sequence targets proteins for export via the endoplasmic reticulum and Golgi (6-8), non-classical protein secretion is less well understood (9-11). Several distinct mechanisms have been revealed for particular proteins such as vesicular transport via lysosomes or exosomes, plasma membrane shedding, and direct translocation across the membrane by specialized transporters. Potential secretory pathways that have been described for interleukin-1β (IL-1β) for instance, include cell surface blebbing and the formation of microvesicles that lyse in the extracellular space (12), and the fusion of endolysosomal vesicles with the cell surface plasma membrane (13). Fibroblast growth factor 2 (FGF-2), which is another unconventionally secreted cytokine, appears to cross the membrane in an ATP- and membrane-potential independent manner (10, 14, 15) and requires the action of phosphatidylinositol phosphate kinases (PIP-K) (16, 17). There is evidence that MIF may be associated with vesicles in particular cell types (18, 19), and a recent report has shown that MIF secretion is reduced by glyburide (5). Glyburide inhibits ATP binding cassette (ABC) transporters; it targets numerous proteins such as ion channels, enzymes and other transporters (20, 21).
To gain insight into the protein components and the mechanisms responsible for MIF release, we performed a yeast two-hybrid interaction study to identify intracellular proteins that might bind MIF and mediate its export. We report herein the identification of the Golgi-associated protein, p115 (22), as a binding partner for MIF, and we provide evidence for the p115-dependent release of MIF from human monocytes/macrophages. We further show that a recently described, small molecule MIF inhibitor, 4-iodo-6-phenylpyrimidine (4-IPP) (23), targets MIF secretion by influencing the interaction between MIF and p115.
Materials and Methods
Yeast Two-hybrid Studies
A human pituitary cDNA library and the CytoTrap yeast two-hybrid system were purchased from Stratagene (La Jolla, CA) and used according to the manufacturer's instructions. Briefly, cdc25H yeast cells were co-transformed with a MIF-bait plasmid (pSOS-MIF) and a library of human pituitary cDNA (pMyr). The expression of library cDNA was controlled by a galactose-inducible promoter. Transformants were grown on selectable minimal glucose plates for 4–6 days at 25 °C and colonies replica-plated onto minimal galactose plates for 5–7 days at 37 °C. Positive colonies exhibiting efficient growth on galactose plates were isolated and tested for galactose-dependent growth at 37 °C. Library plasmids were recovered and the specificity of interactions was tested by re-transformation of cdc25H cells with pSOS-MIF, pSOS-ColI, or pSOS-MAFB. Library plasmids that specifically interacted with MIF were identified by DNA sequencing.
p115 Cloning and Recombinant Protein Preparation
A truncated p115 protein (p115702-962) representing the clone identified in the yeast two-hybrid screen was sub-cloned into the pET22b vector, which contained a carboxy-terminal histidine tag (Novagen, Madison, WI), and expressed in E. coli BL21(DE3). The crude bacterial extract was purified using a HiTrap chelating HP column (Amersham, Piscataway, NJ) with a linear gradient of 20–400 mM imidazole in 50 mM Tris, pH 8.0 and 200 mM NaCl. The peak fractions were subjected to gel filtration chromatography (Superdex-75, HiLoad 16/60, Amersham). Protein purity was verified by SDS-PAGE/Coomassie staining and Western blotting. For the p115 capture assay, recombinant p115702-962 was biotinylated using EZ-LinkTM-sulfo-NHS-LC biotin (Pierce, Rockford, IL). Human MIF was prepared as described previously (4) and biotinylated using the Biotin Protein Labeling Kit from Roche (Indianapolis, IN). The chemical modification of MIF by N-acetyl-p-benzoquinone imine (NAPQI) (24) and 4-iodo-phenylpyrimidine (4-IPP) was achieved by incubation of MIF with a 10× molar excess of compound at room temperature overnight (23). The modified MIF was dialysed against PBS. For expression in mammalian cells, full length p115 was cloned into pcDNA3.1 (Invitrogen, Carlsbad, CA).
MIF/p115 Binding Studies
Ninety-six well Nunc Immuno-Module plates (Rochester, NY) were coated with 1 μM p115702-962 per well. The plates were washed with TBST (Tris-buffered saline, 0.05% Tween 20) and blocked with Superblock (Pierce). 150 nM biotinylated MIF was added in duplicate to wells together with increasing concentrations of unlabeled MIF or lyzozyme as control. Incubation was continued overnight at 4 °C, followed by washing with TBST. The bound, biotinylated MIF was detected by adding streptavidin-conjugated alkaline phosphatase (R&D, Minneapolis, MN) for 1 h at room temperature, followed by washing and detection of the alkaline phosphatase with p-nitrophenyl phosphate (Sigma, St. Louis, MO). For a reverse binding assay, 1 μM MIF was coated onto wells and 150 nM biotinylated p115702-962 added together with increasing concentrations of unlabeled p115702-962. Values are plotted as percent absorbance at 405 nm relative to wells containing biotinylated MIF or p115702-962 alone.
For measurement of MIF's immunoreactivity in an ELISA system, the two monoclonal antibody clones MAB289 (5) and 3H2F (25) were used as capture antibodies.
Cell Culture Studies
Cell lines were obtained from ATCC (Manassas, VA), with the exception of HEK293A cells, which were purchased from Invitrogen. For differentiation of THP-1 monocytes into macrophages, 50 ng/ml of phorbol 12-myristate 13-acetate (Sigma) was added for 24 h, following which the medium was changed and the cells were cultured for another additional 48 h (26).
The MIF secretion assay was performed as described previously (5). Briefly, cells first were synchronized for 3 h in medium containing 1% FCS. THP-1 monocytes or primary human PBMC-derived macrophages (1×106/ml) were stimulated with 10 μg/ml LPS 0111:B4 (Sigma) and phorbol 12-myristate 13-acetate (PMA)-differentiated THP-1 macrophages were stimulated with 0.1 μg/ml LPS. For studies of MIF inhibitors, the test compounds were added 1 h prior to LPS stimulation. Cytokine levels in the supernatants were analyzed by ELISA (eBioscience, San Diego, CA), with the exception of the MIF ELISA, which was performed as described previously (25). Unless stated otherwise, background immunoreactivity was subtracted from the stimulated cytokine level. Cell viability was verified using an LDH assay (Roche, Indianapolis, IN).
RNA Isolation and quantitative PCR
RNA was isolated using RNeasy from Qiagen (Valencia, CA). Reverse transcription was performed using StrataScript according to the manufacturer's instructions (Stratagene). For quantitative PCR, the following primers were used: MIF-forward: 5′-CGGACAGGGTCTACATCAA-3′; MIF-reverse: 5′-CTTAGGCGAAGGTGGAGTT-3′; β-actin-forward: 5′-GGATGCAGAAGGAGATCA-CTG-3′; β-actin-reverse: 5′-CGATCCACACGGAGTACT TG-3′. The amplification profile included denaturation at 95 °C for 5 min followed by annealing and elongation for 1 min at 60 °C for a total of 40 cycles. For the p115 quantitative PCR, primers for p115 and β-actin were designed and synthesized by SuperArray (Frederick, MD). The quantitative PCR profile was done according SuperArray's manual and specific product amplification was verified with a melting curve.
Co-immunoprecipitation and Western blotting
COS-7 cells were transiently transfected with a full-length p115 expression vector (pcDNA3.1/V5) using FuGENE (Roche). Cells were lysed in RIPA buffer (50 mM Tris-HCl pH 7.4,150 mM NaCl, 1% Nonident P-40, 0.5% sodium deoxycholate, 0.1% SDS, 2 mM EDTA and protease inhibitors) and then incubated with 1 μg recombinant MIF or 1 μg GST-MIF beads. Monoclonal anti-MIF antibody (R&D) was used to immunoprecipitate MIF. Precipitates were run on a 4-12% Bis-Tris NuPage gel (Invitrogen) and transferred onto PVDF membrane. The interaction between MIF and p115 was visualized using an anti-V5 antibody to detect the V5 tag on the p115 C-terminus. For endogenous co-immunoprecipitation, LNCap cell lysates were prepared using RIPA buffer and utilized as a source of endogenous p115 and MIF (27). Endogenous MIF was immunoprecipitated and protein complexes were resolved by SDS page and visualized with anti-p115 antibody (Calbiochem, CB1009).
Confocal immunofluorescence
PBMCs (2.5×106/ml) were cultured and differentiated on glass slides (BD Falcon, Bedford, MA), washed in PBS, and fixed and permeabilized using 3.7% formaldehyde and 0.1% Triton-X-100 in PBS (30 min at 37 °C). After blocking in 5% goat serum (Sigma) for 20 min, the slides were incubated overnight (at 4 °C) with anti-p115 and anti-MIF antibodies at a dilution of 1:200. After washing with PBS, the cells were stained with a mixture of Alexa488-conjugated goat anti-rabbit IgG and Alexa568-conjugated goat anti-mouse IgG (Invitrogen) at a dilution of 1:200 for 1 h at room temperature, To-Pro3 was used as nuclear dye (Invitrogen). Slides were mounted with slowfade (Invitrogen) and the cells were visualized using a confocal laser-scanning microscope (LSM510, Carl Zeiss, Thornwood, NY). To confirm that RNAi mediated knockdown of p115 did not result in fragmentation of the Golgi, THP-1 monocytes were differentiated with PMA and p115 depleted directly on culture slides. Cells then were stained with anti-p115 and anti-GM130 (Abcam, Cambridge, MA) antibodies, and Golgi structure observed by fluorescent microscopy. GM130 served as marker for the Golgi (28).
p115 RNA interference (RNAi)
The Block-it RNAi designer (Invitrogen) was used to design a short hairpin RNA molecule specific to human p115. Two constructs were designed: p115-1 from position 2625 to 2645 (5′-GCAGCTGGATTCATCTAATAG-3′) and p115-2 from position 1601 to 1620 (5′-GCAGTTGGTCCAAGGCTTAT-3′). Neither construct shares sequence homology with other genes as analyzed by BLAST search. For negative controls, shRNA constructs against lacZ were generated. Oligonucleotides were cloned into the RNA expression vector pENTR/U6 and confirmed by sequencing. shRNAs against p115 and control were transfected into human THP-1 monocytes (1×106/ml) using electroporation (Amaxa, Gaithersburg, MD) or HeLa cells (1×105/well) using lipofectamine 2000 (Invitrogen). GM130 was targeted using a RNAi construct directed against the sequence ATGAGAACATGGAGATCACC (29).
Generation and Propagation of Adenovirus
The shRNA constructs p115-2 and lacZ in the pENTR/U6 vector were recombined with the adenoviral vector pAd/BLOCK-iT-DEST (Invitrogen) using Gateway LR Clonase according to the manufacturer's protocol. The resulting p115/pAd plasmid was transfected into HEK293A and amplified following the standard procedure described in the manual. The viral titer was determined using AdenoX Rapid Titer Kit (Clontech, Palo Alto, CA). PMA-differentiated THP-1 macrophages were transfected with a multiplicity of infection (MOI) of 50 and incubated over night; cells were additionally cultivated for 96 h after infection. For infection of primary macrophages, cells were transfected with an MOI of 100 for 2 h and then cultured for additional 48 h.
Primary Cell Cultures
Human PBMCs were isolated by Ficoll-Hypaque gradient centrifugation. The cells were resuspended in RPMI 1640 medium supplemented with 20% human AB serum (Cambrex, Walkersville, MD) and plated at 2.5×106 cells per well in 24-well tissue culture plates. After 2 h of culture, the adherent cells were washed extensively with PBS and cultured for 1 week to allow differentiation into monocyte-derived macrophages (MDMs). THP-1 cells (1×106/ml) were infected with C. trachomatis serovar L1 at an MOI of 10 for 1 h at room temperature by slow rotation in RPMI medium. Cells were washed with medium and resuspended in RPMI containing 10% FBS without antibiotics and cultivated at 1×106 cells per ml in 12 well plates. The infection rate was analyzed by flow cytometry and staining with anti-Ct LPS FITC antibody (ViroStat, Portland, ME) (30). Bone marrow-derived macrophages were isolated from C57BL/6 mice as reported previously (31). For experiments, the cells were replated at a concentration of 0.5×106/ml.
Fluorescence-based Quantification of FGF export
HeLa cells were genetically modified to express FGF-2-GFP and FGF-4-GFP fusion proteins in a doxycycline-dependent manner (32). To quantify the secretion of FGF fusion proteins, cells were transfected with knockdown constructs targeting either p115, PIP-K (Temmerman et al. in press), or a control sequence. After 96 h, fusion protein expression was induced by doxycycline for 24 h. Cells were prepared for flow cytometry to detect cell surface associated FGF-2-GFP and FGF-4-GFP, respectively. Primary antibodies were decorated with APC-conjugated secondary antibodies and GFP fluorescence (intracellular and cell surface) and APC fluorescence (cell surface) were determined employing FACSCalibur flow cytometer (BD Bioscience). Untransduced HeLa cells were used to determine autofluorescence signals.
Results
Identification of p115 as an Intracellular MIF Binding Protein
To identify intracellular proteins that mediate MIF secretion, we utilized the CytoTrap yeast two-hybrid screen, which relies on the cytoplasmic rescue of a temperature-sensitive mutation in the Ras signalling pathway (33). MIF cDNA served as “bait” and a human pituitary cDNA library was the “prey” (34). After co-transformation of bait and prey plasmids into Saccharomyces cerevisia strain cdc25H, 90 colonies were identified in a primary screen. The secondary and tertiary screenings ruled out 75 colonies as false positives that showed growth in the absence of the library protein. Prey proteins in the remaining 15 colonies bound specifically to MIF (Fig. 1A). By DNA sequencing, one of the positive clones contained the carboxy-terminal 780 bases of the cDNA for p115. The remaining 14 colonies all contained the coding sequence of MIF; these may be regarded as an internal positive control because MIF forms a homotrimer (35, 36). p115 was characterized initially as a vesicle-docking protein that is localized predominantly to the cytosolic side of vesicular tubular intermediate clusters and the cis-Golgi (22, 37, 38). The carboxy-terminal region of p115 consists of four coiled-coiled domains and a short acidic tail (37) (Fig. 1B).
Fig. 1. Identification of p115 as a new binding partner of MIF.
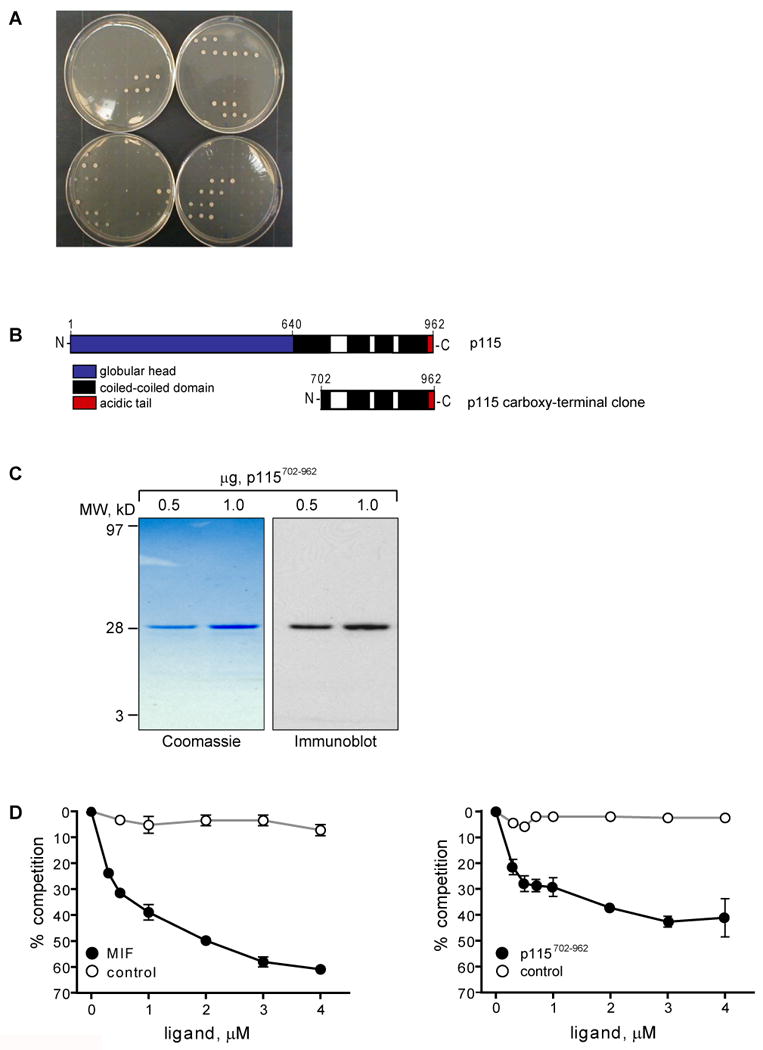
A) Tertiary screen confirming the specific binding of 15 cDNA clones with MIF. Yeast were co-transformed with the 20 putative binding partners identified in the secondary screen plus either MIF or control plasmids. Top left plate shows the positive and negative controls. Remaining plates show galactose-dependent growth of 15 MIF specific interacting clones (plated in triplicates). B) Schematic structure of p115. Top: Full length p115 with its globular head (hatched), four coiled-coiled domains (black), and an acidic tail (grey). Bottom: Carboxy-terminus of p115 (residues 702-962) that interacts with MIF. C) Purified recombinant p115702-962 revealed by SDS-PAGE followed by Coomassie staining (left panel) and Western blot (right panel). D) MIF binding to p115 demonstrated by a competitive binding assay. Left panel: MIF binds to immobilized p115702-962 in a concentration-dependent manner. Right panel: p115702-962 binds to immobilized MIF in a concentration-dependent manner. Lysozyme served as a negative control protein. Results are expressed as mean±S.D. of duplicate measurements and are representative for three independent experiments. E) Co-immunoprecipitation of MIF and p115. Upper panel: V5 epitope-tagged p115 was expressed in COS-7 cells and the cells lysed 48 h later. p115 containing protein complexes were co-precipitated with the addition of recombinant MIF and anti-MIF antibody, or after the addition of GST-MIF. p115 was visualized by Western blot using an anti-V5 antibody. Control IgG1 and GST did not co-precipitate p115, IB: immunoblot. Lower panel: Endogenous MIF–p115 complexes were co-immunoprecipitated from LNCap prostate carcinoma cells. MIF was immunoprecipitated with a monoclonal anti-MIF antibody; p115 was detected by Western blot using a polyclonal anti-p115 antibody.
In order to biochemically confirm the interaction between MIF and p115, we produced the carboxy-terminal region of p115 (p115702-962) in E. coli (Fig.1C), and evaluated its binding to MIF in an in vitro competition binding assay. As shown in Fig. 1D, increasing concentrations of MIF, but not the equimolar addition of a control protein (lysozyme), inhibited the interaction of biotinylated MIF with p115. Protein-protein interaction was verified by performing the reverse assay, i.e. measuring the binding of biotinylated p115702-962 to immobilized MIF.
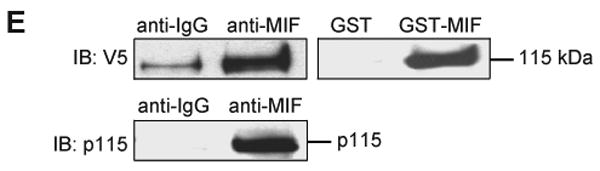
The intracellular interaction between MIF and p115 was further investigated by co-immunoprecipitation. COS-7 cells that were transfected with a V5 epitope-tagged p115 served as a source for p115. After the addition of recombinant MIF, a MIF/p115-complex was co-precipitated using an anti-MIF antibody. p115 also co-precipitated after adding GST-tagged MIF (Fig. 1E). An endogenous interaction between p115 and MIF also was observed by Western blot analysis for p115 after immunoprecipitation with anti-MIF in the prostate carcinoma cell line, LNCap (27). These results, taken together, confirm an interaction between p115 and MIF in mammalian cells.
Macrophage Activation Leads to a Redistribution of Cellular MIF
We studied the cellular distribution of MIF in macrophages in response to LPS stimulation. In unstimulated macrophages, MIF appears present throughout the cytoplasm and p115 is predominantly in the perinuclear area in a pattern that is consistent with the Golgi (Fig. 2A), which is in agreement with prior reports (22, 38). In the time course study of LPS-activated primary macrophages, MIF gradually redistributes from the cytoplasm to a plasma membrane-proximal area, and a portion of p115 now also appears dispersed in the cytoplasm. Furthermore, in the course of LPS stimulation a co-localization of MIF and p115 at the area of the Golgi and in the cytoplasm can be observed (Fig. 2B).
Fig. 2. Immunostaining of p115 and MIF in macrophages.
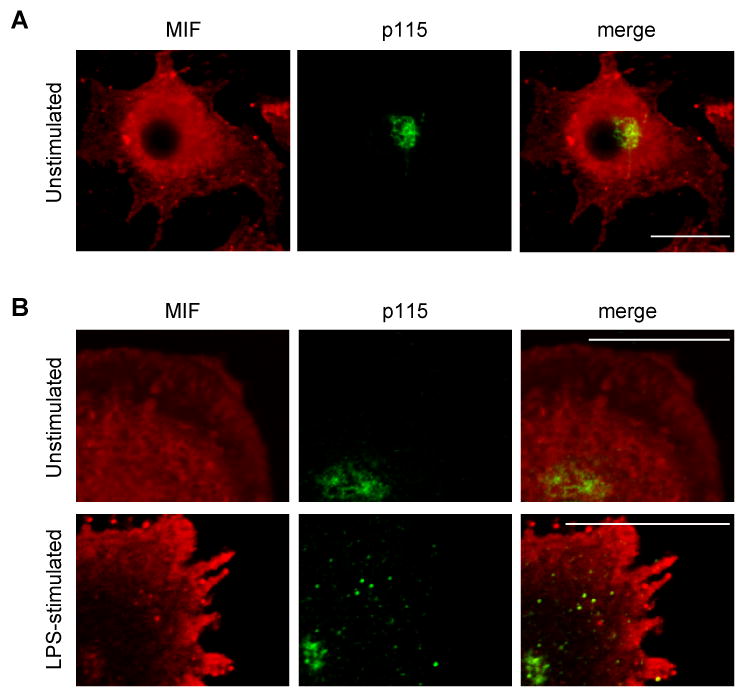
A) Freshly isolated PBMCs were plated on a four-chamber culture slide (2.5×106 cells/chamber). The cells were cultivated for seven days to allow differentiation, fixed with formaldehyde, and permeabilized with Triton-X-100. MIF (red) and p115 (green) were visualized by immunostaining with specific antibodies (1:200). The nucleus was stained using To-Pro3 (blue). B) Macrophages that were stimulated with LPS (10 μg/ml) for 0, 0.5, 1 and 4 h were fixed with formaldehyde, and permeabilized with Triton-X-100. MIF and p115 were visualized by immunostaining with specific antibodies. To-Pro3 was used as a nuclear dye. Images are representative for n=60 cells. Scale bar=10 μm
MIF Release from Human Monocytes
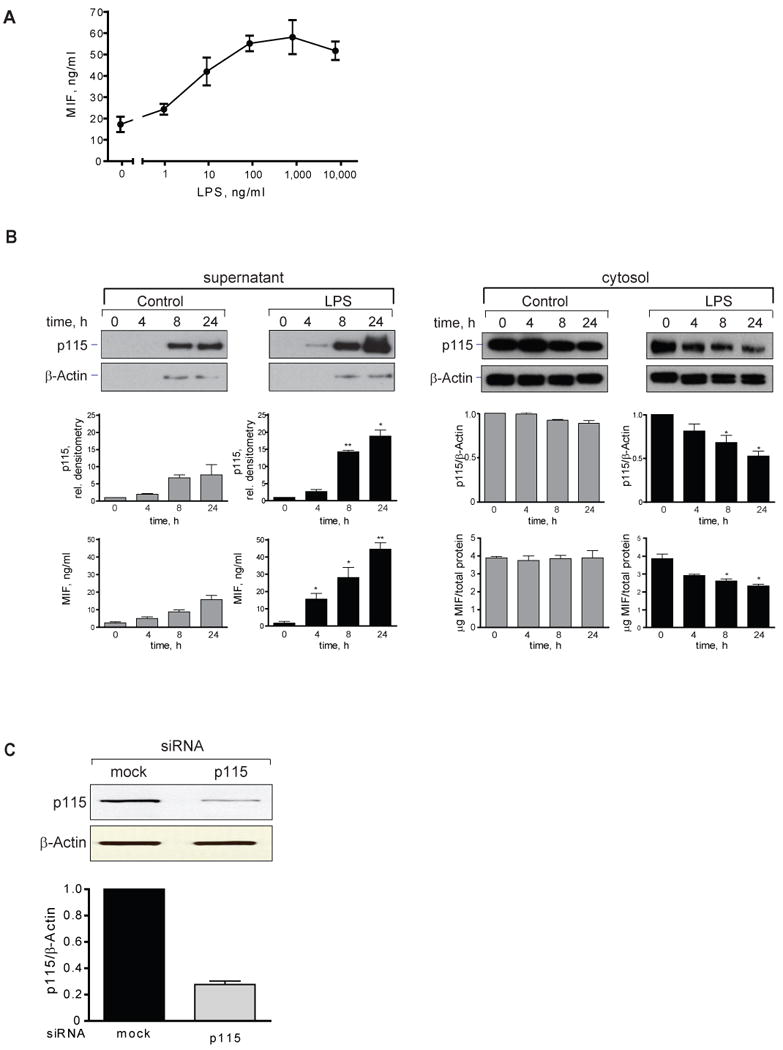
MIF is released from cells in both a regulated and a specific manner (5, 39). We stimulated human THP-1 monocytes with lipopolysaccharide (LPS) and observed an increase in the concentration of MIF in supernatants that was detectable at 2 h and reached a plateau at 4 h (Fig. 3A). The observed increase in the supernatant content of MIF was not the result of cell death as assessed by LDH analysis, and it was not accompanied by an increase in the level of MIF or p115 mRNA measured over 6 h (Fig. 3B and data not shown). These data indicate that the initial release of MIF from THP-1 monocytes occurs from preformed pools and does not require the transcription of MIF mRNA. The results are in accord with previous reports that were based on the immunostaining and in situ hybridization of murine tissues after LPS administration in vivo (2, 3).
Fig. 3. Cell stimulation increases MIF secretion without affecting MIF or p115 mRNA levels.

A) Time course of MIF secretion from LPS-stimulated, THP-1 monocytes. THP-1 cells (1×106/ml) were treated with LPS (10 μg/ml) or PBS (control) and supernatants collected at the indicated time for measurement of MIF by specific ELISA. Results are expressed as mean±S.E. of duplicate measurements from two independent experiments (n=4). P values were calculated by Student's t-test for all time points. *p<0.01 for stimulated vs. unstimulated cells. B) MIF and p115 mRNA levels in LPS-stimulated THP-1 monocytes. RNA was isolated at indicated time points and analyzed by quantitative PCR. Results are expressed as mean±S.E. of three independent experiments.
p115 is Necessary for the Release of MIF from THP-1 Monocytes
p115 plays a role in vesicle transfer from the endoplasmic reticulum to the Golgi complex (40). MIF does not enter the Golgi (5), and it was of interest to examine if p115 is necessary for the release of MIF in response to activating stimuli. We generated RNA interference (RNAi) constructs for the gene-specific silencing of p115 and tested their efficacy in human THP-1 cells. As shown in Fig. 4A, two RNAi constructs specific for p115 (p115-1, p115-2) reduced the concentration of p115 protein by 50–70%. Of importance, this level of RNAi-mediated reduction in p115 production was not associated with any detectable alteration in the morphology of the Golgi complex, as assessed by direct observation with immunofluorescent microscopy (data not shown).
Fig. 4. p115 depletion inhibits MIF secretion.
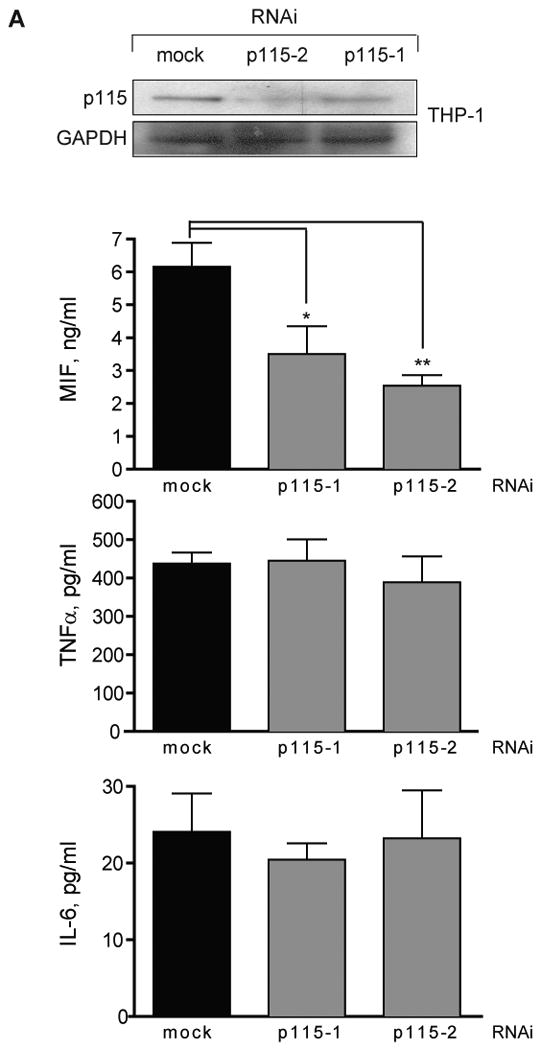
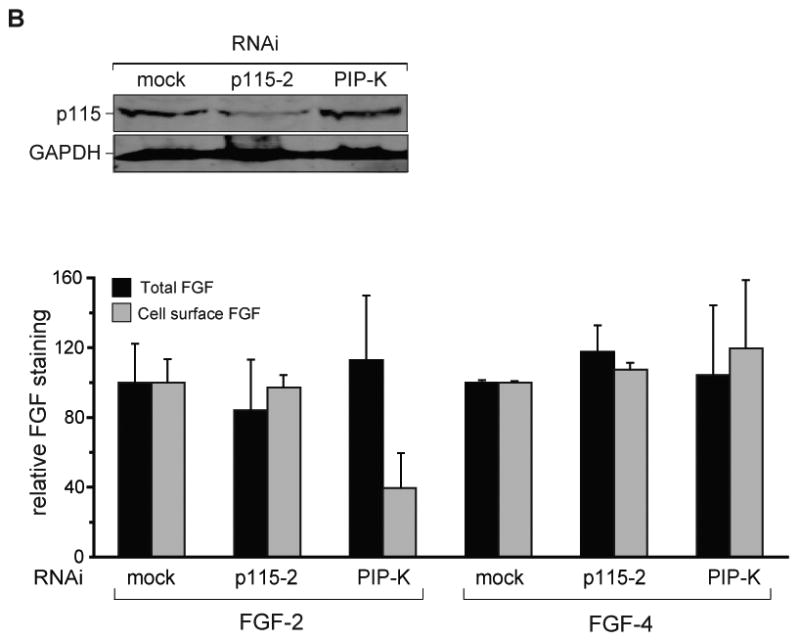
A) Upper Panel: THP-1 cells were transfected with RNAi constructs specific for p115 (p115-1, p115-2) or a control plasmid (mock) and analyzed for cellular p115 content by Western blotting. Lower Panel: THP-1 monocytes (1×106/ml) were transfected with p115 RNAi or mock RNAi plasmid and stimulated with LPS for 4 h. Supernatants were collected and analyzed for MIF, TNFα, and IL-6 by specific ELISA. Results are expressed as the mean±S.E. of four independent experiments and data are cytokine levels after subtraction of baseline. Statistical significance was determined for each p115 RNAi construct against mock siRNA by unpaired Student's t-test, *p<0.05, **p<0.01. B) Upper Panel: HeLa cells were transfected with mock, p115-2, or phosphatidylinositol phosphate kinase (PIP-K) RNAi constructs and analyzed for cellular p115 content by Western blotting. Lower Panel: FGF-GFP fusion protein production was induced by doxycycline 96 h after transfection of cells with mock, p115-2 or PIP-K RNAi. FGF-GFP fusion proteins were measured after an additional 24 h as total or cell surface fluorescence. Data are shown relative to the background level of GFP.
We next examined if p115 knockdown reduced the amount of MIF produced by LPS-stimulated THP-1 monocytes. Transfection of the p115-1 or p115-2 RNAi plasmids resulted in a 40–60% decrease in MIF release in response to LPS (Fig. 4A). Of note, the LPS-stimulated secretion of two conventionally-secreted cytokines, TNFα and IL-6, which have hydrophobic leader sequences, was not affected by a reduction in cellular p115 protein. These data point to the potential specificity of p115 in mediating MIF secretion. TNFα and IL-6 secretion also affirms that the depletion of p115 by the RNAi approach used herein does not appreciably affect the functioning of the ER/Golgi pathway. We further studied the effect of depleting GM130, which is a p115 binding partner that interacts with the carboxy-terminal acidic tail of p115 (22), but there was no measureable effect of GM130 depletion on MIF secretion (data not shown).
To gain a broader understanding of the role of p115 in unconventional protein secretion, we studied the export of the two structurally-related cytokines, FGF-2 and FGF-4. While both proteins are released from cells in response to tissue injury (41-43), FGF-4 is secreted conventionally whereas FGF-2 translocates directly across the plasma membrane (15). Using a previously established, doxycycline-inducible expression system (32), we measured both total and cell surface-associated FGF-2 or FGF-4 protein (Fig. 4B). Cellular depletion of p115 did not influence the export of either FGF-2 or FGF-4, while the secretion of FGF-2 was inhibited by depletion of phosphatidylinositol kinase (PIP-K), as previously reported (16). The finding that the release of both conventionally secreted cytokines (TNFα, IL-6, FGF-4) and the unconventionally secreted cytokine, FGF-2, is not affected by p115 depletion suggests a specific role for p115 in the stimulated export of MIF.
p115 Mediates the Release of MIF from Infected Macrophages
Human THP-1 monocytes acquire many of the characteristics of mature, primary macrophages upon differentiation with phorbol 12-myristate 13-acetate (PMA), such as an increase in the expression of the LPS co-receptor, CD14 (26). Differentiated THP-1 macrophages thus respond to low doses of LPS and show a plateau in the MIF secretion response at an LPS concentration of 0.1 μg/ml (Fig. 5A). Differentiated THP-1 macrophages also secrete ∼3 fold more MIF protein than undifferentiated THP-1 monocytes. Of note, ELISA and Western blot analyses of LPS-stimulated THP-1 macrophages showed a coordinated increase in secreted MIF and p115, and a corresponding decrease in the cellular protein levels over the observed time period of 24 h (Fig. 5B). Moreover, the supernatants of unstimulated macrophages contained only a modest amount of MIF and p115, and no apparent reduction in the cellular MIF and p115 content was measurable. These results taken together suggest that MIF and p115 are co-secreted. Further evidence that MIF and p115 are co-secreted comes from the observation that glyburide, an pharmacological inhibitor known to inhibit the secretion of MIF (5), also shows an inhibitory effect on the secretion of p115 (data not shown). Of note, glyburide is also known to inhibit the release of other unconventionally secreted proteins (44, 45).
Fig. 5. MIF and p115 are secreted from differentiated THP-1 macrophages.
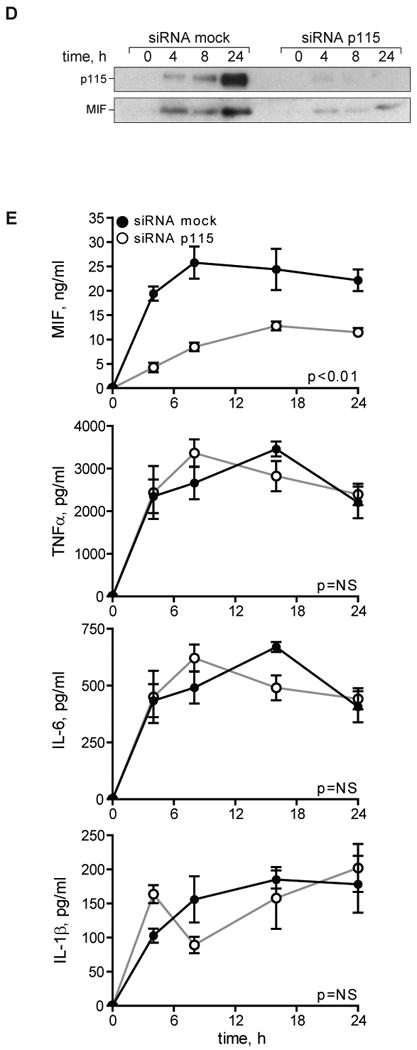
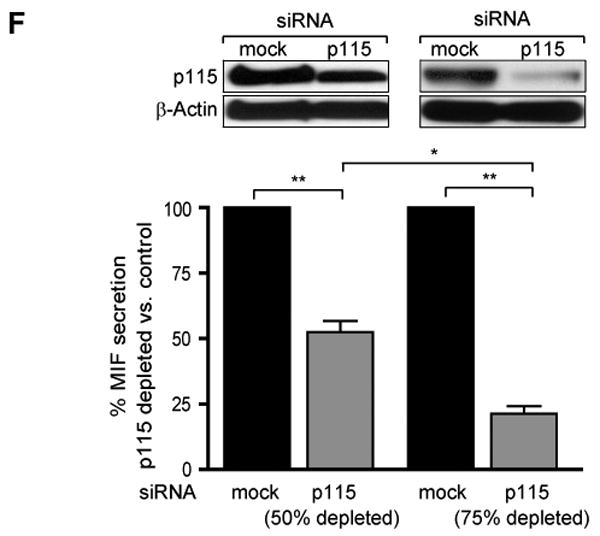
A) Dose-dependent secretion of MIF from LPS-stimulated THP-1 macrophages. Cells (1×106/ml) were stimulated with the indicated dose of LPS for 4 h and the supernatants assayed for MIF content by ELISA. Results are expressed as mean±S.D. of duplicate experiments, each measured in duplicate (n=4). B) Left Panel: Differentiated THP-1 macrophages were stimulated with 0.1 μg/ml LPS or PBS (control) and the supernatants were collected at the indicated time points. MIF concentration was measured by ELISA; p115 release was determined by immunoblotting and subsequent densitometric analysis. Right Panel: Cell lysates of THP-1 macrophages stimulated with LPS or control (PBS) were prepared at the indicated time points. Intracellular MIF concentration was measured by ELISA, and intracellular p115 level was determined by Western blot and densitometric analysis. The ELISA results are expressed as mean values ±S.D. of two independent experiments measured in duplicate. Densitometric results are expressed as mean values ±S.D. of at least two independent experiments. Statistical significance was determined by Student's t-test comparing LPS-stimulated vs. control time point, *p<0.05, **p<0.01. C) Differentiated THP-1 macrophages were transfected with an MOI of 50 of adenovirus encoding siRNA against p115 or a mock control siRNA. 96 h after transfection, the cells were lysed and the protein content analyzed by Western blot and densitometry. The results are expressed as mean values ±S.E. of three independent experiments, statistical significance was determined by Student's t-test, *p<0.001. D) LPS-stimulated secretion of MIF in p115 depleted or mock-treated THP-1 macrophages. Supernatants were collected at the indicated time points and analyzed for export of MIF and p115 by Western blot. E) Time course of LPS-stimulated MIF secretion in control or p115 siRNA treated THP-1 macrophages. Supernatants were collected at the indicated time points and MIF content was measured by ELISA. Supernatants also were assayed for TNFα, IL-6 and IL-1β by ELISA. The results are expressed as mean values ±S.E. of triplicate measurements of two independent experiments (n=6). Data are cytokine levels after subtraction of baseline level. Statistical significance was determined by comparing the curves obtained in p115 knockdown vs. mock control by two-way ANOVA analysis. F) Dose-dependent effect of p115 depletion on MIF secretion. Differentiated THP-1 macrophages were transfected with 50 MOI of adenovirus encoding siRNA against p115 or mock control siRNA. 48 h after transfection (50% depletion of p115 compared to mock treated cells) and 96 h after transfection (75% depletion of p115 compared to mock treated cells), the cells were stimulated for 4 h with 0.1 μg/ml LPS. The supernatants were analyzed for MIF release by ELISA, and data are shown relative to mock treated cells. Results are representative for two independent experiments measured in duplicates. Statistical significance was determined by Student's t-test, *p<0.05, **p<0.01.
We utilized an adenovirus construct to deliver p115 siRNA to differentiated THP-1 macrophages and observed that adenoviral transfection reduced significantly intracellular p115 protein levels (Fig. 5C). The transfection of adenovirus encoding p115 siRNA, but not a control adenovirus, significantly reduced LPS-stimulated MIF release over the 24 h experimental period as demonstrated by Western blot analysis (Fig. 5D) and ELISA (Fig. 5E). Of note, the decrease in MIF secretion in p115 depleted cells is not due to cell death or permeablization as assessed by LDH assay. LDH levels were measured over the course of the experiment; LDH concentration never exceeded 14 mU/ml (<5% total cellular LDH), and no statistical differences between mock-treated and p115-depleted cells were detectable. By contrast, the release of TNFα, IL-6, or IL-1β was not affected by adenovirus encoded p115 siRNA, confirming that partial p115 reduction did not interfere with the functional integrity of the Golgi apparatus (Fig. 5E). The inhibition of MIF secretion also appeared to be dose-dependent with respect to the level of p115 depletion (Fig. 5F). When compared to controls, a ∼50% depletion of p115 was associated with a ∼50% inhibition of MIF release, while a ∼75% depletion of p115 was associated with a ∼75% inhibition of MIF release. Adenoviral-mediated delivery of p115 siRNA also reduced p115 protein production in human peripheral blood monocyte-derived macrophages (Fig. 6A), and reduced by almost 80% MIF release after stimulation with LPS (Fig. 6B). Finally, the LPS stimulation of bone-marrow derived macrophages leads to the export of both MIF and p115 protein (Fig. 6C), thus affirming the results obtained from THP-1 macrophages.
Fig. 6. MIF and p115 are secreted from primary macrophages.
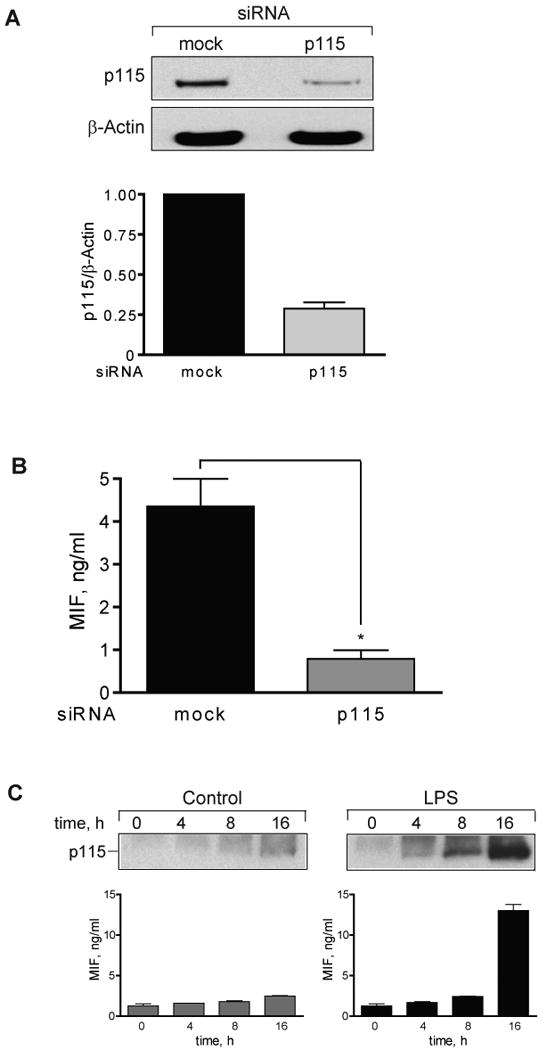
A) Adenovirus-mediated depletion of p115 reduces p115 protein levels in primary human macrophages. Human peripheral blood monocyte-derived macrophages (1×106/ml) were transfected with 100 MOI of adenovirus carrying siRNA against p115 or a mock control. The cells then were analyzed 48 h later for p115 content by Western blot. Knockdown of p115 was evaluated by densitometry. The results are expressed as mean values ±S.D. of three independent experiments, statistical significance was determined by Student's t-test, *p<0.001. B) LPS-stimulated MIF secretion is reduced in p115 siRNA, but not mock siRNA treated primary macrophages. MIF secretion was stimulated with 10 μg/ml LPS for 4 h and the supernatants analyzed for MIF content by ELISA. The results are expressed as mean values ±S.D. of two independent experiments each measured in duplicates (n=4). Data are MIF level after subtraction of baseline. Statistical significance was determined by an unpaired Student's t-test, *p<0.001. C) Bone-marrow derived primary macrophages (0.5×106/ml) were stimulated with 1 μg/ml LPS or control (PBS) and the supernatants collected at the indicated time points. MIF concentration was measured by ELISA and p115 content was determined by immunoblotting. Results are representative of three independent experiments. Statistical significance was determined by Student's t-test comparing LPS-stimulated vs. control time point, *p<0.01.
Chlamydiae are gram-negative, obligate intracellular bacteria that infect and replicate within monocytes/macrophages (46, 47). Infection of cultured THP-1 monocytes with C. trachomatis induces the release of pro-inflammatory cytokines (48) including MIF, which was most evident at 48 h (Fig. 7A). We studied the effect of C. trachomatis infection on MIF production in differentiated THP-1 macrophages that had been pre-treated with adenovirus encoding either a mock control or a p115 siRNA. p115 depletion had no effect on the infection or replication rate of organisms in macrophages, as measured by flow cytometry for the C. trachomatis antigen lipopolysaccharide (24 h post infection: mock siRNA: 38±5% versus p115 siRNA: 39±8%, P=NS, data not shown). Nevertheless, macrophages treated with p115 siRNA showed a significant decrease in MIF secretion after C. trachomatis infection. By contrast, the secretion of TNFα, which was evident at 8 h after infection, was unaffected by p115 siRNA (Fig. 7B). These data indicate that cellular p115 plays an important role in mediating the secretion of MIF by macrophages in response both to a defined microbial ligand such as LPS, as well as to live infection by an intracellular pathogen such as C. trachomatis.
Fig. 7. p115 knockdown inhibits MIF secretion in Chlamydia trachomatis infected human monocytes.

A) THP-1 monocytes (1×106/ml) were infected with 10 MOI of C. trachomatis. Supernatants were collected at the indicated time points and analyzed for MIF release by ELISA. B) Differentiated THP-1 macrophages were treated with 50 MOI of adenovirus encoding a p115 or a control (mock) siRNA 96 h prior to infection with C. trachomatis. Supernatants were collected at the indicated time points and the levels of MIF and TNFα were analyzed by ELISA. Results are expressed as mean±S.E. of one experiment measured in triplicates, and the data are total cytokine levels. Results are representative of three independent experiments. Statistical significance was analyzed by an unpaired Student's t-test, *p<0.01.
The Small Molecule Inhibitor, 4-iodo-6-phenylpyrimidine (4-IPP), Inhibits MIF Secretion by Targeting the MIF/p115 Interaction
Prototypic small molecules have been identified that bind to the MIF's amino-terminal, tautomerase region (24, 49). Mitchell and colleagues recently reported on a suicide substrate, 4-iodo-6-phenylpyrimidine (4-IPP). 4-IPP was identified by a computational virtual screening for MIF antagonists, and co-crystallization of MIF and 4-IPP demonstrated that it covalently modifies the MIF amino-terminus and inhibits MIF-dependent cell motility and growth in vitro (23). It was of interest to investigate whether 4-IPP, which is cell-permeable, inhibits the stimulated release of MIF. Because of prior studies showing that the covalent modification of MIF by a small molecule may affect its immunorecognition in a two-antibody, sandwich ELISA (24), we first determined whether 4-IPP influences the detection of MIF by ELISA. Indeed, MIF that had been incubated with 4-IPP showed increased immunorecognition by a MIF-specific monoclonal antibody (clone MAB289) when compared to native MIF (Fig. 8A). By contrast, when analyzing 4-IPP-modified MIF with a different anti-MIF mAb (clone 3H2F), a diminished recognition compared to native MIF was observed (>2 fold) (data not shown). We concluded that 4-IPP significantly alters the native conformation of the MIF protein, and we therefore employed Western blotting for analyzing the effect of 4-IPP on MIF secretion. 4-IPP modification did not affect MIF detection by Western blot (data not shown).
Fig. 8. The small molecule inhibitor 4-IPP inhibits MIF release by targeting the MIF/p115 interaction.
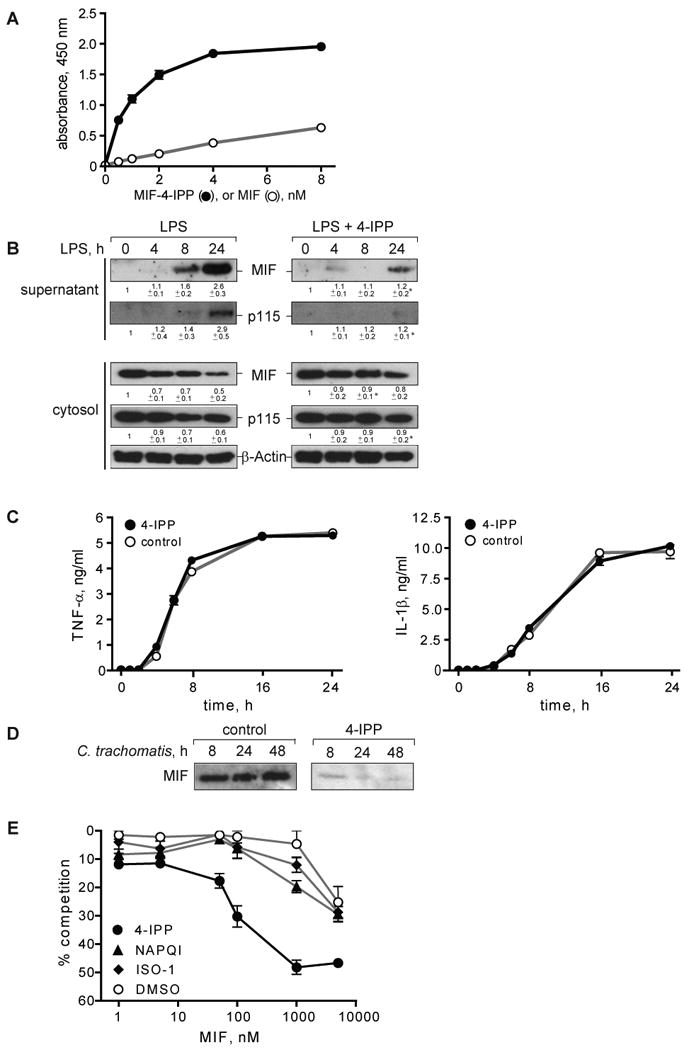
A) Quantification of 4-IPP-modified MIF (MIF-4-IPP) or unmodified MIF (MIF) by ELISA. MIF-4-IPP shows enhanced detection by a monoclonal anti-MIF antibody (MAB289). B) Macrophages (1×106/ml) were pre-treated for 1 h with increasing concentrations of 4-IPP (0–2 μM) and MIF secretion was stimulated with LPS for 4 h. Supernatants were collected and MIF concentration was determined by Western blotting. Western blots were quantified by densitometry and percent of inhibition was calculated compared to control. Results are expressed as mean±S.E. of three independent experiments. C) Unstimulated THP-1 macrophages were treated with 1 μM of 4-IPP or control (DMSO) and supernatants were collected after 16 h. The MIF concentration was measured by Western blotting and densitometric analysis. Results are representative of three independent experiments. Statistical significance for the comparison of 4-IPP vs. DMSO was analyzed by an unpaired Student's t-test, *p<0.05. D) THP-1 macrophages (1×106/ml) were pre-treated with 1 μM of 4-IPP for 1 h prior to stimulation with 0.1 μg/ml LPS, and MIF and p115 was measured in supernatants and cell lysates by Western blotting. Western blots were quantified by densitometry relative to β-actin. Mean±S.E. of at least two independent experiments are stated below the blots. Statistical significance for the comparison of LPS + 4-IPP vs. LPS alone was analyzed by an unpaired Student's t-test, *p<0.05. Control studies showed that 4-IPP did not compromise cell viability, as assessed by LDH assay (not shown). E) Supernatants of LPS-stimulated THP-1 macrophages that had been treated with 4-IPP or control analyzed for the release of TNFα and IL-1β by ELISA. The results are expressed as ±S.D. of one experiment measured in duplicates. Data are representative of two independent experiments. F) THP-1 macrophages were infected with an MOI of 10 of C. trachomatis and treated after infection with 1 μM of 4-IPP or control over the time of infection. Supernatants were collected at the indicated time points and analyzed for MIF content by Western blot. Results are representative for three experiments. G) Binding of biotinylated MIF to immobilized p115702-962 in the presence of increasing concentrations of MIF pre-incubated with the small molecule MIF inhibitors 4-IPP, NAPQI, ISO-1, or solvent control. Results are expressed as mean±S.E. of one experiment measured in duplicates. The data are representative of three independent experiments.
We found that increasing concentrations of 4-IPP dose-dependently block the release of MIF, with 1 μM of 4-IPP (10-fold molar excess) resulting in ∼80% inhibition of MIF secretion (Fig. 8B). 4-IPP also inhibits the basal secretion of MIF (Fig. 8C). Of note, 4-IPP effectively inhibits the release of both MIF and p115 from LPS-stimulated THP-1 macrophages, and correspondingly, the cytoplasmic levels of MIF and p115 did not decrease when LPS-stimulated cells were treated with 4-IPP (Fig. 8D). 4-IPP did not affect the release of TNFα and IL-1β (Fig. 8E), while reducing the secretion of MIF from monocytes infected with C. trachomatis (Fig. 8F). We furthermore studied the influence of the reversible MIF binding molecule (S,R)-3-(4-hydroxyphenyl)-4,5-dihydro-5-isoxazole acetic acid methyl ester (ISO-1) (49) on MIF secretion from stimulated THP-1 macrophages but observed no effect (data not shown). The observation that 4-IPP enhanced the recognition of MIF by an anti-MIF mAb (Fig. 8A) suggests that 4-IPP imparts a significant conformational affect on MIF that may also influence its binding to p115. We tested this possibility by examining the effect of 4-IPP on the competitive binding of MIF to p115 in vitro. As shown in Fig. 8G, 4-IPP, but not the small molecules ISO-1 or NAPQI (24, 49) increased MIF binding to p115. The enhanced binding of MIF to p115 in the presence of 4-IPP thus may play a role in the inhibitory action of 4-IPP on MIF release from cells, perhaps by interfering with additional protein interactions necessary for secretion.
Discussion
MIF plays an important upstream role in the regulation of diverse cellular responses (39, 50-52) and its role in human pathology has been emphasized by the finding that high expression MIF alleles are associated with the incidence or the severity of inflammatory and oncologic diseases (25, 53-56). The precise mechanism underlying the secretion of MIF, which lacks an N-terminal signal sequence, has remained enigmatic (34, 57). MIF has been reported to be associated with vesicles and exosomes (18, 19), and its release from stimulated monocytes is reduced by glyburide, which inhibits ABC transporter-dependent secretion (5).
We utilized a yeast two-hybrid approach to identify intracellular proteins that interact with MIF. The specificity of our screen was confirmed by the finding that 93% of the interacting clones were MIF itself, which associates into a non-covalently associated homotrimer (35, 36). However, we also identified a cDNA clone encoding the carboxyl terminal 260 amino acids of the Golgi-associated protein, p115. Identification of p115 as an interacting partner for MIF was confirmed by in vitro binding assays and endogenous co-immunoprecipitation. The competition binding assay employing recombinant MIF and p115 protein suggests that no other proteins are necessary for the binding of the two proteins in vitro. Nevertheless, the competition curve levels at 50–60% indicating that additional protein interactions may occur in vivo.
The immunofluorescent microscopy studies show that MIF is present in large cytoplasmic pools which disperse towards the periphery upon inflammatory stimulation. Similarly, p115 is also partially redistributed upon LPS stimulation from the Golgi towards the plasma membrane. It remains possible that LPS stimulation induces additional, unknown proteins, which in turn might be necessary for the interaction between MIF and p115 and its export out of the cell.
Because the cellular depletion of p115 reduces stimulated MIF export, p115 may be essential for the transport of MIF from the perinuclear ring to the plasma membrane and then out of the cell. This pathway for release also is supported by the data demonstrating the accumulation of MIF and p115 in the supernatants of stimulated cells.
The partial depletion of p115 by the RNAi technique used in this study neither affected the structural morphology of the Golgi nor reduced the export of ER-dependent proteins such as TNFα, IL-6, and FGF-4. The release of the unconventionally secreted protein, IL-1β, which occurs by vesicles (13), and FGF-2, which translocates across the plasma membrane (15), also were not affected by p115 depletion. That p115 is necessary for the release of MIF but not other cytokines, whether conventionally or unconventionally secreted, suggests that p115 has a specific role in MIF export. It also is possible that p115 mediates a generalized secretory response by activated monocytes/macrophages that results in the release of a p115 macromolecular complex containing numerous proteins, of which MIF is one. Further elucidation of both the signals and the protein-protein interactions underlying this export pathway may be informative and enhance our understanding of the acute secretory response of activated monocytes/macrophages.
That MIF is co-secreted with p115 was affirmed by the finding that the small molecule MIF inhibitor, 4-IPP, reduces the release of both proteins from monocytes/macrophages. These data identify 4-IPP as a potentially selective inhibitor of the p115/MIF secretory pathway. 4-IPP covalently modifies the MIF amino-terminus (23), and it alters the protein's conformational integrity so as to increase its binding interaction with p115. These results indicate that a “native” MIF/p115 interaction is essential for the efficient release of MIF from cells. A non-native MIF/p115 interaction may disrupt trafficking and downstream protein interactions that are necessary for efficient MIF release. 4-IPP also provides proof-of-concept for the possibility of reducing MIF-dependent responses in the earliest phase of cell activation by interfering with MIF's cytoplasmic release. Inhibitors such as 4-IPP may be attractive in clinical application in those settings, such as severe inflammation, in which rapid intervention is desired to prevent the initiation of a tissue-damaging, cytokine cascade (58). We note that our findings do not exclude additional actions for 4-IPP with respect to inhibiting MIF binding with its cell surface receptor or with other effector proteins. Whether p115 is involved exclusively in the stimulated export of MIF or also plays a role in the constitutive release of MIF under basal conditions remains to be answered. The observation that 4-IPP inhibits MIF secretion under basal and stimulated conditions indicates a role for p115 in the constitutive release, but we were unable to observe a significant influence of genetic p115 depletion on MIF release under non-stimulated conditions.
In summary, these data describe a novel function for the Golgi-associated protein p115 in the mediation of MIF unconventional secretion. Pharmacological targeting of the MIF/p115 interaction may offer a powerful approach for inhibiting innate immune responses by interfering with the secretion of the upstream cytokine, MIF.
Acknowledgments
We are grateful to Jason Griffith and Utako Kaneyuki (Yale University School of Medicine) for helpful discussions.
Funding: This work was supported by the NIH (LL, RB, PBK), DFG grant SFB542/A7 (JB), and fellowships from the ‘Studienstiftung des deutschen Volkes’ to Melanie Merk, and the German Academic Exchange Service (DAAD) to Swen Zierow.
Abbreviations
- FGF
fibroblast growth factor
- 4-IPP
4-iodophenylpyrimidine
- ISO-1
(S,R)-3-(4-hydroxyphenyl)-4,5-dihydro-5-isoxazole acetic acid methyl ester
- LPS
lipopolysaccharide
- MDM
monocyte-derived macrophages
- MOI
multiplicity of infection
- NAPQI
N-acetyl-p-benzoquinone imine
- PBMCs
peripheral blood mononuclear cells
- PIP-K
phosphatidylinositol phosphate kinase
- siRNA
short interfering RNA
Footnotes
Competing Interests Statement The University of Louisville has applied for a patent describing the potential therapeutic value of the MIF inhibitor, 4-IPP.
References
- 1.Calandra T, Roger T. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol. 2003;3:791–800. doi: 10.1038/nri1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bacher M, Meinhardt A, Lan HY, Mu W, Metz CN, Chesney JA, Calandra T, Gemsa D, Donnelly T, Atkins RC, Bucala R. Migration inhibitory factor expression in experimentally induced endotoxemia. Am J Pathol. 1997;150:235–246. [PMC free article] [PubMed] [Google Scholar]
- 3.Fingerle-Rowson G, Koch P, Bikoff R, Lin X, Metz CN, Dhabhar FS, Meinhardt A, Bucala R. Regulation of macrophage migration inhibitory factor expression by glucocorticoids in vivo. Am J Pathol. 2003;162:47–56. doi: 10.1016/S0002-9440(10)63797-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bernhagen J, Mitchell RA, Calandra T, Voelter W, Cerami A, Bucala R. Purification, bioactivity, and secondary structure analysis of mouse and human macrophage migration inhibitory factor (MIF) Biochemistry. 1994;33:14144–14155. doi: 10.1021/bi00251a025. [DOI] [PubMed] [Google Scholar]
- 5.Flieger O, Engling A, Bucala R, Lue H, Nickel W, Bernhagen J. Regulated secretion of macrophage migration inhibitory factor is mediated by a non-classical pathway involving an ABC transporter. FEBS Lett. 2003;551:78–86. doi: 10.1016/s0014-5793(03)00900-1. [DOI] [PubMed] [Google Scholar]
- 6.Walter P, Gilmore R, Blobel G. Protein translocation across the endoplasmic reticulum. Cell. 1984;38:5–8. doi: 10.1016/0092-8674(84)90520-8. [DOI] [PubMed] [Google Scholar]
- 7.Rapoport TA, Jungnickel B, Kutay U. Protein transport across the eukaryotic endoplasmic reticulum and bacterial inner membranes. Annu Rev Biochem. 1996;65:271–303. doi: 10.1146/annurev.bi.65.070196.001415. [DOI] [PubMed] [Google Scholar]
- 8.Derby MC, Gleeson PA. New insights into membrane trafficking and protein sorting. Int Rev Cytol. 2007;261:47–116. doi: 10.1016/S0074-7696(07)61002-X. [DOI] [PubMed] [Google Scholar]
- 9.Nickel W. Unconventional secretory routes: direct protein export across the plasma membrane of mammalian cells. Traffic. 2005;6:607–614. doi: 10.1111/j.1600-0854.2005.00302.x. [DOI] [PubMed] [Google Scholar]
- 10.Nickel W. Unconventional secretion: an extracellular trap for export of fibroblast growth factor 2. J Cell Sci. 2007;120:2295–2299. doi: 10.1242/jcs.011080. [DOI] [PubMed] [Google Scholar]
- 11.Cleves AE. Protein transports: the nonclassical ins and outs. Curr Biol. 1997;7:R318–320. doi: 10.1016/s0960-9822(06)00148-5. [DOI] [PubMed] [Google Scholar]
- 12.MacKenzie A, Wilson HL, Kiss-Toth E, Dower SK, North RA, Surprenant A. Rapid secretion of interleukin-1beta by microvesicle shedding. Immunity. 2001;15:825–835. doi: 10.1016/s1074-7613(01)00229-1. [DOI] [PubMed] [Google Scholar]
- 13.Andrei C, Dazzi C, Lotti L, Torrisi MR, Chimini G, Rubartelli A. The secretory route of the leaderless protein interleukin 1beta involves exocytosis of endolysosome-related vesicles. Mol Biol Cell. 1999;10:1463–1475. doi: 10.1091/mbc.10.5.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zehe C, Engling A, Wegehingel S, Schafer T, Nickel W. Cell-surface heparan sulfate proteoglycans are essential components of the unconventional export machinery of FGF-2. Proc Natl Acad Sci U S A. 2006;103:15479–15484. doi: 10.1073/pnas.0605997103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schafer T, Zentgraf H, Zehe C, Brugger B, Bernhagen J, Nickel W. Unconventional secretion of fibroblast growth factor 2 is mediated by direct translocation across the plasma membrane of mammalian cells. J Biol Chem. 2004;279:6244–6251. doi: 10.1074/jbc.M310500200. [DOI] [PubMed] [Google Scholar]
- 16.Temmerman K, Ebert AD, Muller HM, Sinning I, Tews I, Nickel W. A direct role for phosphatidylinositol-4,5-bisphosphate in unconventional secretion of fibroblast growth factor 2. Traffic. 2008;9:1204–1217. doi: 10.1111/j.1600-0854.2008.00749.x. [DOI] [PubMed] [Google Scholar]
- 17.Nickel W, Seedorf M. Unconventional Mechanisms of Protein Transport to the Cell Surface of Eukaryotic Cells. Annu Rev Cell Dev Biol. 2008;24 doi: 10.1146/annurev.cellbio.24.110707.175320. [DOI] [PubMed] [Google Scholar]
- 18.Nishino T, Bernhagen J, Shiiki H, Calandra T, Dohi K, Bucala R. Localization of macrophage migration inhibitory factor (MIF) to secretory granules within the corticotrophic and thyrotrophic cells of the pituitary gland. Mol Med. 1995;1:781–788. [PMC free article] [PubMed] [Google Scholar]
- 19.Eickhoff R, Wilhelm B, Renneberg H, Wennemuth G, Bacher M, Linder D, Bucala R, Seitz J, Meinhardt A. Purification and characterization of macrophage migration inhibitory factor as a secretory protein from rat epididymis: evidences for alternative release and transfer to spermatozoa. Mol Med. 2001;7:27–35. [PMC free article] [PubMed] [Google Scholar]
- 20.Barrett-Jolley R, McPherson GA. Characterization of K(ATP) channels in intact mammalian skeletal muscle fibres. Br J Pharmacol. 1998;123:1103–1110. doi: 10.1038/sj.bjp.0701727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wasada T. Adenosine triphosphate-sensitive potassium (K(ATP)) channel activity is coupled with insulin resistance in obesity and type 2 diabetes mellitus. Intern Med. 2002;41:84–90. doi: 10.2169/internalmedicine.41.84. [DOI] [PubMed] [Google Scholar]
- 22.Nelson DS, Alvarez C, Gao YS, Garcia-Mata R, Fialkowski E, Sztul E. The membrane transport factor TAP/p115 cycles between the Golgi and earlier secretory compartments and contains distinct domains required for its localization and function. J Cell Biol. 1998;143:319–331. doi: 10.1083/jcb.143.2.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Winner M, Meier J, Zierow S, Rendon BE, Crichlow GV, Riggs R, Bucala R, Leng L, Smith N, Lolis E, Trent JO, Mitchell RA. A Novel, Macrophage Migration Inhibitory Factor Suicide Substrate Inhibits Motility and Growth of Lung Cancer Cells. Cancer Res. 2008;68:7253–7257. doi: 10.1158/0008-5472.CAN-07-6227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Senter PD, Al-Abed Y, Metz CN, Benigni F, Mitchell RA, Chesney J, Han J, Gartner CG, Nelson SD, Todaro GJ, Bucala R. Inhibition of macrophage migration inhibitory factor (MIF) tautomerase and biological activities by acetaminophen metabolites. Proc Natl Acad Sci U S A. 2002;99:144–149. doi: 10.1073/pnas.011569399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mizue Y, Ghani S, Leng L, McDonald C, Kong P, Baugh J, Lane SJ, Craft J, Nishihira J, Donnelly SC, Zhu Z, Bucala R. Role for macrophage migration inhibitory factor in asthma. Proc Natl Acad Sci U S A. 2005;102:14410–14415. doi: 10.1073/pnas.0507189102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park EK, Jung HS, Yang HI, Yoo MC, Kim C, Kim KS. Optimized THP-1 differentiation is required for the detection of responses to weak stimuli. Inflamm Res. 2007;56:45–50. doi: 10.1007/s00011-007-6115-5. [DOI] [PubMed] [Google Scholar]
- 27.Meyer-Siegler KL, Iczkowski KA, Leng L, Bucala R, Vera PL. Inhibition of macrophage migration inhibitory factor or its receptor (CD74) attenuates growth and invasion of DU-145 prostate cancer cells. J Immunol. 2006;177:8730–8739. doi: 10.4049/jimmunol.177.12.8730. [DOI] [PubMed] [Google Scholar]
- 28.Kodani A, Sutterlin C. The Golgi Protein GM130 Regulates Centrosome Morphology and Function. Mol Biol Cell. 2008;19:745–753. doi: 10.1091/mbc.E07-08-0847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Puthenveedu MA, Bachert C, Puri S, Lanni F, Linstedt AD. GM130 and GRASP65-dependent lateral cisternal fusion allows uniform Golgi-enzyme distribution. Nat Cell Biol. 2006;8:238–248. doi: 10.1038/ncb1366. [DOI] [PubMed] [Google Scholar]
- 30.Leonhardt RM, Lee SJ, Kavathas PB, Cresswell P. Severe tryptophan starvation blocks onset of conventional persistence and reduces reactivation of Chlamydia trachomatis. Infect Immun. 2007;75:5105–5117. doi: 10.1128/IAI.00668-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, Muramatsu S, Steinman RM. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176:1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Engling A, Backhaus R, Stegmayer C, Zehe C, Seelenmeyer C, Kehlenbach A, Schwappach B, Wegehingel S, Nickel W. Biosynthetic FGF-2 is targeted to non-lipid raft microdomains following translocation to the extracellular surface of CHO cells. J Cell Sci. 2002;115:3619–3631. doi: 10.1242/jcs.00036. [DOI] [PubMed] [Google Scholar]
- 33.Aronheim A, Zandi E, Hennemann H, Elledge SJ, Karin M. Isolation of an AP-1 repressor by a novel method for detecting protein-protein interactions. Mol Cell Biol. 1997;17:3094–3102. doi: 10.1128/mcb.17.6.3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bernhagen J, Calandra T, Mitchell RA, Martin SB, Tracey KJ, Voelter W, Manogue KR, Cerami A, Bucala R. MIF is a pituitary-derived cytokine that potentiates lethal endotoxaemia. Nature. 1993;365:756–759. doi: 10.1038/365756a0. [DOI] [PubMed] [Google Scholar]
- 35.Suzuki M, Sugimoto H, Nakagawa A, Tanaka I, Nishihira J, Sakai M. Crystal structure of the macrophage migration inhibitory factor from rat liver. Nat Struct Biol. 1996;3:259–266. doi: 10.1038/nsb0396-259. [DOI] [PubMed] [Google Scholar]
- 36.Sun HW, Bernhagen J, Bucala R, Lolis E. Crystal structure at 2.6-A resolution of human macrophage migration inhibitory factor. Proc Natl Acad Sci U S A. 1996;93:5191–5196. doi: 10.1073/pnas.93.11.5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sapperstein SK, Walter DM, Grosvenor AR, Heuser JE, Waters MG. p115 is a general vesicular transport factor related to the yeast endoplasmic reticulum to Golgi transport factor Uso1p. Proc Natl Acad Sci U S A. 1995;92:522–526. doi: 10.1073/pnas.92.2.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakamura N, Lowe M, Levine TP, Rabouille C, Warren G. The vesicle docking protein p115 binds GM130, a cis-Golgi matrix protein, in a mitotically regulated manner. Cell. 1997;89:445–455. doi: 10.1016/s0092-8674(00)80225-1. [DOI] [PubMed] [Google Scholar]
- 39.Calandra T, Bernhagen J, Mitchell RA, Bucala R. The macrophage is an important and previously unrecognized source of macrophage migration inhibitory factor. J Exp Med. 1994;179:1895–1902. doi: 10.1084/jem.179.6.1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alvarez C, Fujita H, Hubbard A, Sztul E. ER to Golgi transport: Requirement for p115 at a pre-Golgi VTC stage. J Cell Biol. 1999;147:1205–1222. doi: 10.1083/jcb.147.6.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bikfalvi A, Klein S, Pintucci G, Rifkin DB. Biological roles of fibroblast growth factor-2. Endocr Rev. 1997;18:26–45. doi: 10.1210/edrv.18.1.0292. [DOI] [PubMed] [Google Scholar]
- 42.Nugent MA, Iozzo RV. Fibroblast growth factor-2. Int J Biochem Cell Biol. 2000;32:115–120. doi: 10.1016/s1357-2725(99)00123-5. [DOI] [PubMed] [Google Scholar]
- 43.Powers CJ, McLeskey SW, Wellstein A. Fibroblast growth factors, their receptors and signaling. Endocr Relat Cancer. 2000;7:165–197. doi: 10.1677/erc.0.0070165. [DOI] [PubMed] [Google Scholar]
- 44.Hamon Y, Luciani MF, Becq F, Verrier B, Rubartelli A, Chimini G. Interleukin-1beta secretion is impaired by inhibitors of the Atp binding cassette transporter, ABC1. Blood. 1997;90:2911–2915. [PubMed] [Google Scholar]
- 45.Omer S, Meredith D, Morris JF, Christian HC. Evidence for the role of adenosine 5′-triphosphate-binding cassette (ABC)-A1 in the externalization of annexin 1 from pituitary folliculostellate cells and ABCA1-transfected cell models. Endocrinology. 2006;147:3219–3227. doi: 10.1210/en.2006-0099. [DOI] [PubMed] [Google Scholar]
- 46.Prebeck S, Brade H, Kirschning CJ, da Costa CP, Durr S, Wagner H, Miethke T. The Gram-negative bacterium Chlamydia trachomatis L2 stimulates tumor necrosis factor secretion by innate immune cells independently of its endotoxin. Microbes Infect. 2003;5:463–470. doi: 10.1016/s1286-4579(03)00063-7. [DOI] [PubMed] [Google Scholar]
- 47.Bulut Y, Faure E, Thomas L, Karahashi H, Michelsen KS, Equils O, Morrison SG, Morrison RP, Arditi M. Chlamydial heat shock protein 60 activates macrophages and endothelial cells through Toll-like receptor 4 and MD2 in a MyD88-dependent pathway. J Immunol. 2002;168:1435–1440. doi: 10.4049/jimmunol.168.3.1435. [DOI] [PubMed] [Google Scholar]
- 48.Mpiga P, Mansour S, Morisset R, Beaulieu R, Ravaoarinoro M. Sustained interleukin-6 and interleukin-8 expression following infection with Chlamydia trachomatis serovar L2 in a HeLa/THP-1 cell co-culture model. Scand J Immunol. 2006;63:199–207. doi: 10.1111/j.1365-3083.2006.01734.x. [DOI] [PubMed] [Google Scholar]
- 49.Lubetsky JB, Dios A, Han J, Aljabari B, Ruzsicska B, Mitchell R, Lolis E, Al-Abed Y. The tautomerase active site of macrophage migration inhibitory factor is a potential target for discovery of novel anti-inflammatory agents. J Biol Chem. 2002;277:24976–24982. doi: 10.1074/jbc.M203220200. [DOI] [PubMed] [Google Scholar]
- 50.Bacher M, Metz CN, Calandra T, Mayer K, Chesney J, Lohoff M, Gemsa D, Donnelly T, Bucala R. An essential regulatory role for macrophage migration inhibitory factor in T-cell activation. Proc Natl Acad Sci U S A. 1996;93:7849–7854. doi: 10.1073/pnas.93.15.7849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Atsumi T, Cho YR, Leng L, McDonald C, Yu T, Danton C, Hong EG, Mitchell RA, Metz C, Niwa H, Takeuchi J, Onodera S, Umino T, Yoshioka N, Koike T, Kim JK, Bucala R. The proinflammatory cytokine macrophage migration inhibitory factor regulates glucose metabolism during systemic inflammation. J Immunol. 2007;179:5399–5406. doi: 10.4049/jimmunol.179.8.5399. [DOI] [PubMed] [Google Scholar]
- 52.Miller EJ, Li J, Leng L, McDonald C, Atsumi T, Bucala R, Young LH. Macrophage migration inhibitory factor stimulates AMP-activated protein kinase in the ischaemic heart. Nature. 2008;451:578–582. doi: 10.1038/nature06504. [DOI] [PubMed] [Google Scholar]
- 53.Radstake TR, Sweep FC, Welsing P, Franke B, Vermeulen SH, Geurts-Moespot A, Calandra T, Donn R, van Riel PL. Correlation of rheumatoid arthritis severity with the genetic functional variants and circulating levels of macrophage migration inhibitory factor. Arthritis Rheum. 2005;52:3020–3029. doi: 10.1002/art.21285. [DOI] [PubMed] [Google Scholar]
- 54.Meyer-Siegler KL, Vera PL, Iczkowski KA, Bifulco C, Lee A, Gregersen PK, Leng L, Bucala R. Macrophage migration inhibitory factor (MIF) gene polymorphisms are associated with increased prostate cancer incidence. Genes Immun. 2007;8:646–652. doi: 10.1038/sj.gene.6364427. [DOI] [PubMed] [Google Scholar]
- 55.Wu SP, Leng L, Feng Z, Liu N, Zhao H, McDonald C, Lee A, Arnett FC, Gregersen PK, Mayes MD, Bucala R. Macrophage migration inhibitory factor promoter polymorphisms and the clinical expression of scleroderma. Arthritis Rheum. 2006;54:3661–3669. doi: 10.1002/art.22179. [DOI] [PubMed] [Google Scholar]
- 56.Grigorenko EL, Han SS, Yrigollen CM, Leng L, Mizue Y, Anderson GM, Mulder EJ, de Bildt A, Minderaa RB, Volkmar FR, Chang JT, Bucala R. Macrophage migration inhibitory factor and autism spectrum disorders. Pediatrics. 2008;122:e438–445. doi: 10.1542/peds.2007-3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mitchell R, Bacher M, Bernhagen J, Pushkarskaya T, Seldin MF, Bucala R. Cloning and characterization of the gene for mouse macrophage migration inhibitory factor (MIF) J Immunol. 1995;154:3863–3870. [PubMed] [Google Scholar]
- 58.Sriskandan S, Altmann DM. The immunology of sepsis. J Pathol. 2008;214:211–223. doi: 10.1002/path.2274. [DOI] [PubMed] [Google Scholar]