

Abstract
In Huntington's disease (HD), mutated huntingtin (mhtt) causes striatal neurodegeneration which is paralleled by elevated microglia cell numbers. In vitro cortico-striatal slice and primary neuronal culture models, in which neuronal expression of mhtt fragments drives HD-like neurotoxicity, were employed to examine wild type microglia during both the initiation and progression of neuronal pathology. As neuronal pathology progressed, microglia initially localized in the vicinity of neurons expressing mhtt fragments increased in number, demonstrated morphological evidence of activation, and expressed the proliferation marker, Ki67. These microglia were positioned along irregular neurites, but did not localize with mhtt inclusions nor exacerbate mhtt fragment-induced neurotoxicity. Prior to neuronal pathology, microglia upregulated Iba1, signaling a functional shift. With neurodegeneration, interleukin-6 and complement component 1q were increased. The results suggest a stimulatory, proliferative signal for microglia present at the onset of mhtt fragment-induced neurodegeneration. Thus, microglia effect a localized inflammatory response to neuronal mhtt expression that may serve to direct microglial removal of dysfunctional neurites or aberrant synapses, as is required for reparative actions in vivo.
Keywords: Huntington's disease, microglia, huntingtin, htt, microgliosis, neuron-microglia interaction, complement, interleukin-6, neuroinflammation, protein aggregation, neurite, neurodegeneration, slice culture, neurotoxicity
1. Introduction
Huntington's disease (HD) is an autosomal dominant neurodegenerative disorder involving a gradual loss of medium spiny neurons, the most prevalent cell type in the striatum [Martin and Gusella 1986]. Expansion of a translated trinucleotide (CAG) repeat in the HD gene encoding huntingtin (htt) leads to a polyglutamine expansion at the amino terminus of the resultant 348kD htt protein [HD Collaborative Research Group 1993]. Although loss of normal htt function may contribute to neurodegeneration [Cattaneo, et al. 2005], a growing understanding of HD pathogenesis suggests a gain-of-function neuronal toxicity of mutated htt (mhtt) that involves transcriptional dysregulation, mitochondrial dysfunction, and impaired synaptic transmission [Landles and Bates 2004, Panov, et al. 2002, Ross 2002]. This toxicity appears to be driven by aggregated N-terminal fragments of mhtt, rather than full-length mhtt [Cooper, et al. 1998, DiFiglia, et al. 1997, Wang, et al. 2008].
Normal htt protein contributes to vesicular transport and synaptic transmission and, as such, is highly expressed in dendrites and nerve terminals [Gutekunst, et al. 1995, Trottier, et al. 1995]. In HD, the resistance of mhtt to proteolysis and its tendency to misfold precipitate the formation of inclusion bodies (IBs) in the nucleus, cytoplasm, and neurites [Gutekunst, et al. 1999]. HD patients exhibit an early accumulation of N-terminal fragments of mhtt in non-nuclear regions of the cell [Sapp, et al. 1999] and, throughout disease, IBs in dendrites and axons (neuropil aggregates) appear more frequently than nuclear aggregates [Gutekunst, et al. 1999, Li, et al. 1999]. In support of a primary dysfunction in axons and dendrites, early stages of HD are characterized by dystrophic neurites with fewer dendritic spines and thickened, proximal dendrites [Albin, et al. 1990, Li, et al. 2001]. The progressive appearance of small neuropil aggregates correlates with a disruption in trafficking and synaptic function, as well as mitochondrial damage, microtubule destabilization, neurite retraction, and the eventual development of neurological symptoms [Li, et al. 1999, Li, et al. 2001, Smith, et al. 2005, Trushina, et al. 2004, Trushina, et al. 2003]. However, the actual contribution of aggregate formation to neuronal loss in HD has yet to be identified [Kuemmerle, et al. 1999, Saudou, et al. 1998].
Microglia are necessary for healthy brain function, as these cells clear tissue debris, remove soluble factors and aberrant proteins from the neuronal microenvironment, and respond to brain insults including neurodegenerative disease progression [Gehrmann, et al. 1995, Long-Smith, et al. 2009, Meda, et al. 1995]. A correlation between structural changes in microglia and severity of HD neuropathology has been reported in patients [Pavese, et al. 2006, Sapp, et al. 2001, Singhrao, et al. 1999], suggesting a role in disease progression. Microglial responses in the striatum are characterized by an increased activation state in pre-manifest [Tai, et al. 2007] and manifest HD [Sapp, et al. 2001], as well as the R6/2 mouse model for HD [Tai, et al. 2007].
Although microglia are intimately linked with HD neuropathology, the exact role that they play in regulating the health of mhtt-expressing neurons remains unclear. The functional interaction between diseased neurons in HD and immuno-modulatory microglia appears to represent a critical juncture in the progression and amplification of HD pathology that, if understood, could support the development of anti-inflammatory based patient treatment options. The correlation between increasing microglia cell numbers and the progression of HD pathology suggests that microglia exacerbate the pathology of diseased neurons. In the current study, we evaluated the relationship between neighboring microglia and mhtt-expressing neurons in primary cell and brain slice culture models that possess cortico-striatal neuronal connections shown to be involved in the development of the HD phenotype [Cepeda, et al. 2007]. In these model systems, the timecourse of neurite disruption and neuronal death allowed for the examination of microglial activation state and localization at progressive stages of mhtt-induced neuronal degeneration.
2. Materials and methods
2.1. Animals
Sprague Dawley rats and CD-1 mice (Charles River Labs, Raleigh, NC) were obtained at postnatal day 1-2 or 10 (PND1-2 or 10) and gestational day 15 or 18 (GD15 or 18). Animals were housed in a semi-barrier facility at 21±2°C with 50±5% humidity and 12h light/dark cycle. Food and deionized, reverse osmotic-treated water were available ad libitum. All procedures were conducted in compliance with approved animal protocols from NIEHS/NIH or Duke University Institutional Animal Care and Use Committees.
2.2. Culture preparations and neuronal transfection with HttN90Q73 plasmid (mHtt)
“Composite” cortico-striatal cultures
Cortico-striatal neuronal cultures were prepared from GD18 rats as previously described [Kaltenbach, et al. 2010]. Briefly, dissociated striatal neurons were transfected by electroporation (Lonza, Basel Switzerland) with a fluorescent protein tag (either mCherry or yellow fluorescent protein, YFP), combined with either a control plasmid or a human htt exon-1 (N-terminus) expression construct containing a 73-polyglutamine-encoding repeat (HttN90Q73; mHtt). Cortical neurons were co-transfected with a different fluorescent tag (either YFP or cyan fluorescent protein- CFP) combined with control or mHtt plasmid. All expression plasmids were controlled by the cytomegalovirus promoter. Following electroporation, striatal and cortical neurons were mixed and plated into 96-well plates (60,000 cells per well) on a confluent bed of rat-derived astrocytes (> 97% pure). Cultures were grown in Neurobasal media supplemented with 5% fetal bovine serum (FBS; <0.3EU/mL endotoxin), 2mM Glutamax, KCL, and 5μg/mL gentamycin (all reagents from Invitrogen, Carlsbad, CA). According to preliminary evaluation of NeuN staining, the neuronal plasmid transfection efficiency is approximately 25%.
Biolistic transfection of cortical neurons
Primary cortical neurons were prepared from GD15 mice as previously described [Kraft, et al. 2004]. Cortical cells were filtered (70μm cell strainer; Millipore, Billerica MA) and plated on poly-D-lysine (PDL)-coated plates at 3.0 × 105 cells/cm2 in MEM supplemented with 10% FBS, 10% horse serum, 2mM Glutamax, and 1% penicillin/streptomycin (P/S) for 48h prior to changing to Neurobasal media with B27 supplement containing antioxidants (all reagents from Invitrogen). Dissociated cells were maintained at 37°C under 5% CO2. At 5 days in vitro (DIV), cells were biolistically co-transfected with YFP and CFP (control plasmids) or YFP and HttN90Q73 possessing a CFP tag at the C-terminus (mHtt plasmids). Using particle-mediated gene transfer as previously described [Lo, et al. 1994], gold particles (1.6μm) coated with 12μg of each plasmid were propelled at 95psi into the cultures by a Helios gene gun (Bio-Rad, Hercules, CA) at a height of 2.5cm in the absence of media (conditioned media was replaced immediately afterwards). This resulted in a transfection rate of 1-2 YFP neurons per 30,000μm2 (1-2% transfection efficiency) by 18h.
Acute cortico-striatal slice cultures
Cultures of 250μm-thick coronal slices through the striatum were prepared from PND10 rats as previously described [Crittenden, et al. 2010] and plated on 0.4μm Millipore PET membrane inserts. After a 1h recovery period from sectioning, slices were biolistically co-transfected as described for primary cortical neurons above. Slice cultures were maintained at 32°C under 5% CO2.
2.3. Microglia supplementation to neurons
To examine the influence of microglia on mHtt-transfected neurons, exogenous microglia were added to primary neuronal cultures. Dissociated primary glia were generated according to the method of McCarthy and deVellis [McCarthy and de Vellis 1980] from cortical tissue of PND1-2 rats or mice with trypsin digestion, sequential filtration (165μm and 40μm nylon mesh), and plating in DMEM (Invitrogen) with 10% FBS and 1% P/S. Mixed glia cultures (~10DIV) were shaken at 35rpm for 7h at 37°C. Detached microglia were filtered (70μm) and resuspended in Neurobasal media.
Addition to composite cortico-striatal culture neurons
microglia (0-5000 cells per well) were added to transfected neurons at 2-4h post-electroporation and cultured with the transfected neurons for 7DIV prior to evaluation of cell viability, microglial proliferation, and microglial localization to neurons.
Addition to cortical neurons
microglia were labeled with isolectin B4-594 from Griffonia simplicifolia (IB4; 1:200 dilution; Sigma-Aldritch) during the final 1h of shaking (at 12rpm). Fluorescent microglia were washed with PBS, filtered (70μm cell strainer), and added to neuron cultures 4h post-transfection at 1.5 × 104 cells/cm2 for time-lapse microscopy of localized targeting to neurons.
2.4. Real-time quantitative PCR (qPCR) of brain slice explants
Total RNA was isolated with TRIzol (Invitrogen) after micro-punch (Miltex, York, PA) excision of the striatal tissue (5 cultured slices pooled per sample; n=3). Reverse transcription was performed with 1μg total RNA using SuperScript II Reverse Transcriptase (Invitrogen). qPCR was carried out (Perkin Elmer ABI Prism 7700 Sequence Detector) using 50ng cDNA as template, 1X Power SYBR Green Master Mix (Applied Biosystems; Foster City, CA), and forward and reverse primers (Table 1). Reaction mixtures were held at 50°C for 2-min, 95°C for 10-min, followed by 40 cycles at 95°C for 15-sec and 1-min at 60°C. Amplification curves were generated with Sequence Detection System 1.9.1. Threshold cycle values were determined and mean fold changes over the 2DIV control samples were calculated according to the 2-ΔΔCT method [Livak and Schmittgen 2001] and normalization to RPL32 (see Table 1 for primer sequences).
Table 1.
| Gene | Forward primer | Reverse primer |
|---|---|---|
| Ribosomal protein (RP) L32 (RPL32) | TGTCCTCTAAGAACCGAAAAGCC | CGTTGGGATTGGTGACTCTGA |
| Interleukin (IL)-6 | CGAAAGTCAACTCCATCTGCC | GGCAACTGGCTGGAAGTCTCT |
| IL-1β | CACCTCTCAAGCAGAGCACAG | GGGTTCCATGGTGAAGTCAAC |
| Tumor necrosis factor (TNF)-α | CCAGGTTCTCTTCAAGGGACAA | CTCCTGGTATGAAGTGGCAAATC |
| Complement component 1q, A chain (C1qA) | CGGAATTCGACAAGGTCCTCACCAACCAG | CGGGATCCGGGGTCCTTCTCGATCC |
| C1qB | CGGAATTCCCTTCTCTGCCCTGAGGACGG | CGGGATCCTTTCTGCATGCGGTCTCGGTC |
| C1qC | CCGGGGGAGCCAGGTGTGGAG | GCACAGGTTGGCCGTATGCG |
| Yellow fluorescent protein (YFP) | GTCCAGGAGCGCACCATCT | CGATGCCCTTCAGCTCGAT |
| Ionized calcium binding adaptor molecule 1 (Iba1) | ACAAGCACTTCCTCGATGATCCCA | TCCACCTCCAATTAGGGCAACTCA |
| Cluster of differentiation 68 (CD68) | CATGTGTTCAGCTCCAAGCCCAAA | AGGTCCAGGTGAATTGCTGGAGAA |
| Macrophage scavenger receptor 1 (Msr1) | AATGGCTCCTCCGTTCAGGAGAAA | TTGCCATGCTGAAGTTCTGGAAGC |
| Glial fibrillary acidic protein (GFAP) | TGCCATACAGTGTGAGGGCCTAAA | TGTCTTGCTCCAGCAGCCTATGAA |
2.5. Western blotting from brain slice explants
Pooled slices (n=6 per sample) collected from individual culture preparations were lysed in RIPA buffer with protease inhibitor (Roche, Basel Switzerland), incubated on ice for 30min, and centrifuged at 13,000xg. Thirty μg of protein supernatant was separated by electrophoresis on Novex Bis-Tris gels (4-12%) using NuPage solutions under reduced conditions (Invitrogen). Protein was transferred to PVDF membranes (Invitrogen) and incubated overnight at 4°C with rabbit polyclonal antibodies to Iba1 (1:1000; Wako, Osaka Japan) or β-actin (1:5000, Abcam). Immunoblotting and visualization were performed with WesternBreeze reagents (Invitrogen). Images were acquired (Kodak Image Station 440cf; Kodak, Rochester NY) and Iba1 protein expression level was calculated as mean intensity relative to β-actin.
2.6. Immunostaining
Cultured brain slices were fixed with 4% paraformaldehyde (PFA)/ 0.1% glutaraldehyde in PBS (18h, 4°C). Slices were cryoprotected in 30% sucrose and cryosectioned at 10μm. Dissociated neuronal cultures were fixed in 4% PFA/ 4% sucrose in PBS (15 min). Following blocking (PBS with 2% goat serum, 1% bovine serum albumin, and 0.1% Triton X-100), samples were incubated with primary antibodies (18h, 4°C). Microglia were identified with anti-Iba1 (1:250; rabbit polyclonal; Wako) or IB4 (1:100-1:200; Sigma-Aldritch), proliferative cells with anti-Ki67 (1:200; mouse monoclonal; Leica Microsystems, Bannockburn, IL), and aggregated N-terminal htt fragments [Gutekunst, et al. 1999] with anti-EM48 (1:250; mouse monoclonal; Millipore) followed by Alexa Fluor (594nm or 488nm, Invitrogen) secondary antibodies (1:200-1:500). Dissociated cells were counterstained with 0.1mM Hoescht 33258 (Sigma-Aldritch) and brain slice sections with Prolong + DAPI (Invitrogen). Images were captured by epifluorescence using a Nikon TE2000 inverted microscope (Tokyo, Japan) and processed with Metamorph™ (Universal Imaging Co., Downingtown, PA). Confocal images (LSM 5 Pascal inverted, laser-scanning confocal microscope; Carl Zeiss, Oberkochen, Germany) were processed with LSM 5 software.
2.7. Quantification of neuronal viability and microglia number
Neuronal viability
Neuronal viability in composite cultures was quantified using the Cellomics Arrayscan VTI (Thermo Scientific, Pittsburgh, PA; [Kaltenbach, et al. 2010]). The timing of mHtt-induced loss of healthy YFP+ MSNs beginning at 3DIV in brain slices was confirmed [Crittenden, et al. 2010].
Microglia number
In 7DIV composite cortico-striatal cultures, 70-140 regions for each combination of treatment (control or mHtt transfection) and microglia supplementation (200-1000 microglia) were collected across multiple tissue culture wells and 2-4 individual cell culture preparations. Using an automated x-y stage and pre-programmed, distance-defined coordinates, the regions were selected in a “systematic-random” fashion from random start points within the culture wells and 20x images of YFP, CFP, 594 (microglial markers), and DAPI were acquired using constant exposure times. Images were thresholded and individual, immuno-positive microglia co-expressing DAPI were counted and averaged by culture. Microglia within IB4 or Iba1-stained clusters were counted only if individual cell morphology could be resolved and met cell identification criteria (≥7μm diameter and one DAPI+ nucleus).
2.8. Statistical analysis
Cell viability was analyzed using a one-way ANOVA followed by Dunnett's t-test for comparisons of independent group means. qPCR for mRNA levels, microglial cell counts in non-manipulated dissociated neuronal cultures, and neuronal branching were analyzed using two-tailed Student's t-tests. Microglia morphology and cell counts in acute slice cultures or following addition of exogenous microglia to dissociated neurons were analyzed using a two-way ANOVA with Bonferroni's post-hoc tests for independent group mean analysis. Statistical significance was set at p<0.05.
3. Results
3.1. Microglia increase in number and accumulate at neurites following neuronal mHtt expression
To examine the relationship between microglia and neurons, we visualized cell-cell interactions following neuronal expression of mHtt. Neuronal expression of N-terminal fragments of mhtt protein containing pathological length polyglutamine expansions initiate neuronal cell death mechanisms that are similar to those seen in genetic models [Runne, et al. 2008]. In our “composite” cortico-striatal culture system, mHtt expression results in the degeneration of approximately 60% of the transfected neurons by 7DIV relative to controls [Kaltenbach, et al. 2010]. Concurrent with neurodegeneration, an increased staining for the microglial marker, Iba1, was observed in cultures transfected with mHtt (Fig. 1A-B), correlating with a significant, 26% increase in microglia number (Fig. 1C).
Figure 1. An elevated number of microglia are present after culture with mHtt-expressing neurons.

(A-B) Staining for microglia (Iba1, red) around striatal neurons (yellow, DAPI nuclear counterstain in blue) transfected with control DNA (A) or mHtt DNA (B) in composite cortico-striatal cultures. (C) Iba1 cell counts seven days after transfection with control or mHtt DNA (*p<0.05). (D) Exogenous microglia were added to the composite cultures on the same day as transfection with control or mHtt DNA and microglia counted at 7DIV (*p<0.05). (E-J) Microglial staining around striatal neurons transfected with control DNA (E,G, and I) or mHtt DNA (F,H, and J) and cultured with varying concentrations of exogenously-added microglia: 500 in E-F and 1000 in G-H (scale= 50μm). I and J: higher magnification images of boxes inset in E and F, respectively, to show microglial interactions at neurites and mHtt+ neuronal blebs (arrows; scale= 25μm). Small, YFP-negative, DAPI-positive nuclei are co-cultured cortical neurons and non-transfected striatal neurons. Composite images of the individual fluorochromes (YFP, Iba1, CFP, and DAPI) for E-H are shown in Supplementary Fig. 2.
To further define the relationship between microglia cell number and neuronal expression of mhtt fragments, microglia were added to composite cortico-striatal cultures. An initial seeding of 200, 500, or 1000 exogenous microglia to control cultures resulted in Iba1+ cell number increases of 2.1, 3.9, and 5.8-fold over non-seeded cultures, respectively (Fig. 1D). In comparison, Iba1+ cell numbers in mHtt-transfected cultures were increased 3.6, 7.0, and 9.5-fold. Calculation of the best-fit lines showed an approximate doubling in the slope of Iba1+ cell numbers following neuronal expression of mHtt (Control: y=0.0208x+4.84 versus mHtt: y=0.0464x+8.70). A 2x2 ANOVA revealed significant main effects of transfection and exogenous microglia, and a significant interaction between transfection and exogenous microglia. Although a robust elevation in Iba1 labeling was observed with neuronal mHtt expression, seeding number did not appear to influence the location of microglia as cells remained evenly distributed around transfected striatal neurons (Fig. 1A-B,E-J).
To determine if microglia localize to striatal neurons in a preferential manner, composite cortico-striatal cultures were evaluated. In both control and mHtt-transfected cultures, microglia did not appear to cluster at the neuronal somata; rather, they appeared to display a preferred localization along thin processes and at points of intersection or overlap between neuronal processes (Fig. 1E-J). Relative to controls, mHtt-transfected striatal neurons possessed a reduced number of primary branches and displayed bulbous swellings along fine processes (Supplementary Fig. 1). Iba1+ microglia were often observed in association with process blebs and clustered at the terminal ends of mHtt+ striatal neurites with an irregular morphology (Fig. 1J, arrows).
Proliferation is one component of the microglial response to injury. In composite cortico-striatal cultures, the mhtt fragment-induced increase in Iba1+ microglia at 7DIV appeared to be paralleled by an increase in microglial proliferation, as demonstrated by elevated Ki67 staining in Iba1+ microglia (Fig. 2; quantified in Supplementary Fig. 3A). Both Ki67+, proliferative microglia (arrows) and Ki67- microglia appeared to interact with striatal neurons primarily at neurites at varying distances from the neuronal somata, however, this preference appeared less consistent for Ki67- microglia (Fig. 2).
Figure 2. Proliferative microglia are present in greater numbers around neurons expressing mhtt fragments.
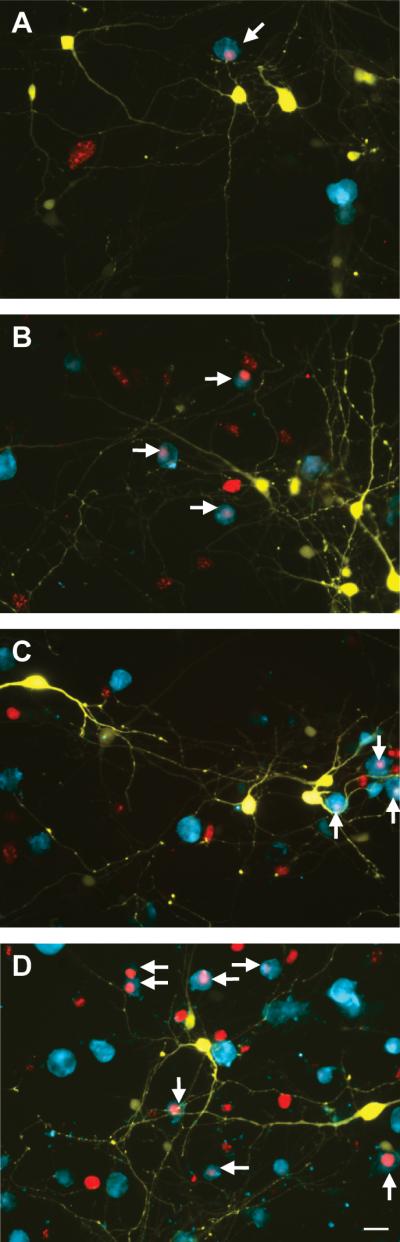
Representative images of microglia (blue, Iba1) and YFP+, transfected striatal neurons (yellow) in control (A, C) and mHtt- transfected (B, D), composite cortico- striatal cultures immunostained for the proliferation marker, Ki67 (red; arrows indicate Iba1 and Ki67 colocalization) at 7DIV. (C-D) 1000 exogenous microglia/ well were added to cultures at transfection (scale= 25μm).
3.2. Microglia-neuron interactions occur at dystrophic neurites and not mHtt aggregation sites
In cortico-striatal cultures, microglial localization at neurites did not appear to be directly related to the location of aggregated mhtt fragments. In striatal neurons, aggregated mHtt labeled with anti-EM48 was observed within fine neurites and thickened processes with bulbous swellings (Fig. 3, arrowheads), as well as within the neuronal somata (Fig. 3, arrows). Microglia appeared to be evenly distributed at aggregate-nonspecific points along fine, YFP+ neurites, rather than at sites of mHtt accumulation (Fig. 3). These microglia-neurite interactions were often some distance from any visible EM48 staining (Fig. 3C-D, inset).
Figure 3. Transplanted microglia associate with neuronal processes at locations devoid of EM48+ aggregates of mhtt.

Seven days following the addition of various concentrations of exogenous microglia to composite cortico-striatal cultures (200 in A-B; 500 in C-D and G-H; and 1000 in E-F; scale= 20μm), the association of striatal neurons expressing mHtt (yellow) with Iba1-labeled microglia (red) and EM48-labeled mHtt aggregates (white in B,D,F, and H; corresponds with panels on left) in the neuropil (arrowheads) and somata (arrows) is represented. The boxed region highlights microglia- neurite contact in the absence of nearby aggregates. DAPI nuclear staining is shown in blue. (G-H) Smaller process aggregates were assessed at higher magnification (scale= 25μm).
Using biolistic transfection, we generated a primary neuronal culture system with a lower expression level of mHtt. This system allowed us to visualize individual processes of transfected neurons and determine whether microglial association with neurites is associated with the onset or progression of neuritic degeneration. Neurons transfected with control plasmids displayed normal neurite morphology for >72h (Fig 4A). Within 24h, mHtt+-transfected neurons displayed characteristic CFP+ mhtt aggregates in the cell nucleus and the proximal and distal neurites (Fig. 4B). The expression and size of aggregates coincided with the appearance of neurite irregularities, such as axonal swelling, re-curving of distal neurites, and deteriorating dendrite morphology by 48h (Fig. 4C). Consistent with the composite cortico-striatal cultures, localization of pre-labeled, exogenous microglia appeared to preferentially occur along neurites (Fig. 4) at mHtt aggregate-nonspecific sites (data not shown). Using time-lapsed microscopy, 24 to 48h after mHtt transfection, microglia were observed at distal processes near to the terminal ends of neurites, at sites of neurite intersection, and at positions along neurites where irregularities form (Fig. 4D-G). The severity of degeneration progressed rapidly after 48h, such that by 72h the majority of mHtt-transformed neurons displayed degenerated processes and/or fragmented cell somata (Fig. 4J). During this period of active neuronal degeneration, microglia-neuron associations appeared to remain committed to the initial neurite contact, as microglia tracked between these timepoints did not appear to migrate to an alternative site (Fig. 4H, J). In many instances, a shift in the morphological phenotype of microglia associating with degenerating neurites was observed (Fig. 4I, K).
Figure 4. Microglia added after neuronal transfection with mHtt are associated with neurites throughout degeneration.

Representative images of control neurons at 72h post-biolistic transfection (A) compared with neurons transfected with mHtt at 24h (B) and 48h (C) in dissociated cortical neuronal cultures (scale= 25μm). mHtt fragments (CFP, green) within neurites proximal (arrows in B-C) and distal (arrowheads) to the cell body. Time-lapse images of neurons expressing mHtt (yellow) and microglia exogenously- added at 2h post-transfection (IB4, red) after 24h (D) and 48h (F; scale= 50μm). Arrows highlight the appearance of microglia at the terminal ends of processes, locations where connections are remodeled, or at process intersections (E and G: higher magnification image of inset in D and F, respectively; scale= 50μm). Time-lapse image of mHtt neurons at 48h (H), and 72h (J; scale= 25μm). Arrows show smooth neurites which microglia interact with that subsequently become stippled and degenerate (I and K: higher magnification image of inset in H and J, respectively; scale= 25μm).
3.3. Microglial association with neurites in slice cultures
To extend these observations to a more intact system, we examined the pattern of microglial changes occurring in a cortico-striatal slice culture system. This slice maintains inputs to striatal cells from cortical sources, but lacks extrinsic afferents from dopaminergic or thalamic sources. Consistent with previous work [Lo, et al. 1994], biolistic transformation of slice cultures resulted in selective gene expression within neurons (Fig. 5A-D) without ancillary trauma or noticeable activation of surrounding glia. Control slices expressed both YFP and CFP throughout the transfected striatal neurons (data not shown). mHtt- transfected slices expressed YFP throughout the neurons and CFP selectively at locations of aggregated mhtt in the neuronal somata and processes (Fig. 5A), as indicated by co-localization with EM48 (data not shown). Consistent with previous studies showing that transfection of slice cultures with mHtt induces progressive neurodegeneration that becomes measurably divergent from controls between 3-5DIV [Crittenden, et al. 2010], striatal YFP mRNA levels were similar between groups at 2DIV (mHtt slices= 81 ± 7.9% of controls, n=3) and decreased by mHtt at 5DIV (mHtt slices= 46 ± 17% of controls, p<0.05, n=3).
Figure 5. Endogenous slice microglia associate with processes of striatal neurons in greater numbers following transfection with mHtt, but appear to do so independently of mhtt aggregation.

(A)Representative images of striatal neurons in slices transfected with mhtt plasmids encoding HttN90Q73-CFP (mHtt; blue) and YFP (yellow) after 5DIV reveal CFP+ aggregates in degenerating neurites (arrows) and the neuronal soma (arrowheads). Representative IB4 staining for microglia (red) at 3DIV (B-C) and 5DIV (D-E) in slice cultures transfected with control plasmids (B and D) or mHtt plasmids (C and E). YFP alone is shown at right (yellow) to display fine processes of transfected neurons and show microglial association with striatal neurites (arrows). (C and E) mHtt aggregates (arrowheads) indicated by CFP fluorescence (white) in mHtt-transfected cultures (scale= 25μm).
The tendency of microglia to distribute and localize to neuronal processes as observed in dissociated cell cultures was maintained in slice cultures. At 3 and 5DIV, microglia appeared uniformly distributed around the somata of labeled neurons (Fig. 5B-E), with an apparent preferential localization of IB4-labeled microglia to neuronal processes (arrows). In the mHtt-transfected slices, microglia were observed at sites of process fragmentation (Fig. 5C, E), however, the localization of microglia to neuronal processes did not appear to demonstrate preferential colocalization to CFP-labeled mhtt aggregates (arrowheads).
In HD, increases in microglial cell number are closely linked with elevations in disease severity [Sapp, et al. 2001]. To quantify and localize the observed increase in microglia endogenous to the slice following neuronal expression of mhtt fragments, IB4-labeled microglia within 100μm of YFP-labeled neuronal somata in the striatum of control and mHtt-transformed cultures were counted using a Sholl analysis (Supplementary Fig. 4A, C). In control cultures, a progressive increase was observed over time in culture, with a significant increase occurring at 5DIV. In mHtt-transformed cultures, this process was accelerated, with a significant increase in the number of microglia observed at 3DIV (Fig. 6A). This accelerated microglia increase coincided with the progression of neuronal degeneration. Similar to the cell death time course, a differential between control and mHtt slices was detected by 3DIV and exacerbated by 5DIV, with a significantly greater number of microglia seen in the mHtt-transfected slices (Fig. 6A,C). Two-way ANOVA revealed significant main effects of DIV and transfection, and a significant DIV by transfection interaction.
Figure 6. Microglia cell numbers are elevated in the vicinity of mhtt fragment-expressing neurons, but their distribution in relation to the cell soma remains constant.

(A) IB +4 cells in cryosections from control and mHtt- transfected slice cultures were quantified within a 100μm radius of transformed striatal neuron somata over DIV. Values are given per neuron analyzed and are averaged across animal (n=3-4 rats; neurons analyzed in control and mHtt cultures, respectively: 13 and 16 at 1DIV; 32 and 34 at 2DIV; 26 and 33 at 3DIV; and 28 and 20 at 5DIV; *p<0.05 from 1DIV counts in control cultures or #p<0.05 from DIV-matched control cultures). (B) IB +4 cells were quantified within random regions encompassing a 100μm radius of the striatum and >250μm of any visible YFP signal in control and mHtt- transfected slice cultures. Values are given per region analyzed and are averaged across animal (12 neuron-free regions analyzed in the striatum of n=3 rats). (C) Sholl-like analysis of microglial distribution around transfected neurons showing fold changes in microglia cell number per region in mHtt-transformed cultures at 3 and 5DIV compared to control cultures (*p<0.05 from sholl and DIV-matched controls). (D) Taking the total number of microglia within 100μm of neuronal somata as 100%, the median percentage across 1, 2, 3, and 5DIV (± range) of those microglia found within each of the distance-defined sholl regions is shown for control and mHtt- transfected cultures.
The increase in microglia was not observed uniformly across the slice but was specific to within the experimentally-defined radius of the neuronal process field. At 3DIV, the number of microglia observed within regions devoid of YFP expression (>250μm of any observed YFP signal) were similar across control and mHtt-transformed cultures (Fig. 6B), indicating that the treatment-related increase in microglia was dependent upon the local presence of neurons expressing mhtt fragments. When the distribution of microglia along the neurite axis was quantified, the pattern of localization in slice cultures confirmed observations of a lack of microglia homing to the cell body of neurons in dissociated cultures. In mHtt-transfected slice cultures, the number of microglia within each of the Sholl regions was significantly increased at 3 and 5DIV, as compared to controls (Fig. 6C). However, the distance-defined localization of microglia around neuronal somata was similar across DIV (data not shown) and transfection (Fig. 6D).
The increase in microglia number could have arisen from increased proliferation of microglia. Similar to observations in composite cortico-striatal cultures, staining of Iba1+ microglia nearby transfected striatal neurons was increased with expression of mhtt fragments (Fig. 7). At 3 and 5DIV, an increase in Iba1+ microglia co-expressing Ki67 (e.g. arrows in Fig. 7E-F) was observed in mHtt-transfected striatal slices, as compared to control slices (Fig. 7; quantified in Supplementary Fig. 3B), suggesting a proliferative response of microglia localized within the dendritic arbor of mHtt-transfected neurons.
Figure 7. Microglia cell numbers in mHtt- transfected cultures are enhanced by proliferation.

Representative images of striatal cryosections from 3DIV (A-B) and 5DIV (C-F) slice cultures transfected with control plasmids (A,C, and E) or mHtt plasmids (B,D, and F) stained for Iba1 (red), Ki67 (green), and DAPI (blue). (A-B) Orthogonal views show nuclear Ki67 within Iba1+ microglia counterstained with DAPI. (C-D) Iba1 and Ki67 fluorescence in the absence of DAPI counterstain (scale= 50μm). (E-F) Arrows indicate proliferative microglia (scale= 20μm).
3.4. Characteristics of microglia in response to neuronal mHtt expression
Cell surface molecules expressed by microglia can reveal select changes in their functional phenotype [Gehrmann, et al. 1995]. In cortico-striatal slice cultures, at 2DIV, prior to observable neuronal injury and prior to any observed increase in microglia cell number, Iba1 mRNA levels were elevated by neuronal expression of mHtt (Fig. 8A). Iba1 is a calcium-binding adaptor molecule that is expressed by microglia at rest and at progressively higher levels with increasing activation state. This increase suggests an early response of these cells prior to microglial proliferation and neuronal death. At 3DIV, when mHtt-induced neurodegeneration is first observed, increased microglia cell numbers coincided with a 50-70% elevation in Iba1 protein levels over controls (Fig. 8B). At 5DIV, Iba1 mRNA levels remained elevated in response to mHtt, as compared to controls (Fig. 8A). mRNA levels for the scavenger receptor, Msr1, were decreased prior to neuronal death (2DIV), but returned to control levels by 5DIV (Fig. 8A). Genes associated with microglial phagocytosis (CD68), migration (CXCR3), and chemokine signaling from neurons (CX3CR1) were unchanged by mHtt transfection at both time points (Fig. 8A and data not shown).
Figure 8. Markers associated with microglial activity are altered following expression of mHtt in slice cultures.

(A) qPCR for various markers common to microglia were analyzed in the striatum of 2DIV and 5DIV control and mHtt- transfected slices. Values are corrected to Rpl32 housekeeping gene expression and displayed according to their % change from DIV- matched control values (n=3 independent pools of 5 slices from an individual animal; *p<0.05). (B) Western blotting for the housekeeping protein, β-Actin (Actin), and Iba1 in slices harvested immediately after preparation (no biolistic transfection; 0DIV); in slices 3DIV after culture preparation and transfection with control plasmids (Control 3DIV); and in slices 3DIV after transfection with mHtt plasmids (mHtt 3DIV).
The activation state and functional demands of microglia can be inferred, in part, according to their morphology, which ranges from process-bearing to amoeboid [Hanisch and Kettenmann 2007]. Given that microglia can be activated by cellular changes in the environment such as neuronal injury, we examined the temporal expression of distinct morphological phenotypes of microglia as a function of mHtt neuronal transfection in slices (Fig. 9). As mHtt- transfected neurons within slice cultures begin to display clear evidence of perturbation at 3DIV, the ramification states of microglia <100μm from transfected striatal neurons were analyzed at 3 and 5DIV. Since the procedure for generating slice cultures induces a level of morphological activation of microglia over subsequent DIV [Huuskonen, et al. 2005], these analyses were expressed relative to DIV- matched microglia in control cultures. Microglia surrounding mHtt- transfected striatal neurons exhibited a reduction in the number and length of their processes in comparison to microglia surrounding control neurons (Fig. 9A). To quantify the shift towards a rounded, or amoeboid, morphological phenotype, form factor (FF) analyses were conducted (Supplementary Fig. 4B, D) to calculate the process complexity of microglia <100μm from transfected neurons based on the cell perimeter, cell area, and the area of the polygon inscribed by the outermost microglial processes, as previously described [Heppner, et al. 1998, Wilms, et al. 1997]. At both time points, a significant increase in the percentage of microglia with amoeboid morphology was observed in the vicinity of mHtt-expressing neurons compared to their YFP counterparts (Fig 9B). Two-way ANOVA showed a significant main effect of both transfection and DIV. No change in the overall size of the individual, IB +4 microglia was observed, although cellular clustering was more frequent in mHtt-transfected cultures (data not shown). No mHtt-related changes in GFAP levels or astrocyte morphology were observed (Supplementary Fig. 5).
Figure 9. Microglia assume a more amoeboid phenotype when surrounding striatal neurons expressing mhtt fragments than when surrounding control neurons.

(A) Camera lucida drawings of microglia representing the median morphology of microglia quantified within a 100μm radius of striatal neurons transfected with control or mHtt plasmids at 3 and 5DIV. (B) Quantification of microglia morphology (within a 100μm radius of transfected neurons) by the FF1 or FF2 method at 3 and 5DIV around control or mHtt-expressing neurons (*p<0.05 from microglia in 3DIV control cultures; #p<0.05 from DIV-matched controls; at 3 and 5DIV, respectively: 187 and 440 microglia were evaluated in control cultures and 338 and 331 microglia in mHtt- transfected cultures).
3.5. Neuroprotection with microglia supplementation
To examine the functional impact of the increased number of microglia within the dendritic arborization of mHtt-transfected neurons, the composite cortico-striatal cultures were used to directly manipulate the number of microglia. Adding exogenous, primary microglia to mHtt-expressing neurons allowed for the assessment of cell type-specific contributions to an HD-like phenotype. Supplementation of cortical and striatal co-cultured neurons with exogenous microglia on the day of electroporation and plating resulted in “dose-dependent” increases in neuronal survival that was proportional to the numbers of microglia added (Fig. 10). While neuroprotection of striatal neurons occurred in both control and mHtt cultures, the effect on mHtt- transfected neurons was significantly greater, with an increase in neuronal viability at 5x lower levels of microglial supplementation (Fig. 10A). Unlike the striatal neurons, viability of cortical neurons in the control cultures was not altered by addition of exogenous microglia. The viability of mHtt-expressing cortical neurons was significantly improved by the addition of microglia (Fig. 10B).
Figure 10. Microglia are neuroprotective to composite cortico-striatal neuronal cultures.

(A) Numbers of striatal neurons surviving after incubation with increasing amounts of exogenously-added microglia in cultures transfected with either control DNA or mHtt DNA. (B) Numbers of cortical neurons surviving in cultures transfected with control DNA or mHtt DNA following incubation with exogenous microglia. Striatal and cortical cell numbers were normalized to the mHtt condition, set to 100%, and represent the average of 7 independent experiments. *p<0.05 from the 0 microglia condition (n=6-8).
3.6. Neuronal mHtt expression elevates IL-6 and complement component 1q with neurodegeneration
Previous associations between HD progression and increases in cytokines such as IL-1β, IL-6, and TNF-α have been reported [Kyrkanides, et al. 2001]. To address the potential influence of microglia on the induction of these pro-inflammatory cytokines, we examined mRNA levels for TNF-α, IL-1β, and IL-6, in the striatum of cortico-striatal slice cultures. The proliferation and increased local reaction of microglia in the mHtt- transfected slices was not accompanied by elevations in TNF-α, IL-1β, or IFNγ mRNA levels (Fig. 11A and data not shown). In response to mHtt transfection, there was no change in the expression of any of these cytokines prior to neuronal damage and, at the onset of neurodegeneration (3DIV), no changes in IL-1ß and IL-6 protein levels were observed (data not shown and Supplementary Fig. 5). However, by 5DIV, mRNA levels for IL-6 were significantly elevated in the striatum of mHtt-transfected slice cultures, as compared to controls (Fig. 11A).
Figure 11. Neuronal mHtt expression reconfigures the molecular profile of striatal slices, resulting in increased IL-6 and C1q expression.

(A) qPCR for select cytokines in the striatum of 2DIV and 5DIV cultures transfected with control or mHtt DNA. (B) qPCR for complement component 1q (C1q) components at 2DIV and 5DIV. Values are corrected to Rpl32 housekeeping gene expression and displayed according to their % change from DIV-matched control cultures (n=3 independent pools of 5 slices from an individual animal; *p<0.05).
The localization of microglia to neurites, independent of mhtt aggregate presence, and the observation in dissociated neuronal culture that microglia retained the initial contact association with neurites, both suggest a role for microglia in contact-dependent relationships with afflicted neurons. Activation of the cellular complement system has been reported to be involved in neuronal dysfunction in HD [Singhrao, et al. 1999] and specific signaling interactions of complement component 1q (C1q) have been implicated in synaptic/dendritic remodeling in neurodegenerative disease [Stevens, et al. 2007]. Functional C1q consists of six A, B, and C chains encoded by the genes C1qA, C1qB, and C1qC, respectively. The expression of mHtt in slice cultures resulted in a significant, 75% increase in mRNA levels for C1qA and a significant, 35% increase in C1qB mRNA levels (p<0.05), as compared to control slices at 5DIV (Fig. 11B). mRNA levels for C1qC were not significantly different between transfection conditions (Fig. 11B) and, at the onset of neurodegeneration (3DIV), C1q protein was marginally increased by 10% in mHtt-transfected cultures, as compared to controls (Supplementary Fig. 5).
4. Discussion
Neuronal expression of N-terminal mhtt fragments containing pathological length polyglutamine expansions is sufficient to initiate HD-like phenotypes and neuronal degeneration [Runne, et al. 2008, Saudou, et al. 1998]. We now demonstrate that expression of mHtt in striatal neurons, within both primary cells and brain slice cultures, initiated a localized response of wild type microglia, including elevated numbers and activated morphological phenotypes. These proliferating microglia appeared to associate preferentially with neurites at time points representing early and late degenerative phenotypes of the neurons, but did not directly contribute to neurodegeneration. Rather, microglial supplementation provided measurable support to mHtt- transfected neurons. Thus, a functional shift of microglia prior to neuronal toxicity appears to occur in response to some unidentified factor that may be produced along dystrophic or dysfunctional neurites and is associated with downstream signaling responses, including IL-6 and C1q.
4.1. Signaling for microglia from mHtt neurons
We now report a stimulatory effect of mHtt-induced neuronal dysfunction on microglia. Microglial interactions with degenerating neurons appeared to occur primarily at neurite intersections and swellings, as well as thinning processes, regardless of the local presence of aggregated mhtt. Although aggregated mhtt fragments can induce toxicity [Yang, et al. 2002], they do not correlate with the progression of HD pathology [Saudou, et al. 1998] and may even represent a protective response of the cell to soluble mhtt [Arrasate, et al. 2004]. From qualitative observations of co-labeling for microglia and mhtt aggregates, the absence of a distinct localization of microglia to sites of visible mhtt aggregation argues against a localized, aggregation-specific signal for microglial activation. However, aggregation-mediated changes which subsequently activate microglia at distant sites or an altered composition of in vitro aggregates cannot be ruled out.
Neurite intersections are unique in that they contain a large proportion of mitochondria and have a high likelihood of synaptic contacts. Thinning neuronal processes can be representative of a severe loss of dendritic spines and the associated loss of synapses, while structural sites of process bleb formation have been correlated with high Ca2+ entry after stimulation of glutamate receptors (GluRs) [Bindokas and Miller 1995]. HD patients exhibit neuronal network remodeling, including dendritic atrophy and sprouting [Graveland, et al. 1985], and one mechanism of mhtt-induced cell death involves excitotoxic overstimulation of GluRs [Tabrizi, et al. 1999]. In general, morphological changes in the neurites of mhtt-expressing neurons can be attributed to metabolic disturbances, including disrupted transcription [Nucifora, et al. 2001] and release of factors that block protein transport [Li, et al. 2001].
In models of brain injury, microglia or brain macrophages can either be recruited to a neuronal injury site [Kurpius, et al. 2007] or proliferate locally [Graeber, et al. 1988]. Depending on the source of the brain macrophages and their capacity for proliferation, the accumulation of these cells can result in differential effects on the neuronal population. Previous studies demonstrate that transient, proliferative microglia can aid in the repair and regeneration of neurons following injury, while induction of non-proliferative microglia can be associated with phagocytic activity and dendrite loss [Graeber, et al. 1998, Rappert, et al. 2004, Rogove, et al. 2002]. In human HD, activated microglia increase with disease severity and contact diseased neurons at both the cell body and processes [Sapp, et al. 2001]. Within the current culture model, neuronal expression of mhtt fragments initiated a proliferation of microglia that correlated with the apparent localization of microglia to morphologically- aberrant and dystrophic neurites, yet there was no evidence of direct neurotoxic actions. Together, the localization of microglia to these structural sites and microglial proliferation preceding overt neurotoxicity suggests that microglia are responding to early, detrimental changes within afflicted neurons rather than initiating processes which directly lead to neuronal dysfunction and death. Early changes in the synaptic health of mHtt- expressing neurons may represent one such activation signal for microglia
4.2. Protective microglia respond to aberrant mHtt neurites
Microglia possess great diversity in their activation phenotypes. The acquisition of these various functional states involves a wide range of cell surface receptors and cytokines whose expression depends on the type and timing of initiating cues in the microenvironment [Perry, et al. 2010]. While microglial activation is typically associated with increased phagocytosis, many other functional and adaptive changes are employed prior to, and during, cellular activation [Napoli and Neumann 2009]. Several of these changes involve limiting or counteracting neuronal damage and dysfunction, including repair or regeneration of damaged tissue, turnover of persistent proteins, removal of glutamate, and support of neurogenesis [Hanisch and Kettenmann 2007, Neumann, et al. 2009]. Additionally, microglia are known to be involved in monitoring and pruning neuronal synapses, both on a regular basis and with injury, the latter occurring, presumably, as a neuroprotective action [Trapp, et al. 2007, Wake, et al. 2009].
Our data support the induction of an early phenotypic change in microglia in response to neuronal expression of mhtt fragments. Msr1, a scavenger receptor expressed by phagocytic microglia, is decreased, and Iba1, a microglial marker that is upregulated in response to cellular activation, is increased, at 2DIV. These events occurred prior to neuronal degenerative events and any observable difference in microglia cell numbers or proliferation around mHtt- transfected neurons. Alterations to these markers indicate a changing phenotype of microglia in association with neurons lacking any mHtt- induced neuropathological features, which precedes microglial proliferation and morphological activation.
Despite these early alterations in cell surface molecules and close association with sites along mHtt neurites, the microglia gave no evidence of actively contributing to degenerative phenotypes in mHtt-transfected neurons. Microglia were often localized to neuronal processes in both control and mHtt-transfected cultures, indicative of a normal process of neurite stabilization and remodeling. The observation that microglia were neuroprotective/ neurosupportive to primary striatal neurons, but not cortical neurons, following transfection with control plasmids suggests that a differential sensitivity to microglial actions may exist across neuronal populations derived from different brain regions. An amplification of this normal repair process appears to be required following neuronal expression of mhtt fragments, as supplementation with microglia provided neuroprotection for both striatal and cortical, mHtt-transfected neurons. Similar to observations of trophic support of normal neurons by non-stimulated microglia in culture [Zhang and Fedoroff 1996], microglia in co-culture with neurons expressing mhtt fragments were not toxic, but rather, displayed protective phenotypes that seemed to be initiated along neurites. In their study, Zhang and Federoff noted that microglial interactions improved the arborization and thickness of contacted neurites, despite the acquisition of “phagocytic” phenotypes. This observation suggested that morphological activation of microglia does not preclude their capacity for trophic support of neurons.
While normal microglia were supportive of mHtt- transfected neurons in the current studies, microglia expressing mhtt may possess a different phenotype. As microglia in HD have been shown to express mhtt [Shin, et al. 2005], it is important to recognize that the models used in the current studies recapitulate only a select facet of the disease: namely, the acute response to neuronal expression of mhtt. In the R6/2 mouse model of HD, with mhtt expression in all cells, there is a reported decrease in the overall number of microglia, with many displaying structural changes such as process swellings and fragmentation [Ma, et al. 2003]. Microglial expression of mhtt has been reported to induce a “functional overactivity” involving altered iron metabolism, perturbed kynurenine pathway regulation, and the production of excess IL-6 upon challenge [Bjorkqvist, et al. 2008, Giorgini, et al. 2008, Simmons, et al. 2007]. Such alterations would significantly alter the normal capacity of microglia to regulate the neuronal environment. Thus, it is possible that targeting the regulation of microglia toward a more normal responsive phenotype can significantly contribute to the protection of HD neurons.
Abnormalities in synapse proteins and synaptic transmission occur prior to neuron loss and contribute to early HD symptoms [DiProspero, et al. 2004, Li, et al. 2003]. Microglial processes directly contact neuronal synapses in vivo and these contacts increase in frequency with elevated neuronal activity [Wake, et al. 2009]. During the early stages of neuronal injury, signals occur at synapses to orchestrate synapse stripping and remodeling. Both activated microglia [Blinzinger and Kreutzberg 1968, Trapp, et al. 2007] and complement factors [Stevens, et al. 2007] are now shown to be essential for this process, which occurs in the absence of neuropathology [Trapp, et al. 2007]. Complement is a critical component of the innate immune response against pathogens. Upon injury, C1q produced by microglia [Lynch, et al. 2004] binds to neurons, promoting phagocytosis through its interaction with membranes of apoptotic cells, blebs, or structures otherwise identified as abnormal [Fraser, et al. 2010, Ogden, et al. 2001], including excess or aberrant synapses targeted for elimination [Stevens, et al. 2007]. Local production of complement factors by reactive microglia has been observed in the caudate of HD patients [Singhrao, et al. 1999]. We now report that mHtt expression in striatal neurons resulted in the appearance of degenerative features such as neuronal blebs, which coincided with an increased expression of C1q. This concurrent timing suggests that C1q serves as a signal to target microglia to neurites for clearance of aberrantly-functioning synapses.
Changes to the pro-inflammatory cytokine profile of microglia can have a significant impact on the health of the neurons with which they associate [Kim and de Vellis 2005]. Reciprocally, microglial production of these same cytokines (e.g. IL-6 and TNF-α) can be increased by the release of factors such as ATP from damaged or stressed neurons [Diaz-Hernandez, et al. 2009, Inoue 2002]. In physical lesion models, brain macrophages can lead to secondary neuronal and dendritic damage [Eyupoglu, et al. 2003] and neurodegeneration initiated by microglia can be driven through TNF-α signaling [Iliev, et al. 2004]. TNF-α is predominantly produced by microglia [Hetier, et al. 1990, Kraft, et al. 2009] and has been observed to be inversely associated with their phagocytic activation [Magnus, et al. 2001]. Recent work has demonstrated a dual role of C1q in modulating the cytokine environment and facilitating microglial clearance of injury-generated apoptotic neurons or neuronal blebs [Fraser, et al. 2010]. During this specific process, and in the absence of activation of the entire complement cascade, C1q elicits enhanced microglial phagocytosis and concomitantly suppresses the pro-inflammatory cytokines IL-1α, IL-1β, TNF-α, and IL-6. The absence of elevations in TNF-α or IL-1β mRNA levels within the mHtt-transfected slice cultures is consistent with suppression by C1q.
In the current slice culture model, we now demonstrate that the proliferation and apparent targeting of microglia to neurites does not involve an elevation in pro-inflammatory TNF-α. The absence of TNF-α induction suggests that microglia fail to mount a cytotoxic, pro-inflammatory response to neuronal expression of mHtt and the subsequent neurite degeneration. Although HD has previously been shown to induce production of IL-1β [Ona, et al. 1999], no elevation in this cytokine was observed in our model. CSF and plasma from HD patients shows increases in cytokines involved with the innate immune response, including IL-6, at early stages of disease [Bjorkqvist, et al. 2008, Dalrymple, et al. 2007]. IL-6 has a dual role as cytokine and growth factor and can be regulated by complement factors [Hajishengallis and Lambris 2010]. Importantly, in our model, IL-6 is not changed until well after the onset of neuronal degeneration initiated by mHtt. Rather than a microglial pro-inflammatory response, the elevations in IL-6 may represent a protective response of stressed or damaged neurons, as has been shown with injuries such as axotomy [Murphy, et al. 1999] and cerebral ischemia [Suzuki, et al. 1999].
4.3. Conclusions: amplified responses of normal microglia after neuronal expression of mHtt
We observed commonalities between the reaction of microglia to control neurons and to neurons expressing mHtt; however, a clear amplification of these normal responses occurred following neuronal expression of mhtt fragments. We propose that, in the early stages of the disease, microglia are stimulated and targeted to neuronal processes, where they proliferate. This targeting occurs similar to the normal microglial response to synapse loss or dysfunction and process degeneration, and is not specifically driven by the aggregation of mhtt fragments within the neurites. The suggested sequence of events involves microglial proliferation, morphological progression towards an amoeboid phenotype, the recognition of C1q on neuronal synaptic terminals and somata for phagocytosis and clearance, and an elevation in IL-6 as a potential component of the repair process. In the context of HD therapy research, the neuroprotection facilitated by these cells cautions against the assumption that microglia implicitly pose a detrimental threat to neurons. Future detailed examination into mhtt fragment-induced changes in synaptic health and the resultant amplification of normal microglial processes will contribute to defining the role of these cells during the initiating stage of disease.
Supplementary Material
Acknowledgements
The authors wish to thank Gregory Turmel, M. McLean Bolton, Bijal Shah, and Denise Dunn for their technical assistance, Drs. William Mundy and Rebekah Jakel for their review of the manuscript, and Dr. Andrew Tenner (University of California, Irvine) for her gift of C1q antibody. This research was partially funded by the Division of Intramural Research of the National Institute of Environmental Health Sciences, National Institutes of Health, Department of Health and Human Services Z# ES101623 and ES021164, the Cure Huntington's Disease Initiative Foundation, Inc., and the Hereditary Disease Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
None of the authors have any actual or potential conflicts of interest
References
- Albin RL, Young AB, Penney JB, Handelin B, Balfour R, Anderson KD, Markel DS, Tourtellotte WW, Reiner A. Abnormalities of striatal projection neurons and N-methyl-D-aspartate receptors in presymptomatic Huntington's disease. N Engl J Med. 1990;322:1293–8. doi: 10.1056/NEJM199005033221807. [DOI] [PubMed] [Google Scholar]
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–10. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- Bindokas VP, Miller RJ. Excitotoxic degeneration is initiated at non-random sites in cultured rat cerebellar neurons. J Neurosci. 1995;15:6999–7011. doi: 10.1523/JNEUROSCI.15-11-06999.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkqvist M, Wild EJ, Thiele J, Silvestroni A, Andre R, Lahiri N, Raibon E, Lee RV, Benn CL, Soulet D, Magnusson A, Woodman B, Landles C, Pouladi MA, Hayden MR, Khalili-Shirazi A, Lowdell MW, Brundin P, Bates GP, Leavitt BR, Moller T, Tabrizi SJ. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington's disease. J Exp Med. 2008;205:1869–77. doi: 10.1084/jem.20080178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blinzinger K, Kreutzberg G. Displacement of synaptic terminals from regenerating motoneurons by microglial cells. Z Zellforsch Mikrosk Anat. 1968;85:145–57. doi: 10.1007/BF00325030. [DOI] [PubMed] [Google Scholar]
- Cattaneo E, Zuccato C, Tartari M. Normal huntingtin function: an alternative approach to Huntington's disease. Nat Rev Neurosci. 2005;6:919–30. doi: 10.1038/nrn1806. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Wu N, Andre VM, Cummings DM, Levine MS. The corticostriatal pathway in Huntington's disease. Progress in neurobiology. 2007;81:253–71. doi: 10.1016/j.pneurobio.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JK, Schilling G, Peters MF, Herring WJ, Sharp AH, Kaminsky Z, Masone J, Khan FA, Delanoy M, Borchelt DR, Dawson VL, Dawson TM, Ross CA. Truncated N-terminal fragments of huntingtin with expanded glutamine repeats form nuclear and cytoplasmic aggregates in cell culture. Hum Mol Genet. 1998;7:783–90. doi: 10.1093/hmg/7.5.783. [DOI] [PubMed] [Google Scholar]
- Crittenden JR, Dunn DE, Merali FI, Woodman B, Yim M, Borkowska AE, Frosch MP, Bates GP, Housman DE, Lo DC, Graybiel AM. CalDAG-GEFI down-regulation in the striatum as a neuroprotective change in Huntington's disease. Human molecular genetics. 2010;19:1756–65. doi: 10.1093/hmg/ddq055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalrymple A, Wild EJ, Joubert R, Sathasivam K, Bjorkqvist M, Petersen A, Jackson GS, Isaacs JD, Kristiansen M, Bates GP, Leavitt BR, Keir G, Ward M, Tabrizi SJ. Proteomic profiling of plasma in Huntington's disease reveals neuroinflammatory activation and biomarker candidates. J Proteome Res. 2007;6:2833–40. doi: 10.1021/pr0700753. [DOI] [PubMed] [Google Scholar]
- Diaz-Hernandez M, Diez-Zaera M, Sanchez-Nogueiro J, Gomez-Villafuertes R, Canals JM, Alberch J, Miras-Portugal MT, Lucas JJ. Altered P2X7-receptor level and function in mouse models of Huntington's disease and therapeutic efficacy of antagonist administration. Faseb J. 2009;23:1893–906. doi: 10.1096/fj.08-122275. [DOI] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–3. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- DiProspero NA, Chen EY, Charles V, Plomann M, Kordower JH, Tagle DA. Early changes in Huntington's disease patient brains involve alterations in cytoskeletal and synaptic elements. J Neurocytol. 2004;33:517–33. doi: 10.1007/s11068-004-0514-8. [DOI] [PubMed] [Google Scholar]
- Eyupoglu IY, Bechmann I, Nitsch R. Modification of microglia function protects from lesion-induced neuronal alterations and promotes sprouting in the hippocampus. Faseb J. 2003;17:1110–1. doi: 10.1096/fj.02-0825fje. [DOI] [PubMed] [Google Scholar]
- Fraser DA, Pisalyaput K, Tenner AJ. C1q enhances microglial clearance of apoptotic neurons and neuronal blebs. J Neurochem. 2010;112:733–43. doi: 10.1111/j.1471-4159.2009.06494.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehrmann J, Matsumoto Y, Kreutzberg GW. Microglia: intrinsic immuneffector cell of the brain. Brain Res Brain Res Rev. 1995;20:269–87. doi: 10.1016/0165-0173(94)00015-h. [DOI] [PubMed] [Google Scholar]
- Giorgini F, Moller T, Kwan W, Zwilling D, Wacker JL, Hong S, Tsai LC, Cheah CS, Schwarcz R, Guidetti P, Muchowski PJ. Histone deacetylase inhibition modulates kynurenine pathway activation in yeast, microglia, and mice expressing a mutant huntingtin fragment. J Biol Chem. 2008;283:7390–400. doi: 10.1074/jbc.M708192200. [DOI] [PubMed] [Google Scholar]
- Graeber MB, Lopez-Redondo F, Ikoma E, Ishikawa M, Imai Y, Nakajima K, Kreutzberg GW, Kohsaka S. The microglia/macrophage response in the neonatal rat facial nucleus following axotomy. Brain Res. 1998;813:241–53. doi: 10.1016/s0006-8993(98)00859-2. [DOI] [PubMed] [Google Scholar]
- Graeber MB, Tetzlaff W, Streit WJ, Kreutzberg GW. Microglial cells but not astrocytes undergo mitosis following rat facial nerve axotomy. Neurosci Lett. 1988;85:317–21. doi: 10.1016/0304-3940(88)90585-x. [DOI] [PubMed] [Google Scholar]
- Graveland GA, Williams RS, DiFiglia M. Evidence for degenerative and regenerative changes in neostriatal spiny neurons in Huntington's disease. Science. 1985;227:770–3. doi: 10.1126/science.3155875. [DOI] [PubMed] [Google Scholar]
- Gutekunst CA, Levey AI, Heilman CJ, Whaley WL, Yi H, Nash NR, Rees HD, Madden JJ, Hersch SM. Identification and localization of huntingtin in brain and human lymphoblastoid cell lines with anti-fusion protein antibodies. Proc Natl Acad Sci U S A. 1995;92:8710–4. doi: 10.1073/pnas.92.19.8710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutekunst CA, Li SH, Yi H, Mulroy JS, Kuemmerle S, Jones R, Rye D, Ferrante RJ, Hersch SM, Li XJ. Nuclear and neuropil aggregates in Huntington's disease: relationship to neuropathology. J Neurosci. 1999;19:2522–34. doi: 10.1523/JNEUROSCI.19-07-02522.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G, Lambris JD. Crosstalk pathways between Toll-like receptors and the complement system. Trends in immunology. 2010;31:154–63. doi: 10.1016/j.it.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–94. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- HD Collaborative Research Group T. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell. 1993;72:971–83. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Heppner FL, Roth K, Nitsch R, Hailer NP. Vitamin E induces ramification and downregulation of adhesion molecules in cultured microglial cells. Glia. 1998;22:180–8. [PubMed] [Google Scholar]
- Hetier E, Ayala J, Bousseau A, Denefle P, Prochiantz A. Amoeboid Microglial Cells and not Astrocytes Synthesize TNF-alpha in Swiss Mouse Brain Cell Cultures. Eur J Neurosci. 1990;2:762–8. doi: 10.1111/j.1460-9568.1990.tb00466.x. [DOI] [PubMed] [Google Scholar]
- Huuskonen J, Suuronen T, Miettinen R, van Groen T, Salminen A. A refined in vitro model to study inflammatory responses in organotypic membrane culture of postnatal rat hippocampal slices. J Neuroinflammation. 2005;2:25. doi: 10.1186/1742-2094-2-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliev AI, Stringaris AK, Nau R, Neumann H. Neuronal injury mediated via stimulation of microglial toll-like receptor-9 (TLR9). Faseb J. 2004;18:412–4. doi: 10.1096/fj.03-0670fje. [DOI] [PubMed] [Google Scholar]
- Inoue K. Microglial activation by purines and pyrimidines. Glia. 2002;40:156–63. doi: 10.1002/glia.10150. [DOI] [PubMed] [Google Scholar]
- Kaltenbach LS, Bolton MM, Shah B, Kanju PM, Lewis GM, Turmel GJ, Whaley JC, Trask OJ, Jr., Lo DC. Composite Primary Neuronal High-Content Screening Assay for Huntington's Disease Incorporating Non-Cell-Autonomous Interactions. Journal of biomolecular screening : the official journal of the Society for Biomolecular Screening. 2010;15:806–19. doi: 10.1177/1087057110373392. [DOI] [PubMed] [Google Scholar]
- Kim SU, de Vellis J. Microglia in health and disease. J Neurosci Res. 2005;81:302–13. doi: 10.1002/jnr.20562. [DOI] [PubMed] [Google Scholar]
- Kraft AD, Johnson DA, Johnson JA. Nuclear factor E2-related factor 2-dependent antioxidant response element activation by tert-butylhydroquinone and sulforaphane occurring preferentially in astrocytes conditions neurons against oxidative insult. J Neurosci. 2004;24:1101–12. doi: 10.1523/JNEUROSCI.3817-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft AD, McPherson CA, Harry GJ. Heterogeneity of microglia and TNF signaling as determinants for neuronal death or survival. Neurotoxicology. 2009;30:785–93. doi: 10.1016/j.neuro.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuemmerle S, Gutekunst CA, Klein AM, Li XJ, Li SH, Beal MF, Hersch SM, Ferrante RJ. Huntington aggregates may not predict neuronal death in Huntington's disease. Ann Neurol. 1999;46:842–9. [PubMed] [Google Scholar]
- Kurpius D, Nolley EP, Dailey ME. Purines induce directed migration and rapid homing of microglia to injured pyramidal neurons in developing hippocampus. Glia. 2007;55:873–84. doi: 10.1002/glia.20509. [DOI] [PubMed] [Google Scholar]
- Kyrkanides S, O'Banion MK, Whiteley PE, Daeschner JC, Olschowka JA. Enhanced glial activation and expression of specific CNS inflammation-related molecules in aged versus young rats following cortical stab injury. J Neuroimmunol. 2001;119:269–77. doi: 10.1016/s0165-5728(01)00404-0. [DOI] [PubMed] [Google Scholar]
- Landles C, Bates GP. Huntingtin and the molecular pathogenesis of Huntington's disease. Fourth in molecular medicine review series. EMBO Rep. 2004;5:958–63. doi: 10.1038/sj.embor.7400250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Li SH, Cheng AL, Mangiarini L, Bates GP, Li XJ. Ultrastructural localization and progressive formation of neuropil aggregates in Huntington's disease transgenic mice. Hum Mol Genet. 1999;8:1227–36. doi: 10.1093/hmg/8.7.1227. [DOI] [PubMed] [Google Scholar]
- Li H, Li SH, Yu ZX, Shelbourne P, Li XJ. Huntingtin aggregate-associated axonal degeneration is an early pathological event in Huntington's disease mice. J Neurosci. 2001;21:8473–81. doi: 10.1523/JNEUROSCI.21-21-08473.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JY, Plomann M, Brundin P. Huntington's disease: a synaptopathy? Trends Mol Med. 2003;9:414–20. doi: 10.1016/j.molmed.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Lo DC, McAllister AK, Katz LC. Neuronal transfection in brain slices using particle-mediated gene transfer. Neuron. 1994;13:1263–8. doi: 10.1016/0896-6273(94)90412-x. [DOI] [PubMed] [Google Scholar]
- Long-Smith CM, Sullivan AM, Nolan YM. The influence of microglia on the pathogenesis of Parkinson's disease. Prog Neurobiol. 2009;89:277–87. doi: 10.1016/j.pneurobio.2009.08.001. [DOI] [PubMed] [Google Scholar]
- Lynch NJ, Willis CL, Nolan CC, Roscher S, Fowler MJ, Weihe E, Ray DE, Schwaeble WJ. Microglial activation and increased synthesis of complement component C1q precedes blood-brain barrier dysfunction in rats. Molecular immunology. 2004;40:709–16. doi: 10.1016/j.molimm.2003.08.009. [DOI] [PubMed] [Google Scholar]
- Ma L, Morton AJ, Nicholson LF. Microglia density decreases with age in a mouse model of Huntington's disease. Glia. 2003;43:274–80. doi: 10.1002/glia.10261. [DOI] [PubMed] [Google Scholar]
- Magnus T, Chan A, Grauer O, Toyka KV, Gold R. Microglial phagocytosis of apoptotic inflammatory T cells leads to down-regulation of microglial immune activation. J Immunol. 2001;167:5004–10. doi: 10.4049/jimmunol.167.9.5004. [DOI] [PubMed] [Google Scholar]
- Martin JB, Gusella JF. Huntington's disease. Pathogenesis and management. N Engl J Med. 1986;315:1267–76. doi: 10.1056/NEJM198611133152006. [DOI] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meda L, Cassatella MA, Szendrei GI, Otvos L, Jr., Baron P, Villalba M, Ferrari D, Rossi F. Activation of microglial cells by beta-amyloid protein and interferon-gamma. Nature. 1995;374:647–50. doi: 10.1038/374647a0. [DOI] [PubMed] [Google Scholar]
- Murphy PG, Borthwick LS, Johnston RS, Kuchel G, Richardson PM. Nature of the retrograde signal from injured nerves that induces interleukin-6 mRNA in neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1999;19:3791–800. doi: 10.1523/JNEUROSCI.19-10-03791.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napoli I, Neumann H. Microglial clearance function in health and disease. Neuroscience. 2009;158:1030–8. doi: 10.1016/j.neuroscience.2008.06.046. [DOI] [PubMed] [Google Scholar]
- Neumann H, Kotter MR, Franklin RJ. Debris clearance by microglia: an essential link between degeneration and regeneration. Brain. 2009;132:288–95. doi: 10.1093/brain/awn109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nucifora FC, Jr., Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, Troncoso J, Dawson VL, Dawson TM, Ross CA. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science. 2001;291:2423–8. doi: 10.1126/science.1056784. [DOI] [PubMed] [Google Scholar]
- Ogden CA, deCathelineau A, Hoffmann PR, Bratton D, Ghebrehiwet B, Fadok VA, Henson PM. C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J Exp Med. 2001;194:781–95. doi: 10.1084/jem.194.6.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ona VO, Li M, Vonsattel JP, Andrews LJ, Khan SQ, Chung WM, Frey AS, Menon AS, Li XJ, Stieg PE, Yuan J, Penney JB, Young AB, Cha JH, Friedlander RM. Inhibition of caspase-1 slows disease progression in a mouse model of Huntington's disease. Nature. 1999;399:263–7. doi: 10.1038/20446. [DOI] [PubMed] [Google Scholar]
- Panov AV, Gutekunst CA, Leavitt BR, Hayden MR, Burke JR, Strittmatter WJ, Greenamyre JT. Early mitochondrial calcium defects in Huntington's disease are a direct effect of polyglutamines. Nat Neurosci. 2002;5:731–6. doi: 10.1038/nn884. [DOI] [PubMed] [Google Scholar]
- Pavese N, Gerhard A, Tai YF, Ho AK, Turkheimer F, Barker RA, Brooks DJ, Piccini P. Microglial activation correlates with severity in Huntington disease: a clinical and PET study. Neurology. 2006;66:1638–43. doi: 10.1212/01.wnl.0000222734.56412.17. [DOI] [PubMed] [Google Scholar]
- Perry VH, Nicoll JA, Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol. 2010;6:193–201. doi: 10.1038/nrneurol.2010.17. [DOI] [PubMed] [Google Scholar]
- Rappert A, Bechmann I, Pivneva T, Mahlo J, Biber K, Nolte C, Kovac AD, Gerard C, Boddeke HW, Nitsch R, Kettenmann H. CXCR3-dependent microglial recruitment is essential for dendrite loss after brain lesion. J Neurosci. 2004;24:8500–9. doi: 10.1523/JNEUROSCI.2451-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogove AD, Lu W, Tsirka SE. Microglial activation and recruitment, but not proliferation, suffice to mediate neurodegeneration. Cell Death Differ. 2002;9:801–6. doi: 10.1038/sj.cdd.4401041. [DOI] [PubMed] [Google Scholar]
- Ross CA. Polyglutamine pathogenesis: emergence of unifying mechanisms for Huntington's disease and related disorders. Neuron. 2002;35:819–22. doi: 10.1016/s0896-6273(02)00872-3. [DOI] [PubMed] [Google Scholar]
- Runne H, Regulier E, Kuhn A, Zala D, Gokce O, Perrin V, Sick B, Aebischer P, Deglon N, Luthi-Carter R. Dysregulation of gene expression in primary neuron models of Huntington's disease shows that polyglutamine-related effects on the striatal transcriptome may not be dependent on brain circuitry. J Neurosci. 2008;28:9723–31. doi: 10.1523/JNEUROSCI.3044-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapp E, Kegel KB, Aronin N, Hashikawa T, Uchiyama Y, Tohyama K, Bhide PG, Vonsattel JP, DiFiglia M. Early and progressive accumulation of reactive microglia in the Huntington disease brain. J Neuropathol Exp Neurol. 2001;60:161–72. doi: 10.1093/jnen/60.2.161. [DOI] [PubMed] [Google Scholar]
- Sapp E, Penney J, Young A, Aronin N, Vonsattel JP, DiFiglia M. Axonal transport of N-terminal huntingtin suggests early pathology of corticostriatal projections in Huntington disease. J Neuropathol Exp Neurol. 1999;58:165–73. doi: 10.1097/00005072-199902000-00006. [DOI] [PubMed] [Google Scholar]
- Saudou F, Finkbeiner S, Devys D, Greenberg ME. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998;95:55–66. doi: 10.1016/s0092-8674(00)81782-1. [DOI] [PubMed] [Google Scholar]
- Shin JY, Fang ZH, Yu ZX, Wang CE, Li SH, Li XJ. Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J Cell Biol. 2005;171:1001–12. doi: 10.1083/jcb.200508072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons DA, Casale M, Alcon B, Pham N, Narayan N, Lynch G. Ferritin accumulation in dystrophic microglia is an early event in the development of Huntington's disease. Glia. 2007;55:1074–84. doi: 10.1002/glia.20526. [DOI] [PubMed] [Google Scholar]
- Singhrao SK, Neal JW, Morgan BP, Gasque P. Increased complement biosynthesis by microglia and complement activation on neurons in Huntington's disease. Exp Neurol. 1999;159:362–76. doi: 10.1006/exnr.1999.7170. [DOI] [PubMed] [Google Scholar]
- Smith R, Brundin P, Li JY. Synaptic dysfunction in Huntington's disease: a new perspective. Cell Mol Life Sci. 2005;62:1901–12. doi: 10.1007/s00018-005-5084-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–78. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Tanaka K, Nogawa S, Nagata E, Ito D, Dembo T, Fukuuchi Y. Temporal profile and cellular localization of interleukin-6 protein after focal cerebral ischemia in rats. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 1999;19:1256–62. doi: 10.1097/00004647-199911000-00010. [DOI] [PubMed] [Google Scholar]
- Tabrizi SJ, Cleeter MW, Xuereb J, Taanman JW, Cooper JM, Schapira AH. Biochemical abnormalities and excitotoxicity in Huntington's disease brain. Ann Neurol. 1999;45:25–32. doi: 10.1002/1531-8249(199901)45:1<25::aid-art6>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Tai YF, Pavese N, Gerhard A, Tabrizi SJ, Barker RA, Brooks DJ, Piccini P. Imaging microglial activation in Huntington's disease. Brain Res Bull. 2007;72:148–51. doi: 10.1016/j.brainresbull.2006.10.029. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Wujek JR, Criste GA, Jalabi W, Yin X, Kidd GJ, Stohlman S, Ransohoff R. Evidence for synaptic stripping by cortical microglia. Glia. 2007;55:360–8. doi: 10.1002/glia.20462. [DOI] [PubMed] [Google Scholar]
- Trottier Y, Devys D, Imbert G, Saudou F, An I, Lutz Y, Weber C, Agid Y, Hirsch EC, Mandel JL. Cellular localization of the Huntington's disease protein and discrimination of the normal and mutated form. Nat Genet. 1995;10:104–10. doi: 10.1038/ng0595-104. [DOI] [PubMed] [Google Scholar]
- Trushina E, Dyer RB, Badger JD, 2nd, Ure D, Eide L, Tran DD, Vrieze BT, Legendre-Guillemin V, McPherson PS, Mandavilli BS, Van Houten B, Zeitlin S, McNiven M, Aebersold R, Hayden M, Parisi JE, Seeberg E, Dragatsis I, Doyle K, Bender A, Chacko C, McMurray CT. Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol Cell Biol. 2004;24:8195–209. doi: 10.1128/MCB.24.18.8195-8209.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trushina E, Heldebrant MP, Perez-Terzic CM, Bortolon R, Kovtun IV, Badger JD, 2nd, Terzic A, Estevez A, Windebank AJ, Dyer RB, Yao J, McMurray CT. Microtubule destabilization and nuclear entry are sequential steps leading to toxicity in Huntington's disease. Proc Natl Acad Sci U S A. 2003;100:12171–6. doi: 10.1073/pnas.2034961100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wake H, Moorhouse AJ, Jinno S, Kohsaka S, Nabekura J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci. 2009;29:3974–80. doi: 10.1523/JNEUROSCI.4363-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CE, Tydlacka S, Orr AL, Yang SH, Graham RK, Hayden MR, Li S, Chan AW, Li XJ. Accumulation of N-terminal mutant huntingtin in mouse and monkey models implicated as a pathogenic mechanism in Huntington's disease. Hum Mol Genet. 2008;17:2738–51. doi: 10.1093/hmg/ddn175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilms H, Hartmann D, Sievers J. Ramification of microglia, monocytes and macrophages in vitro: influences of various epithelial and mesenchymal cells and their conditioned media. Cell Tissue Res. 1997;287:447–58. doi: 10.1007/s004410050769. [DOI] [PubMed] [Google Scholar]
- Yang W, Dunlap JR, Andrews RB, Wetzel R. Aggregated polyglutamine peptides delivered to nuclei are toxic to mammalian cells. Hum Mol Genet. 2002;11:2905–17. doi: 10.1093/hmg/11.23.2905. [DOI] [PubMed] [Google Scholar]
- Zhang SC, Fedoroff S. Neuron-microglia interactions in vitro. Acta Neuropathol. 1996;91:385–95. doi: 10.1007/s004010050440. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.