

Abstract
A strategy for the ortho-silylation of aryl ketone, benzaldehyde and benzyl alcohol derivatives has been developed in which a hydroxyl group formally serves as the directing element for Ir-catalyzed arene C-H bond activation. One-pot generation of a (hydrido)silyl ether from the carbonyl compound or alcohol is followed by dehydrogenative cyclization at 80–100 °C in the presence of norbornene as hydrogen acceptor and the combination of 1 mol % [Ir(cod)OMe]2 and 1,10-phenanthroline as catalyst to form benzoxasiloles. The synthetic utility of the benzoxasilole products is demonstrated by conversion to phenol or biaryl derivatives by Tamao-Fleming oxidation or Hiyama cross-coupling. Both of these transformations of the C-H silylation products exploit the Si-O bond in the system and proceed by activation of the silyl moiety with hydroxide, rather than fluoride.
The site-selective functionalization of traditionally inert carbon-hydrogen bonds with transition-metal catalysts is creating new strategies for the construction of functionalized organic molecules.1,2 Among these strategies, the silylation of C-H bonds is particularly attractive because silanes are usually inexpensive, non-toxic, and environmentally benign, and appropriately substituted organosilanes undergo a variety of transformations to form useful derivatives. Since seminal reports on intermolecular dehydrogenative arene silylation first appeared in the 1980s,3 significant advances have been documented, and a variety of directing groups have been identified in recent years that lead to the regioselective ortho-silylation of arenes with hydrosilane coupling partners.4 Despite the progress that has been made, current ortho-silylation processes are typically limited in three ways: 1) Specialized directing groups are often needed that are not commonly found in the final desired product and are not easily installed and removed; 2) mixtures of mono- and bissilylated products are often observed from reactions of substrates containing two ortho C-H bonds; and 3) triorganosilanes are often required for the catalytic process, and these reagents generate tetraorganosilane products, which do not undergo coupling and oxidation processes. We report the development of a regioselective ortho-silylation of benzyl alcohols to form silylated products. The regioselectivity of the silylation process is controlled by a simple hydroxyl group, and the products possess an Si-O bond. Therefore, this reaction can be used as a mild, catalytic method to access arylsilane reagents that undergo cross-coupling and oxidation processes to form C-C and C-O bonds.
The new ortho-silylation of arenes described here originates from the combination of two prior observations from our laboratory (Figure 1, top). First, we reported platinum-catalyzed intramolecular silylation of β-phenethyl dimethylsilane at 200 °C.5 Although this reaction was the first intramolecular arene silylation, the synthetic utility of this reaction was limited by the harsh reaction conditions, lack of scope, and limited reactivity of the tetraorganosilane product. Second, we reported the silyl-directed borylation of aromatic alcohols.6 In this case, the reactions did not occur with benzylic alcohols and led to organoboron, not organosilicon, products.
Figure 1.

(Hydrido)silyl-directed arene C-H bond activation
With these results in mind, however, we envisioned that a hydroxyl group might direct7 the ortho-silylation of arenes by initial formation of a (hydrido)silyl ether (3), followed by a catalytic intramolecular dehydrogenative cyclization (Figure 1, bottom). Because the C-H bond functionalization step is intramolecular, the formation of bis-silylated products is avoided. Furthermore, the presence of the Si-O bond in the resulting benzoxasilole (4)8,9 would allow the product to undergo synthetically useful processes, such as Tamao-Fleming oxidation10 and Hiyama cross-coupling.11
 |
(1) |
We initiated our studies by examining conditions to generate the requisite (hydrido)silyl ethers. Phenol derivatives were reported previously to form the corresponding diethyl(hydrido)silyl ethers by treatment with Et2SiH2 and catalytic [Ir(cod)Cl]2 (0.5 mol %) in benzene.6a,12 We found that [Ir(cod)X]2 (X=Cl, OMe) also catalyzes the hydrosilylation of various carbonyl compounds (5) with Et2SiH2 (1.2 equiv) in THF at room temperature and that these reactions occur with a catalyst loading of just 0.05 mol % (eq 1).13 These conditions provide the corresponding diethyl(hydrido)silyl ethers (6) from a wide range of ketones, aldehydes and alcohols without the need for purification of the silyl ether product.
Having established this route to (hydrido)silyl ethers, we sought to identify conditions for intramolecular dehydrogenative silylation. We began by testing reactions with silyl ether 6a (eq 2) catalyzed by the combination of [Ir(cod)OMe]2 and 4,4'-di-tert-butylbipyridine (dtbpy), which catalyzes the borylation of arenes.14 After heating a solution of 6a (0.25 mmol) and 1 mol % of this catalyst in THF for 16 h at 120 °C, 73% conversion of 6a and 31% yield of the corresponding cyclized product (7a) was observed by GC. However, reactions catalyzed by [Ir(cod)OMe]2 and 1,10-phenanthroline (phen) under otherwise identical conditions occurred with higher conversion (88%) and substantially higher yield (66%) of 7a. Although dehydrogenative cyclization proceeds in the absence of any added H2 scavenger, reactions with 1.2 equiv of added norborene and phen as ligand occurred to full conversion after 16 h at 120 °C to give 87% yield of 7a.15 Moreover, the same reaction at the lower temperature of 80 °C for 16 h gave a comparable 90% yield of 7a. Faster rates were also observed when norbornene was added to reactions conducted with dtbpy as ligand, but the yields for these reactions were lower than those for reactions conducted with phen as ligand. Under our optimized conditions, benzoxasilole (7a) was obtained in 73% overall isolated yield from acetophenone (1.0 mmol scale) by hydrosilylation and intramolecular dehydrogenative arene silylation, followed by standard silica gel chromatography.16
 |
(2) |
The scope of the directed silylation of arenes is shown in Tables 1 and 2. As shown in Table 1, a range of substituted benzoxasilole products were obtained in good yields by cyclization of secondary and tertiary (hydrido)silyl ethers, which were prepared in situ from the corresponding alcohol or ketone precursors. Monoalkyl (7a–d), dialkyl (7e) and aryl (7f) substituents were all tolerated at the α-position of the silyl ether. Formation of a five-membered ring was much faster than formation of a six-membered ring, as exemplified by benzyl-substituted benzoxasilole 7d. In addition to simple benzoxasiloles, tricyclic (7g) and heteroaromatic (7h) products were also accessible by this procedure. Halogen substituents (7i–j) were tolerated under the reaction conditions, as was a boronate ester group (7k). In addition, substrates bearing silyl ether, ester and dialkylamino groups underwent cyclization in good yield (7m–o). However, functional groups that are potentially sensitive to silane-mediated reduction, such as a nitro group, were incompatible with the reaction conditions. Although (hydrido)silyl ethers containing a terminal double bond did not undergo high-yielding cyclization, presumably due to competitive oligomerization from intermolecular reaction of the silane with the alkene, substrates containing cis or trans 1,2-disubstituted alkenes were cyclized in good yield (7p–q), with >95% retention of olefin geometry (as determined by GC and 1H NMR analysis). The selectivity from reactions of substrates that can undergo cyclization to form two constitutional isomers (7l–q) was consistently greater than 20:1, favoring reaction at the less sterically hindered C-H bond. This selectivity mirrors the high selectivity from sterically controlled C-H borylation catalyzed by Ir-bipyridine complexes.14 Finally, we found that the silylation reaction could be conducted with 5 mmol of substrate to provide the corresponding benzoxasilole products (7a, 7c, 7i, 7o) in yields similar to those of reactions conducted on 1 mmol scale.
Table 1.
Benzoxasilole products derived from 2° and 3° silyl ethersa
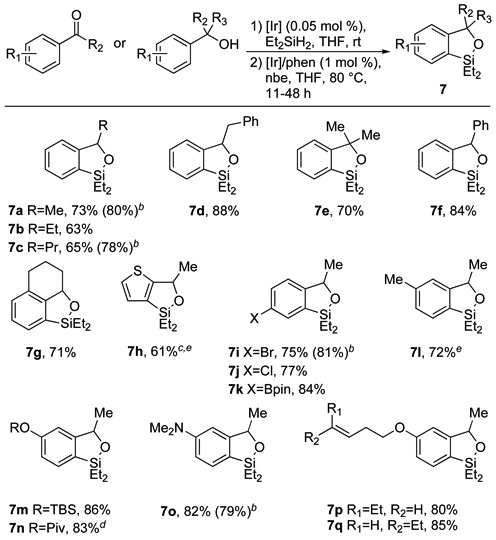 |
Isolated yields for reactions conducted on 1.0 mmol scale following purification by silica gel chromatography.
Isolated yield for reaction conducted on 5.0 mmol scale (using 0.5 mol % [Ir]/phen at 100 °C).
Cyclization conducted at 50 °C.
Cyclization conducted at 100 °C.
Product isolated by bulb-to-bulb distillation.
Table 2.
Benzoxasilole products derived from 1° silyl ethersa
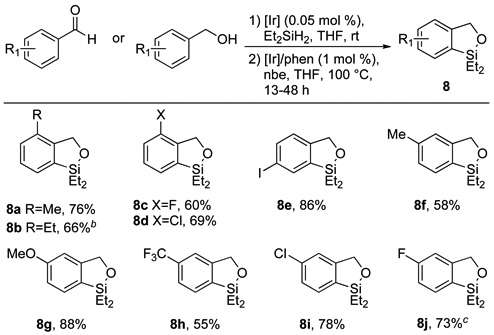 |
Isolated yields for reactions conducted on 1.0 mmol scale following purification by bulb-to-bulb distillation.
Cyclization conducted at 120 °C.
Obtained as an inseparable mixture of constitutional isomers in a 1.0:0.79 ratio, as determined by 1H NMR.
Cyclization of primary (hydrido)silyl ethers derived from benzaldehyde and benzyl alcohol precursors also occurred, and these reactions are summarized in Table 2.17 Reactions of these substrates required a slightly higher temperature of 100 °C to occur to completion, but full conversion was still observed with just 1 mol % catalyst. Benzoxasiloles possessing ortho alkyl groups (8a–b) or small halogens (8c–d) were accessible by the cyclization of primary (hydrido)silyl ethers. These results contrast with those from reactions of ortho-substituted secondary (hydrido)silyl ethers, which did not provide the corresponding benzoxasiloles in high yield. Like reactions of ortho-substituted secondary (hydrido)silyl ethers, reactions of primary (hydrido)silyl ethers containing ortho-bromo and ortho-iodo substituents were low-yielding, likely due the larger size of these substituents. However, a para-iodo substituent was well tolerated by the silylation process (8e). To probe further the origin of the high regioselectivity observed for the cyclization of meta-substituted silyl ethers, a series of primary (hydrido)silyl ethers bearing meta substituents with varying steric and electronic properties were prepared and allowed to cyclize to the corresponding benzoxasiloles (8f–j). Only the reaction with the substrate containing the less sterically-biased 3-fluoro substituent yielded a mixture (1.0:0.79) of constitutional isomers (8j). All others generated a >20:1 ratio of product from silylation at the less hindered position to product from silylation at the more hindered position. This result is consistent with the hypothesis that the regioselectivity of this cyclization is governed by steric, rather than electronic factors.
To develop the synthetic utility of the benzoxasilole products, we sought to identify conditions to transform the Ar-Si moiety into a C-O or C-C bond. A Tamao-Fleming oxidation would correspond to a net ortho-oxygenation of the initial benzylic alcohol.8m,10,18,19 As illustrated in Table 3, this transformation proceeded upon treatment of benzoxasiloles (7 or 8) with H2O2 and KHCO3 in THF/MeOH at room temperature. The fact that oxidation of these substrates occurs without the typical need for fluoride additives20 both increases the operational simplicity of this procedure and also allows for the preparation of silyl ether derivatives such as 9e without any detectible desilylation of the TBS moiety. Furthermore, we were able to use these conditions to prepare oxygenated aryl iodide 9g, which would be difficult to obtain by classical DoM methodology due to competitive metal-halogen exchange.19,21 These transformations can all be conducted in one pot. A sequence of hydrosilylation, ortho-silylation and oxidation in the same flask beginning with acetophenone on 1.0 mmol scale provided 9a in 57% overall isolated yield.22
Table 3.
Tamao-Fleming oxidation of benzoxasilolesa
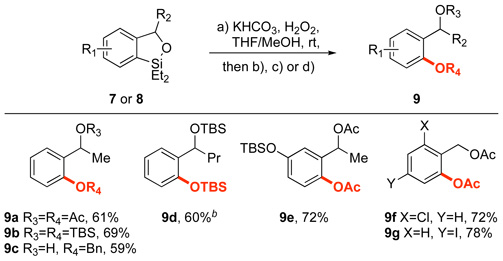 |
Conditions: a. 7 or 8 (1 equiv), KHCO3 (2.0 equiv), 30% H2O2 (8.0 equiv), 1:1 THF/MeOH, rt; b. Ac2O (3.0 equiv), 2:1 CH2Cl2/Et3N, rt; c. TBSCl (2.5 equiv), imidazole (3.0 equiv), DMF, rt; d. BnBr (1.25 equiv), K2CO3 (1.5 equiv), TBAI (0.1 equiv), acetone, 65 °C; isolated yields (over 2 steps) for reactions conducted on 0.50 mmol scale following purification by silica gel chromatography.
Oxidation conducted at 40 °C.
In addition to developing conditions for the Tamao-Fleming oxidation of the benzoxasiloles, we developed conditions for Hiyama-type coupling of the benzoxasiloles with aryl halides. Based on the recent success of Denmark and co-workers in conducting Hiyama couplings with organosilanolate nucleophiles,11a we hypothesized that base-mediated hydrolysis of the benzoxasilole at the Si-O bond might generate an aryl silanolate species capable of undergoing transmetallation.23 After evaluating a series of reaction conditions, we found that the combination of Pd(OAc)2 and 1,2-bis(dicyclohexylphosphino)-ethane (dcpe) in dioxane at 65 °C, in the presence of 5 equiv of 2 M aq NaOH, mediated the coupling of a series of aryl iodides with benzoxasiloles (7) derived from secondary alcohols. As shown in Table 4, a range of biaryl products were obtained in good yield from cross-coupling with electron-neutral (10a), electron-rich (10c, 10e) and electron-deficient (10d, 10f) aryl iodides, as well as heteroaryl coupling partners (10b). The conditions shown in Table 4 were also effective for the cross-coupling of benzoxasiloles with aryl bromides and chlorides. The biaryl products 10a, 10e, 10f were isolated from reactions of these aryl halides in moderate to good yields. Finally, a one-pot sequence of hydrosilylation, ortho-silylation and Hiyama coupling of acetophenone on 1.0 mmol scale provided 10a in 63% overall isolated yield.22
Table 4.
Hiyama coupling of benzoxasilolesa
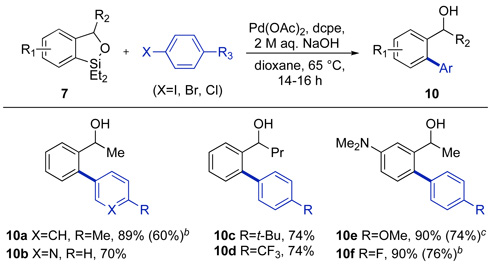 |
Conditions: 7 (1 equiv), aryl iodide (1.2 equiv), Pd(OAc)2 (4 mol %), dcpe (4.5 mol %), 2 M aq. NaOH (5 equiv), dioxane, 65 °C; isolated yields for reactions conducted on 0.25 mmol scale following purification by silica gel chromatography.
Isolated yield for reaction conducted using aryl bromide (1.2 equiv).
Isolated yield for reaction conducted using aryl chloride (1.2 equiv).
During the course of our studies to optimize the Hiyama coupling of benzoxasiloles, we discovered that these species are unreactive in the presence of weak bases, such as phosphate or carbonate. Since such bases are commonly used to activate boronic acid derivatives toward transmetallation, we investigated the possibility of conducting an orthogonal Suzuki-Miyaura coupling with a halogen-containing benzoxasilole. As shown in Scheme 1, coupling of bromo-benzoxasilole 7i with 3,5-dimethylphenylboronic acid promoted by K2CO3 proceeded smoothly to generate biaryl benzoxasilole 11 in 89% yield. Subsequent Hiyama coupling of 11 with 4-iodoanisole promoted by NaOH provided triaryl benzyl alcohol 12 in 79% yield. This sequence could also be run as a one-pot procedure, giving 12 in 65% overall yield from 7i.22 These results illustrate the potential of the hydroxyl-directed ortho-silylation to build molecular complexity.
Scheme 1.

Orthogonal cross-coupling of benzoxasilole 7i. a
a Conditions: a. 7i (1 equiv), 3,5-dimethylphenylboronic acid (1.2 equiv), K2CO3 (2.5 equiv), Pd(PCy3)2Cl2 (2 mol %), THF/H2O, 65 °C; b. 4-iodoanisole (1.2 equiv), Pd(OAc)2 (4 mol %), dcpe (4.5 mol %), 2 M aq. NaOH (5 equiv), dioxane, 65 °C.
Although the mechanistic details of the present silylation have yet to be elucidated, some insight into the C-H bond activation step may be gained from the regioselectivity observed for the silylation of 4-chlorobenzophenone, which produced a mixture of constitutional isomers 13a and 13b in a 71:29 ratio (eq 3). Although the regioselectivity is only modest, the slight preference for silylation of the more electron-deficient C-H bond is consistent with C-H bond cleavage proceeding by an oxidative addition mechanism, rather than an electrophilic metallation pathway.
 |
(3) |
In summary, we report a new strategy for arene ortho-silylation in which a hydroxyl group serves as the directing element for Ir-catalyzed C-H bond activation via dehydrogenative cyclization of in situ-generated diethyl(hydrido)silyl ethers. This transformation proceeds under relatively mild conditions (80–100 °C) with low catalyst loadings (1 mol %), and a range of functional groups were found to be compatible with the reaction conditions. The ability to conduct C-H bond functionalization with a substrate containing an Si-O bond is unusual and makes possible the synthetically useful conversions of the benzoxasilole products to phenol and biaryl derivatives by Tamao-Fleming oxidation and Hiyama cross-coupling. Both of these transformations of the silylated products can be conducted as a part of a one-pot orthosilylation/functionalization procedure, without the need for isolation of the intermediate benzoxasilole. Further studies to expand the substrate scope and to gain insight into the C-H activation and C-Si bond-forming steps of this transformation, are currently in progress.
Supplementary Material
Acknowledgement
We thank the NIH (GM087901) for funding of this work and Johnson Matthey for a gift of [Ir(cod)OMe]2 and Pd(OAc)2. E.M.S. thanks Dan Robbins and Carl Liskey for insightful discussions.
Footnotes
Supporting Information Available: Complete experimental details and characterization of new products. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For general C-H activation reviews, see: Dick AR, Sanford MS. Tetrahedron. 2006;62:2439–2463. Goldberg KI, Goldman AS, editors. Activation and Functionalization of C-H Bonds. Vol. 885. Washington, D.C.: American Chemical Society; 2004. Kakiuchi F, Chatani N. Adv. Synth. Catal. 2003;345:1077–1101. Shilov AE, Shul'pin GB. Chem. Rev. 1997;97:2879–2932. doi: 10.1021/cr9411886.
- 2.For recent applications of C-H activation to complex molecule synthesis, see: Fischer DF, Sarpong R. J. Am. Chem. Soc. 2010;132:5926–5927. doi: 10.1021/ja101893b. Feng Y, Chen G. Angew. Chem. Int. Ed. 2010;49:958–961. doi: 10.1002/anie.200905134. Chen K, Baran PS. Nature. 2009;459:824–828. doi: 10.1038/nature08043. Bowie AL, Jr, Trauner D. J. Org. Chem. 2009;74:1581–1586. doi: 10.1021/jo801791j. For a review, see: Godula K, Sames D. Science. 2006;312:67–72. doi: 10.1126/science.1114731.
- 3.(a) Gustavson WA, Epstein PS, Curtis MD. Organometallics. 1982;1:884–885. [Google Scholar]; (b) Sakakura T, Tokunaga Y, Sodeyama T, Tanaka M. Chem. Lett. 1987:2375–2378. [Google Scholar]
- 4.For examples using hydrosilanes, see: Kakiuchi F, Igi K, Matsumoto M, Chatani N, Murai S. Chem. Lett. 2001:422–423. Kakiuchi F, Igi K, Matsumoto M, Hayamizu T, Chatani N, Murai S. Chem. Lett. 2002:396–397. Kakiuchi F, Matsumoto M, Tsuchiya K, Igi K, Hayamizu T, Chatani N, Murai S. J. Organomet. Chem. 2003;686:134–144. Ihara H, Suginome M. J. Am. Chem. Soc. 2009;131:7502–7503. doi: 10.1021/ja902314v. For examples using other silane species, see: Kakiuchi F, Matsumoto M, Sonoda M, Fukuyama T, Chatani N, Murai S, Furukawa N, Seki Y. Chem. Lett. 2000:750–751. Tobisu M, Ano Y, Chatani N. Chem.-Asian J. 2008;3:1585–1591. doi: 10.1002/asia.200800090.
- 5.Tsukada N, Hartwig JF. J. Am. Chem. Soc. 2005;127:5022–5023. doi: 10.1021/ja050612p. [DOI] [PubMed] [Google Scholar]
- 6. Boebel TA, Hartwig JF. J. Am. Chem. Soc. 2008;130:7534–7435. doi: 10.1021/ja8015878. For an extension of this approach to the silylation of indoles, see: Robbins DW, Boebel TA, Hartwig JF. J. Am. Chem. Soc. 2010;132:4068–4069. doi: 10.1021/ja1006405.
- 7.For recent examples of hydroxyl-directed C-H bond activation, see: Wang X, Lu Y, Dai H-X, Yu J-Q. J. Am. Chem. Soc. 2010;132:12203–12205. doi: 10.1021/ja105366u. Watson AJA, Maxwell AC, Williams JMJ. Org. Lett. 2010;12:3856–3859. doi: 10.1021/ol101548a. Lu Y, Wang D-H, Engle KM, Yu J-Q. J. Am. Chem. Soc. 2010;132:5916–5921. doi: 10.1021/ja101909t.
- 8.For previous examples of structurally related benzoxasiloles, see: Ando W, Sekiguchi A, Migita T. J. Am. Chem. Soc. 1975;97:7159–7160. Ando W, Sekiguchi A. J. Organomet. Chem. 1977;133:219–230. Ando W, Ikeno M, Sekiguchi A. J. Am. Chem. Soc. 1977;99:6447–6449. Kang KT, Song HY, Seo HC. Chem. Lett. 1985:617–620. Farnham WB, Dixon DA, Middleton WJ, Calabrese JC, Harlow RL, Whitney JF, Jones GA, Guggenberger LJ. J. Am. Chem. Soc. 1987;109:476–483. Yamamoto Y, Takeda Y, Akiba K. Tetrahedron Lett. 1989;30:725–728. Sieburth SM, Fensterbank L. J. Org. Chem. 1992;57:5279–5281. Belzner J, Ihmels H, Pauletto L, Noltemeyer M. J. Org. Chem. 1996;61:3315–3319. Fitch JW, Cassidy PE, Ahmed MJ. J. Organomet. Chem. 1996;522:55–57. Hijji YM, Hudrlik PF, Hudrlik AM. Chem. Commun. 1998:1213–1214. Studer A, Steen H. Chem. Eur. J. 1999;5:759–773. Hudrlik PF, Arango JO, Hijji YM, Okoro CO, Hudrlik AM. Can. J. Chem. 2000;78:1421–1427. Bashiardes G, Chaussebourg V, Laverdan G, Pornet J. Chem. Commun. 2004:122–123. doi: 10.1039/b311804e. Chouraqui Gl, Petit M, Aubert C, Malacria M. Org. Lett. 2004;6:1519–1521. doi: 10.1021/ol049484k. Levin S, Nani RR, Reisman SE. Org. Lett. 2010;12:780–783. doi: 10.1021/ol902848k.
- 9.For the use of benzoxasiloles as reusable aryl and alkenyl transfer agents, see: Nakao Y, Imanaka H, Sahoo AK, Yada A, Hiyama T. J. Am. Chem. Soc. 2005;127:6952–6953. doi: 10.1021/ja051281j. Nakao Y, Sahoo AK, Yada A, Chen JS, Hiyama T. Sci. Technol. Adv. Mater. 2006;7:536–543. Nakao Y, Imanaka H, Chen JS, Yada A, Hiyama T. J. Organomet. Chem. 2007;692:585–603. Nakao Y, Ebata S, Chen JS, Imanaka H, Hiyama T. Chem. Lett. 2007;36:606–607. Nakao Y, Chen J, Imanaka H, Hiyama T, Ichikawa Y, Duan W-L, Shintani R, Hayashi T. J. Am. Chem. Soc. 2007;129:9137–9143. doi: 10.1021/ja071969r. Nakao Y, Chen J, Tanaka M, Hiyama T. J. Am. Chem. Soc. 2007;129:11694–11695. doi: 10.1021/ja074728s. Shintani R, Ichikawa Y, Hayashi T, Chen J, Nakao Y, Hiyama T. Org. Lett. 2007;9:4643–4645. doi: 10.1021/ol702125q. Nakao Y, Takeda M, Chen JS, Hiyama T, Ichikawa Y, Shintani R, Hayashi T. Chem. Lett. 2008;37:290–291. Nakao Y, Takeda M, Chen J, Hiyama T. Synlett. 2008:774–776.
- 10.Jones GR, Landais Y. Tetrahedron. 1996;52:7599–7662. [Google Scholar]
- 11.(a) Denmark SE, Regens CS. Acc. Chem. Res. 2008;41:1486–1499. doi: 10.1021/ar800037p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Denmark SE, Sweis RF. In: Metal-Catalyzed Cross-Coupling Reactions. Second Edition. De Meijere A, Diederich F, editors. New York: Wiley-VCH; 2004. pp. 163–216. [Google Scholar]; (c) Hiyama T, Shirakawa E. In: Cross-Coupling Reactions. Miyaura N, editor. Vol. 219. Springer Berlin / Heidelberg: 2002. pp. 61–85. [Google Scholar]
- 12.Field LD, Messerle BA, Rehr M, Soler LP, Hambley TW. Organometallics. 2003;22:2387–2395. [Google Scholar]
- 13.For examples of Rh-catalyzed ketone hydrosilylation with Et2SiH2, see: Reyes C, Prock A, Giering WP. J. Organomet. Chem. 2003;671:13–26. Ojima I, Kogure T, Kumagai M, Horiuchi S, Sato T. J. Organomet. Chem. 1976;122:83–97.
- 14.Mkhalid IAI, Barnard JH, Marder TB, Murphy JM, Hartwig JF. Chem. Rev. 2010;110:890–931. doi: 10.1021/cr900206p. [DOI] [PubMed] [Google Scholar]
- 15.The accelerating effect of norbornene on Ir-catalyzed C-H activation has been observed previously, see: Lu B, Falck JR. Angew. Chem. Int. Ed. 2008;47:7508–7510. doi: 10.1002/anie.200802456. Lu B, Falck JR. J. Org. Chem. 2010;75:1701–1705. doi: 10.1021/jo902678p.
- 16.Although purification of the diethyl(hydrido)silyl ethers was found to be unnecessary, removal of the volatiles (by placing the reaction mixture under vacuum) was necessary to prevent poisoning of the second charge of [Ir(cod)OMe]2 by residual Et2SiH2, in line with previous observations (see ref. 6a).
- 17.The benzoxasilole products derived from primary silyl ethers proved sensitive to silica gel chromatography, but were readily purified by bulb-to-bulb distillation of the concentrated reaction mixture.
- 18.For recent examples of arene ortho-oxygenation by directed C-H activation, see: Zhang Y-H, Yu J-Q. J. Am. Chem. Soc. 2009;131:14654–14655. doi: 10.1021/ja907198n. Desai LV, Malik HA, Sanford MS. Org. Lett. 2006;8:1141–1144. doi: 10.1021/ol0530272. Dick AR, Hull KL, Sanford MS. J. Am. Chem. Soc. 2004;126:2300–2301. doi: 10.1021/ja031543m.
- 19.For arene ortho-oxygenation by a directed ortho-lithiation/silylation/oxidation sequence, see: Bracegirdle S, Anderson EA. Chem. Commun. 2010;46:3454–3456. doi: 10.1039/b924135c.
- 20.For previous examples of fluoride-free Tamao-Fleming oxidation see: (a) ref 19. Tamao K, Ishida N. J. Organomet. Chem. 1984;269:c37–c39.
- 21.Snieckus V. Chem. Rev. 1990;90:879–933. [Google Scholar]
- 22.See the Supporting Information for details.
- 23.For the NaOH-promoted cross-coupling of aryl- and alkenyl(dichloro)-silanes, in which in situ-generated organosilanolates are likely intermediates, see: Hagiwara E, Gouda K-i, Hatanaka Y, Hiyama T. Tetrahedron Lett. 1997;38:439–442.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.