

Abstract
Multiple cell death pathways are activated in cerebral ischemia. Much of the initial injury, especially in the core of the infarct where cerebral blood flow is severely reduced, is necrotic and secondary to severe energy failure. However there is considerable evidence that delayed cell death continues for several days, primarily in the penumbral region. As reperfusion therapies grow in number and effectiveness, restoration of blood flow early after injury may lead to a shift towards apoptosis. It is important to elucidate what are the key mediators of apoptotic cell death after stroke, as inhibition of apoptosis may have therapeutic implications. There are two well described pathways that lead to apoptotic cell death; the caspase pathway and the more recently described caspase-independent pathway triggered by Poly-ADP Ribose Polymers (PARP) activation. Caspase-induced cell death is initiated by release of mitochondrial cytochrome C, formation of the cytosolic apoptosome, and activation of endonucleases leading to a multitude of small randomly cleaved DNA fragments. In contrast caspase-independent cell death is secondary to activation of apoptosis inducing factor (AIF). Mitochondrial AIF translocates to the nucleus, where it induces peripheral chromatin condensation, as well as characteristic high-molecular-weight (50 kbp) DNA fragmentation. Although caspase independent cell death has been recognized for some time and is known to contribute to ischemic injury, the upstream triggering events leading to activation of this pathway remain unclear. The two major theories are that ischemia leads to NAD+ depletion and subsequent energy failure, or alternatively that cell death is directly triggered by a pro-apoptotic factor produced by activation of the DNA repair enzyme PARP. PARP activation is robust in the ischemic brain producing variable lengths of PAR polymers as byproducts of PARP activation. PAR polymers may be directly toxic by triggering mitochondrial AIF release independently of NAD+ depletion. Recently, sex differences have been discovered that illustrate the importance of understanding these molecular pathways, especially as new therapeutics targeting apoptotic cell death are developed. Cell death in females proceeds primarily via caspase activation whereas caspase-independent mechanisms triggered by the activation of Poly-ADP-Ribose Polymerase (PARP) predominate in the male brain. This review summarizes the current literature in an attempt to clarify the roles of NAD+ and PAR polymers in caspase-independent cell death, and discuss sex specific cell death to provide an example of the possible importance of these downstream mediators.
Keywords: Energy Metabolism, NAD+, PARP-1, PAR Polymers, Caspase-Independent Cell Death
Introduction
Stroke affects 15 million people worldwide each year and is the leading cause of long-term disability (Rosamond et al., 2008). Tissue Plasminogen Activator (tPA) is the only approved treatment, which can only be administered within a short treatment window, recently extended to 4.5 hours after stroke onset (Lansberg et al., 2009). However cell death continues beyond this point and targeting specific components of this delayed cell death may provide an opportunity for therapeutic intervention (Siegel et al., 2010). Ischemic cell death can proceed through several well documented molecular cascades, one of which includes activation of caspases. Caspases are cysteine proteases expressed as pro-enzymes and cleaved upon activation (Schulz et al., 1999). Caspase mediated cell death can be triggered by either intrinsic mechanisms, via loss of mitochondrial membrane potential and cytoplasmic cytochrome c release, or from extrinsic signals, usually ligand binding to an cell membrane death receptor such as FasL. Over the past ten years, caspase independent cell death has gained attention and has been shown to be a target for neuroprotective interventions (Lang and McCullough, 2008). These include apoptotic cell death induced by apoptosis-inducing factor (AIF; see below) and neuronal cell death induced by autophagy (Puyal et al., 2009).
A well-studied pathway that does not require caspase activation to induce apoptosis is triggered by rising levels of neuronal nitric oxide synthase (nNOS), poly ADP-Ribose polymerase (PARP-1) activation, and Apoptosis Inducing Factor (AIF) (Figure 1). Interestingly, the mechanism by which PARP-1 activation causes cell death is not completely clear. The literature has suggested that either depletion of Nicotinamide Adenine Dinucleotide (NAD+) or activation of PARP-1 with subsequent Poly ADP-Ribose (PAR) polymer formation (Chiarugi, 2002, Jagtap and Szabo, 2005) are critical steps in the induction of cell death. Whether these pathways act in concert, or are independent processes has been a subject of much debate. Does NAD+ depletion lead to energy failure with the subsequent activation of PARP-1, or does the activation of PARP-1 lead to loss of NAD+ with the formation of cytotoxic PAR polymers? This becomes a critical issue when developing therapies for neuroprotection. Can cell death be halted by repletion of NAD+, or is the inhibition of PARP-1 necessary and sufficient? As PARP-1 inhibitors such as Minocycline are entering clinical trials (Lampl et al., 2007), understanding the molecular sequence of cell death has become a priority. This review will discuss the current literature regarding PARP-mediated cell death and discuss the evidence for NAD+ depletion versus PAR polymer formation as the critical step in triggering caspase-independent ischemic cell death. The importance of this cell death pathway to cell death in the male and female brain, which may have different molecular” executioners”, will also be discussed.
Figure 1.
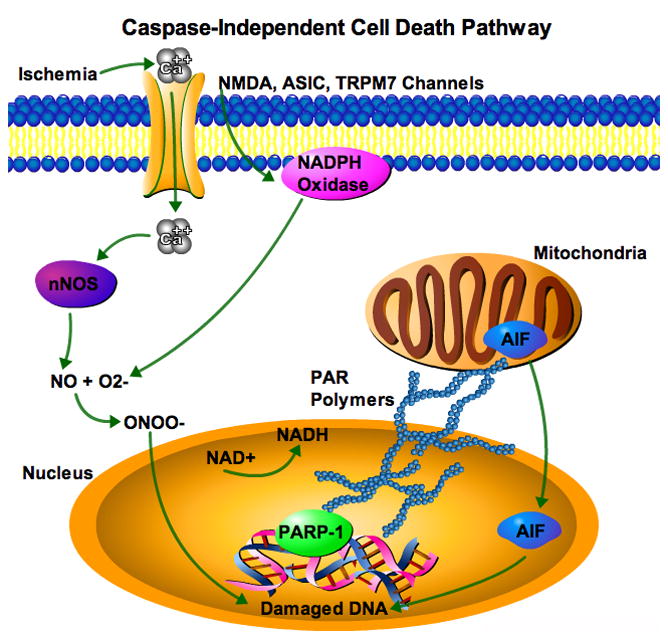
Caspase-Independent Cell Death Cascade. Ischemia causes calcium influx through multiple channels including, NMDA, ASIC, and TRPM7, which leads to increased neuronal nitric oxide synthase (nNOS), and generation of peroxynitrite (ONOO−) from nitric oxide (NO) and superoxide (O2−), damagings DNA. O2− is generated from NMDA receptor activation of NADPH oxidase. The DNA repair enzyme PARP-1 is activated utilizing cellular NAD+ forming PAR polymers, which cause nuclear translocation of AIF further damaging DNA. Cell death occurs from AIF-mediated DNA degradation and NAD+ depletion causing energy failure. What plays the role of executioner?
PARP-1 Mediated Cell Death
PARP-1 is a 116kDa DNA repair enzyme comprised of an N-terminal DNA-binding domain, an automodification domain, and a C-terminal catalytic domain (Hong et al., 2004). PARP-1 binds to damaged DNA and cleaves NAD+ into nicotinamide and ADP-Ribose, which forms PAR polymers on histones, neighboring proteins, and PARP-1 itself (Figure 2a) (Pacher and Szabo, 2008). PAR polymers are formed by PARP-1 activity, which suggests that any substrate of PARP-1 is vulnerable to the effects of PAR polymers. Poly ADP-Ribosylation adds a large structure, which can inhibit protein function, as well as alter charge. PAR has also been linked to p53 and caspases, which play important roles in apoptosis (Hong et al., 2004). PAR polymers have been shown to bind to transcription factors, proteins of the high mobility group, topoisomerases, and PARP-1. Excessive amounts of PAR polymers work in cooperation with acetylation, methylation, and phosphorylation to remodel chromatin. PAR polymers also have a negative charge, which cause repulsion and transcriptional repression (Chiarugi, 2002). Excessive PAR polymer formation is toxic to the cell and mediates nuclear translocation of AIF leading to DNA damage and cell death (Andrabi et al., 2006, Yu et al., 2006). AIF in the cytosol causes depolarization of the mitochondria and exposure of phosphatidylserine on the plasma membrane. AIF in the mitochondria also causes release of cytochrome-c and caspase-9 demonstrating the inter-relation between cell death pathways. AIF in the nucleus induces condensation of chromatin and cleaves DNA into characteristic 50kb fragments, inducing cell death (Susin et al., 1999).
Figure 2.

PAR Polymer Formation and Degradation. (A) PARP-1 encounters DNA strand breaks. PARP-1 breaks down NAD+ into Nicotinamide (NA) and forms PAR polymers while repairing DNA. PAR polymers are toxic, and attach to neighboring proteins and PARP-1 itself. PARG degrades PAR Polymers, and under normal conditions keeps toxic PAR polymers at low levels. (B) Western blot illustrating sex differences in PAR polymer formation. Male mice have increased PAR Polymer formation compared to shams and females 2, 6, 12, and 24 hours after 90 minute MCAO (Yuan et. al., 2009). Note AIF translocation is equivalent between the sexes after injury.
Ischemic cell death is initiated by Ca2+ influx into neurons through activated NMDA receptors, acid sensing ion channels (ASICs) (Xiong et al., 2008), and activation of TRPM7 channels (MacDonald et al., 2006). Ca2+ influx activates neuronal nitric oxide synthase (nNOS) leading to increased nitric oxide (NO) production (Beckman and Crow, 1993). NO combines with the free radical superoxide forming peroxynitrite (ONOO−), a major cause of oxidative and nitrosative DNA damage (Crow and Beckman, 1995). Superoxide formation is induced by NMDA receptor activation of the cytoplasmic enzyme, NADPH oxidase. Inhibition of NADPH oxidase by apocynin or deletion of gp91−/−, a subunit of NADPH oxidase reduces damage in stroke models (Chen et al., 2009). DNA damage secondary to rising levels of oxygen free radicals such as ONOO-activates the DNA repair enzyme PARP-1, which requires both NAD+ and ATP for its function. During stroke, excessive DNA damage leads to over-activation of PARP-1 depleting both NAD+ and ATP in the cell (Ying et al., 2005). PARP-1 activation also results in the formation of PAR polymers (Figure 2) that are implicated in the translocation of the pro-apoptotic molecule AIF from the mitochondria to the nucleus (Andrabi et al., 2006, Yu et al., 2006). AIF subsequently exacerbates DNA damage by effecting nuclease activity, and combined with PARP-1 over-activation depleting the cell’s energy stores leads to cell death (Figure 1) (Yu et al., 2003). These effects are not blocked by caspase inhibitors, leading to the characterization of this pathway as “caspase-independent” (Yu et al., 2000), although there is cross talk between these cell death pathways. Caspase independent cell death has been well characterized with the utilization of different knockout mice including nNOS−/−, PARP-1 −/−, and Harlequin mice (which have an 85% knockdown of AIF) (Ayata et al., 1997, Eliasson et al., 1997, Klein et al., 2002). In these studies, examination of mice with these targeted deletions all yielded the same results: decreased susceptibility to infarction as compared to wild-type littermates (McCullough et al., 2005, Yuan et al., 2009). The beneficial effects of the loss of nNOS, PARP-1 or AIF have been confirmed in neuronal cultures derived from fetal brain (Cao et al., 2003, Eliasson et al., 1999, Eliasson et al., 1997, Huang et al., 1994). Neuroprotection was also seen in mice treated with pharmacological inhibitors of nNOS and PARP-1 (McCullough et al., 2005). Neuronal cultures treated with these same inhibitors resulted in a similar neuroprotection (Abdelkarim et al., 2001, Virag and Szabo, 2002, Yoshida et al., 1994). These data suggest both PARP-1 mediated PAR polymer induced AIF play an integral role in caspase-independent ischemic cell death.
PAR Polymer Mediated AIF Translocation in Cell Death
Stroke-induced PAR polymer formation has been observed in both in vivo (adult) and in vitro (neonatal) models of ischemia (Eliasson et al., 1997, Endres et al., 1997, Tokime et al., 1998). This can be ameliorated by PARP-1 inhibition or deletion (Hagberg et al., 2004, McCullough et al., 2005, Takahashi et al., 1999, Yuan et al., 2009). PAR accumulation is present in the ischemic human brain (Love et al., 2000), suggesting that this may be an appropriate therapeutic target in clinical models. Cell death can be induced by either increasing the production of PAR, or by decreasing its degradation. PAR polymers are cleaved into ADP-ribose by the enzyme poly ADP-Ribose Glycohydrolase (PARG). Complete PARG gene deletion is embryonic lethal (Koh et al., 2004), but animals with deletion of the 110kDa isoform of PARG developed normally. However, when this animal is exposed to stroke, excessive PAR polymers form, leading to an increase in stroke damage compared to WT mice. These data suggest that rising levels of PAR play an important role in the induction of caspase-independent cell death. This is further confirmed in mice overexpressing PARG, which have lower levels of cellular PAR and a reduction in stroke-induced injury (Andrabi et al., 2006). It is unclear if these animals also have metabolic derangements or changes in NAD+ levels after injury. In addition, direct injection of PAR polymers into cells in vitro, leads to rapid and robust nuclear AIF translocation with subsequent cell death that is correlated with the length of the polymer (Andrabi et al., 2006). It is not known if this is also accompanied by NAD+ loss, but this work suggests that PAR accumulation is the final executioner in ischemic cell death.
Energy Metabolism and NAD+ Depletion
Glycolysis produces pyruvate from glucose through a multi-step process including the conversion of NAD+ to NADH. Under pathological conditions cytosolic NAD+, which is normally utilized during glycolysis, is depleted by the over activation of PARP-1. This impairs both anaerobic glycolysis and mitochondrial aerobic respiration. NAD+ is synthesized de novo from exogenous tryptophan or nicotinic acid, or endogenously via recycling of the NAD+ byproduct nicotinamide (Figure 3) (Yang and Sauve, 2006). Nicotinamide Phosphoribosyltranferase (Nampt) is the rate-limiting enzyme in the pathway producing NAD+ from nicotinamide. Nampt converts nicotinamide into nicotinamide mononucleotide (NMN) (Garten et al., 2009, Imai, 2009, Magni et al., 2004). The NAD+ recycling pathway is activated under cellular stress when exogenous precursors are not readily available. Activation of NAD+ recycling functions to restore NAD+ levels, but as cellular stores of ATP are depleted, the cell cannot support NAD+ salvage leading to further energy failure and cell death (Chiarugi, 2002, Suh et al., 2007, Yang and Sauve, 2006).
Figure 3.

NAD+ Biosynthesis. De Novo NAD+ synthesis starts with the conversion of Nicotinic Acid to Nicotinic Acid Mononucleotide (NAMN) by nicotinic acid phosphoribosyltransferase (npt). NAMN is then converted to Nicotinic acid adenine dinucleotide (NAAD) by nicotinic acid/nicotinamide mononucleotide adenylyltransferase (Nmnat). NAAD is finally converted to NAD+ by NAD synthetase. The salvage pathway utilizes Nicotinamide, which is converted to Nicotinamide mononucleotide (NMN) by nicotinamide phosphoribosyltranferase (Nampt). Nmnat then converts NMN to NAD+. The salvage pathway is continually active as NAD+ is degraded by PARP-1 and other proteins (adapted from Yang and Sauve, 2006).
As described earlier, PARP-1 requires NAD+ to perform its functions as a DNA repair enzyme. A family of enzymes known as Sirtuins are NAD+ dependent deacetylases that are involved in both longevity and the response to stroke (Garten et al., 2009, Tang and Chua, 2008). Sirtuins function by hydrolyzing NAD+ into acetyl-ADP ribose, nicotinamide, and deacetylated proteins (histones H3 and H4, NFkB, p53, and FOXO for example) (Fulco and Sartorelli, 2008, Saunders and Verdin, 2007). The role played by Sirt-1 in ischemic cell death and how it interacts with NAD+ is complex. Sirt-1 is a known inducer of longevity, regulates stress responses and metabolism (Howitz et al., 2003, Wood et al., 2004). Sirt-1 has also been implicated in the sequestration of the pro-apoptotic Bax protein, which leads to decreased cell death (Cohen et al., 2004). Transgenic mice overexpressing Sirt-1 have a phenotype similar to that of an animal experiencing caloric restriction (Bordone et al., 2007), which also extends lifespan and reduces stroke injury (Longo, 2009, Ungvari et al., 2008). Resveratrol, a polyphenol found in red wine and Sirt-1 activator, mimics ischemic preconditioning where exposure to a sub-lethal dose of ischemia leads to neuroprotection in part by Sirt-1 activation (Della-Morte et al., 2009) when the brain is subsequently challenged with lethal ischemia (Cadet and Krasnova, 2009). The neuroprotection seen in these studies was abolished by sirtinol, a Sirt1 inhibitor (Chong et al., 2005, Della-Morte et al., 2009, Raval et al., 2006). It is important to remember that Sirt-1 is an NAD+ dependent enzyme. Activation of Sirt-1 is neuroprotective, but when NAD+ is not readily available, activation of Sirt-1 can further deplete NAD+, which is detrimental to energy homeostasis in the cell (Liu et al., 2009a).
Examining PARP-1 mediated cell death from a different angle
A study performed in neuronal cultures found that N-methyl-N-nitro-N-nitrosoguanidine (MNNG), a strong alkylating agent resulting in DNA double-strand breaks, led to robust AIF translocation but only partial loss of NAD+ and ATP (Fossati et al., 2007). This suggests that it is PAR polymer mediated AIF translocation that leads to cell death as opposed to PARP-1 mediated NAD+ depletion. However, other studies have found that MNNG induction of cell death depleted cytosolic NAD+, inhibiting glycolysis (Ying et al., 2005). This is especially relevant to stroke as cytosolic NAD+ depletion renders glucose unusable, and glucose is the only metabolic substrate utilized by the brain under normal (non-hypoxic) conditions. Although constituting only 2% of body mass, the brain utilizes up to 20% of the body’s supply of oxygen and glucose (Alle et al., 2009, Magistretti and Pellerin, 1999, Pellerin et al., 1998). Consumption increases in proportion to neuronal activity, as energy is required to restore membrane potentials after postsynaptic potentials (Alle et al., 2009, Magistretti and Pellerin, 1999). Given the high metabolic energy demands of the brain and its relative intolerance to ischemia, hypoxia, and energy depletion, the role of NAD+ is critical to neuronal function and survival. During ischemia, activation of glycolysis occurs via increased phosphofructokinase (PFK-2) the enzyme that catalyzes the rate-limiting step in glycolysis, increasing fatty acid oxidation and inhibiting glycogen synthesis (Almeida et al., 2004). This leads to increases in ATP, providing energy for energy-depleted neurons. However, unlike peripheral tissues, there is minimal, if any, activation of PFK-2 in neurons (Vilchez et al., 2007), making them unable to produce ATP via glycolysis, the major ATP-generating pathway activated during hypoxia. In addition, neurons do not oxidize fatty acids efficiently, and have no glycogen stores, explaining the exquisite sensitivity of neurons to ischemic damage. Astrocytes can metabolize glucose into lactate through glycolytic pathways, oxidize fatty acids to form ketones and store some glycogen (Bouzier-Sore et al., 2002) providing a short-term energy supply for ischemic neurons (Pellerin and Magistretti, 1994, Tsacopoulos and Magistretti, 1996). However these compensatory strategies are easily exhausted during the severe ischemia seen in stroke.
Although astrocytes initially protect neurons, increasing glycolysis and producing lactate for use by neighboring neurons, providing fuel for synaptic activity (Magistretti et al., 1993, Pellerin and Magistretti, 1994, Tsacopoulos and Magistretti, 1996), with ongoing ischemia, increasing acidosis and lactate production inhibits astroglial glycolysis. These complex metabolic interactions between glia and neurons illustrate the importance of examining the effects of NAD+ repletion in vivo. Administration of Nicotinamide, a precursor to NAD+ is protective after an induced stroke in both adult male mice and neonatal rat models (Klaidman et al., 2003, Sadanaga-Akiyoshi et al., 2003, Feng et al., 2006). Nicotinamide has been shown to prevent NAD+ depletion and reduce ischemic damage in rat cortical neuron cultures (Liu et al., 2009a). NAD+ has also been examined in primary astrocyte culture models. Astrocytes challenged with the DNA alkylating agent MNNG had decreased intracellular NAD+ levels leading to an increase in cell death. However, simply administering exogenous NAD+ or NADH was able to rescue astrocytes from cell death (Ying et al., 2003, Zhu et al., 2005). NAD+ repletion allowed the cell to continue glycolysis with subsequent ATP production. This suggests that energy failure is the primary cause of cell death, and if this can be aborted other pathways (i.e., PARP1) became less relevant. Besides affecting glycolysis NAD+ repletion inhibited mitochondrial membrane depolarization, mitochondrial permeability transition pore (MPT) formation, and AIF translocation (Alano et al., 2004). PARP-1 inhibition with NU1025 reduced infarction, PAR polymer formation, and NAD+ depletion (Kaundal et al., 2006). These studies implicate NAD+ depletion in cell death, but did not directly examine levels of PAR polymer. If NAD+ levels do not drop DNA damage may not occur and PARP will not be activated, making it difficult to determine if NAD or PARP is the executioner of caspase independent cell death. However, recent studies have tried to separate these two mechanisms.
What plays the role of executioner in ischemic cell death?
The majority of the studies presented in this review have focused on NAD+ and PARP-1 vs. PAR and AIF as the primary trigger for non-caspase mediated cell death. It is important to remember that there are multiple regulatory steps in these cascades and that caspase-dependent and caspase-independent cell death is not mutually exclusive. AIF and caspases work together in the cell death cascade, and their contribution may depend on the specific apoptosis-inducing stimulus and perhaps the cell type. In several cases, it appears that the simultaneous neutralization of caspases and AIF is required to prevent hallmarks of apoptosis such as chromatin condensation.
A recent study combined the two in an attempt to determine which mechanism primarily executes caspase-independent cell death. NAD+ repletion prevented glycolytic inhibition, mitochondrial depolarization, and AIF translocation despite an ongoing increase in PAR polymer formation. It was also shown that AIF translocation occurred in PARP-1 −/− animals (which have much less PAR polymer formation due to the lack of PARP) as a result of NAD+ depletion (Alano et al., 2010). This demonstrates that NAD+ depletion can, independently of PARP elevation and PAR formation, induce AIF translocation and subsequent caspase independent cell death. NAD+ may be the critical molecule regulating cell death in this pathway, with PAR formation simply reflecting an “epiphenomenon” of injury rather than a causal factor to cell death. However, others have shown direct injections of PAR polymers are directly cytotoxic, with increasing toxicity with increasing length and complexity of the polymer (Andrabi et al., 2006). Importantly, NAD+ levels were not examined, and it is possible that these polymers caused NAD+ loss with subsequent AIF translocation and cell death.
Can sex differences clarify the role of NAD+ depletion vs. PAR Polymer formation in stroke-induced cell death?
More recently, studies have shown that sex differences exist in the “sensitivity” of male and female brain to specific cell death pathways activated by ischemic injury (Lang and McCullough, 2008, Turtzo and McCullough, 2008). In cell culture models, neurons derived from female embryos were more vulnerable to induction of cell death by cytochrome-c and caspases, while male-derived cortical neurons were more susceptible to cell death by the induction of PARP-1 and translocation of AIF (Du et al., 2004). This has been further supported by subsequent studies in neonatal hypoxic injury and adult MCAO models. The pan caspase inhibitor Q-VD-OPH was neuroprotective in female animals, but had no effect in males (Liu et al., 2009b, Renolleau et al., 2007). In contrast, genetic deletion or pharmacological inhibition of nNOS or PARP-1 was neuroprotective in males, but exacerbated stroke induced damage in neonatal, adult, and aging females (Hagberg et al., 2004, McCullough et al., 2005, Zhu et al., 2006). These data suggest that the female brain is exquisitely sensitive to caspase dependent cell death and that of males by caspase independent mechanisms involving PARP-1 activation and AIF translocation.
Both NAD+ depletion and PAR polymer formation occur in stroke, and both could be the final “executioner” for caspase-independent cell death. However, our work in the female brain has shed some light on this issue. We have shown that although PAR polymer formation is higher in the male brain (Figure 2b); downstream AIF translocation occurs to an equal extent in both sexes (Yuan et al., 2009). Genetic loss of PARP-1, with associated amelioration of PAR polymer formation and AIF translocation led to neuroprotection in males, but exacerbated injury in females (Yuan et al., 2009). Why females seem to be more tolerant to activation of PARP-1, PAR polymer formation, or AIF translocation is not yet known, but it is clear that reducing PAR or AIF has no effect on stroke outcome or paradoxically worsens injury. This suggests that neither AIF nor PAR polymers are the final executioners in caspase-independent cell death, at least in females. As inhibition or deletion of PARP-1 leads to reduction in both PAR formation and AIF translocation and neuroprotection in males, the contribution of PARP activation or NAD+ loss to cell death is more difficult to separate. Obviously the next series of experiments that should be performed to directly answer these questions are to examine NAD+ levels in ischemic brains of PARP knockout and wild-type male and female mice. If pronounced NAD+ loss is seen in PARP knockout females, or if the detrimental phenotype of PARP female mice can be rescued with NAD+ repletion, then it is likely that NAD+ loss is the trigger for caspase independent cell death. We would expect that PARP knockout males would have preservation of intra-ischemic NAD+, thus leading to neuroprotection. Preliminary findings in our lab suggest that PARP-1 −/− females have significantly more rapid and severe ischemia-induced NAD+ depletion compared to WT females, consistent with the hypothesis that NAD+ loss is the causative factor in caspase independent cell death.
It is not yet known if sex differences exist in NAD+ levels. Emerging work suggests that males have a rapid reduction in NAD+ after an induced stroke, which is blunted in the female brain. If NAD+ loss is the critical factor inducing cell death, and females have higher levels of NAD+, retain NAD+ longer, or recycle NAD+ more efficiently, this could explain the relative “insensitivity” of females to PARP-1 (polymer) mediated cell death, as activation of PARP-1 (which occurs to the same levels as in males) would not lead to as dramatic of a NAD+ loss, reducing the main trigger for ischemia-induced cell death. It is also possible that loss of PARP-1 could “shunt” cells into a more detrimental pathway in the female brain, in this case, caspases. Caspase activation is increased in WT females, and caspases cleave PARP-1 (Garnier et al., 2003). We have recently found (unpublished results) that female PARP-1 knockout mice demonstrate higher levels of caspase activation after injury, and that the detrimental effect of PARP-1 deletion can be rescued by administration of a pan-caspase inhibitor. However, how levels of NAD+ change in these models has yet to be evaluated. NAD+ and PAR polymer levels must also be examined to confirm the hypothesis that NAD+ depletion is the important triggering event leading to caspase-independent cell death (Figure 4).
Figure 4.

NAD+ vs. PAR Polymers in Caspase-Independent Cell Death. This cartoon depicts the dichotomy in the caspase-independent cell death cascade (exaggerated for clarity as significant overlap likely occurs in vivo). There is cross talk between PARP-1 and NAD+ depletion, but NAD+ depletion caused glycolytic inhibition, mitochondrial depolarization, AIF translocation, and cell death without PARP-1 activation. Although PARP-1 activation causes PAR polymer formation, AIF translocation and cell death does not necessarily follow. The reviewed literature suggests that NAD+ depletion is necessary and plays the role of executioner in the caspase-independent cell death pathway.
Summary
The PARP-1 inhibitor minocycline is in clinical trial to treat stroke patients, but preclinical data suggest it may be more efficacious in males (Lampl et al., 2007). NAD+ repletion in vivo leads to neuroprotection, but NAD+ or the NAD+ precursor Nicotinamide has only been evaluated when given prior to the induction of ischemia and in male animals. Obviously, a more translational approach is to supplement NAD+ after the ischemic event, as many stroke patients do not come to medical attention for several hours. Whether NAD+ repletion can still salvage ischemic neurons at this late time-point, when metabolic failure and PARP-1 activation is in full swing, is not yet known. In vitro studies suggest that NAD+ repletion is effective even when given after injury (Alano, et. al., 2010) and ongoing studies in our lab are investigating this in MCAO models in animals of both sexes. If females have delayed loss of NAD+, or higher baseline levels of NAD+, the effect of NAD+ repletion may differ compared to males. Perhaps females will respond better to this therapy, whereas males, if they have very rapid loss of NAD+, may not nave any “salvageable NAD+” remaining. Neuronal uptake of NAD+ occurs via the P2X7 receptor, and ongoing studies are examining expression and function of this receptor in experimental stroke. The effects of NAD+ repletion on energy metabolism and NAD+ dependent enzymes in the brain also remain unknown. The current literature suggests that NAD+ is the critical mediator of the caspase-independent cell death, which plays an important role in the male brain. Future studies focusing on this pathway must take sex differences into account when designing experiments and possible therapeutic interventions.
Acknowledgments
Funding: This work was funded by the National Institutes of Health: 1R01-NS055215 and NSO50505 to LDM, 5F31-NS062608-02 to CS, and 5T32-NS041224-08
Footnotes
Conflicts of Interest: There are no conflicts of interest to disclose.
References
- Abdelkarim GE, Gertz K, Harms C, Katchanov J, Dirnagl U, Szabo C, Endres M. Protective effects of PJ34, a novel, potent inhibitor of poly(ADP-ribose) polymerase (PARP) in in vitro and in vivo models of stroke. Int J Mol Med. 2001;7:255–60. [PubMed] [Google Scholar]
- Alano CC, Garnier P, Ying W, Higashi Y, Kauppinen TM, Swanson RA. NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J Neurosci. 2010;30:2967–78. doi: 10.1523/JNEUROSCI.5552-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alano CC, Ying W, Swanson RA. Poly(ADP-ribose) polymerase-1-mediated cell death in astrocytes requires NAD+ depletion and mitochondrial permeability transition. J Biol Chem. 2004;279:18895–902. doi: 10.1074/jbc.M313329200. [DOI] [PubMed] [Google Scholar]
- Alle H, Roth A, Geiger JR. Energy-efficient action potentials in hippocampal mossy fibers. Science. 2009;325:1405–8. doi: 10.1126/science.1174331. [DOI] [PubMed] [Google Scholar]
- Almeida A, Moncada S, Bolanos JP. Nitric oxide switches on glycolysis through the AMP protein kinase and 6-phosphofructo-2-kinase pathway. Nat Cell Biol. 2004;6:45–51. doi: 10.1038/ncb1080. [DOI] [PubMed] [Google Scholar]
- Andrabi SA, Kim NS, Yu SW, Wang H, Koh DW, Sasaki M, Klaus JA, Otsuka T, Zhang Z, Koehler RC, Hurn PD, Poirier GG, Dawson VL, Dawson TM. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci U S A. 2006;103:18308–13. doi: 10.1073/pnas.0606526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayata C, Ayata G, Hara H, Matthews RT, Beal MF, Ferrante RJ, Endres M, Kim A, Christie RH, Waeber C, Huang PL, Hyman BT, Moskowitz MA. Mechanisms of reduced striatal NMDA excitotoxicity in type I nitric oxide synthase knock-out mice. J Neurosci. 1997;17:6908–17. doi: 10.1523/JNEUROSCI.17-18-06908.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckman JS, Crow JP. Pathological implications of nitric oxide, superoxide and peroxynitrite formation. Biochem Soc Trans. 1993;21:330–4. doi: 10.1042/bst0210330. [DOI] [PubMed] [Google Scholar]
- Bordone L, Cohen D, Robinson A, Motta MC, van Veen E, Czopik A, Steele AD, Crowe H, Marmor S, Luo J, Gu W, Guarente L. SIRT1 transgenic mice show phenotypes resembling calorie restriction. Aging Cell. 2007;6:759–67. doi: 10.1111/j.1474-9726.2007.00335.x. [DOI] [PubMed] [Google Scholar]
- Bouzier-Sore AK, Merle M, Magistretti PJ, Pellerin L. Feeding active neurons: (re)emergence of a nursing role for astrocytes. J Physiol Paris. 2002;96:273–82. doi: 10.1016/s0928-4257(02)00016-5. [DOI] [PubMed] [Google Scholar]
- Cadet JL, Krasnova IN. Cellular and molecular neurobiology of brain preconditioning. Mol Neurobiol. 2009;39:50–61. doi: 10.1007/s12035-009-8051-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao G, Clark RS, Pei W, Yin W, Zhang F, Sun FY, Graham SH, Chen J. Translocation of apoptosis-inducing factor in vulnerable neurons after transient cerebral ischemia and in neuronal cultures after oxygen-glucose deprivation. J Cereb Blood Flow Metab. 2003;23:1137–50. doi: 10.1097/01.WCB.0000087090.01171.E7. [DOI] [PubMed] [Google Scholar]
- Chen H, Song YS, Chan PH. Inhibition of NADPH oxidase is neuroprotective after ischemia-reperfusion. J Cereb Blood Flow Metab. 2009;29:1262–72. doi: 10.1038/jcbfm.2009.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarugi A. Poly(ADP-ribose) polymerase: killer or conspirator? The ‘suicide hypothesis’ revisited. Trends Pharmacol Sci. 2002;23:122–9. doi: 10.1016/S0165-6147(00)01902-7. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Lin SH, Li F, Maiese K. The sirtuin inhibitor nicotinamide enhances neuronal cell survival during acute anoxic injury through AKT, BAD, PARP, and mitochondrial associated “anti-apoptotic” pathways. Curr Neurovasc Res. 2005;2:271–85. doi: 10.2174/156720205774322584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, Howitz KT, Gorospe M, de Cabo R, Sinclair DA. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004;305:390–2. doi: 10.1126/science.1099196. [DOI] [PubMed] [Google Scholar]
- Crow JP, Beckman JS. Reactions between nitric oxide, superoxide, and peroxynitrite: footprints of peroxynitrite in vivo. Adv Pharmacol. 1995;34:17–43. doi: 10.1016/s1054-3589(08)61079-0. [DOI] [PubMed] [Google Scholar]
- Della-Morte D, Dave KR, DeFazio RA, Bao YC, Raval AP, Perez-Pinzon MA. Resveratrol pretreatment protects rat brain from cerebral ischemic damage via a sirtuin 1-uncoupling protein 2 pathway. Neuroscience. 2009;159:993–1002. doi: 10.1016/j.neuroscience.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du L, Bayir H, Lai Y, Zhang X, Kochanek PM, Watkins SC, Graham SH, Clark RS. Innate gender-based proclivity in response to cytotoxicity and programmed cell death pathway. J Biol Chem. 2004;279:38563–70. doi: 10.1074/jbc.M405461200. [DOI] [PubMed] [Google Scholar]
- Eliasson MJ, Huang Z, Ferrante RJ, Sasamata M, Molliver ME, Snyder SH, Moskowitz MA. Neuronal nitric oxide synthase activation and peroxynitrite formation in ischemic stroke linked to neural damage. J Neurosci. 1999;19:5910–8. doi: 10.1523/JNEUROSCI.19-14-05910.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliasson MJ, Sampei K, Mandir AS, Hurn PD, Traystman RJ, Bao J, Pieper A, Wang ZQ, Dawson TM, Snyder SH, Dawson VL. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med. 1997;3:1089–95. doi: 10.1038/nm1097-1089. [DOI] [PubMed] [Google Scholar]
- Endres M, Wang ZQ, Namura S, Waeber C, Moskowitz MA. Ischemic brain injury is mediated by the activation of poly(ADP-ribose)polymerase. J Cereb Blood Flow Metab. 1997;17:1143–51. doi: 10.1097/00004647-199711000-00002. [DOI] [PubMed] [Google Scholar]
- Feng Y, Paul IA, LeBlanc MH. Nicotinamide reduces hypoxic ischemic brain injury in the newborn rat. Brain Res Bull. 2006;69:117–22. doi: 10.1016/j.brainresbull.2005.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fossati S, Cipriani G, Moroni F, Chiarugi A. Neither energy collapse nor transcription underlie in vitro neurotoxicity of poly(ADP-ribose) polymerase hyper-activation. Neurochem Int. 2007;50:203–10. doi: 10.1016/j.neuint.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Fulco M, Sartorelli V. Comparing and contrasting the roles of AMPK and SIRT1 in metabolic tissues. Cell Cycle. 2008;7:3669–79. doi: 10.4161/cc.7.23.7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnier P, Ying W, Swanson RA. Ischemic preconditioning by caspase cleavage of poly(ADP-ribose) polymerase-1. J Neurosci. 2003;23:7967–73. doi: 10.1523/JNEUROSCI.23-22-07967.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garten A, Petzold S, Korner A, Imai S, Kiess W. Nampt: linking NAD biology, metabolism and cancer. Trends Endocrinol Metab. 2009;20:130–8. doi: 10.1016/j.tem.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagberg H, Wilson MA, Matsushita H, Zhu C, Lange M, Gustavsson M, Poitras MF, Dawson TM, Dawson VL, Northington F, Johnston MV. PARP-1 gene disruption in mice preferentially protects males from perinatal brain injury. J Neurochem. 2004;90:1068–75. doi: 10.1111/j.1471-4159.2004.02547.x. [DOI] [PubMed] [Google Scholar]
- Hong SJ, Dawson TM, Dawson VL. Nuclear and mitochondrial conversations in cell death: PARP-1 and AIF signaling. Trends Pharmacol Sci. 2004;25:259–64. doi: 10.1016/j.tips.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL, Scherer B, Sinclair DA. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–6. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- Huang Z, Huang PL, Panahian N, Dalkara T, Fishman MC, Moskowitz MA. Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase. Science. 1994;265:1883–5. doi: 10.1126/science.7522345. [DOI] [PubMed] [Google Scholar]
- Imai S. Nicotinamide phosphoribosyltransferase (Nampt): a link between NAD biology, metabolism, and diseases. Curr Pharm Des. 2009;15:20–8. doi: 10.2174/138161209787185814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagtap P, Szabo C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat Rev Drug Discov. 2005;4:421–40. doi: 10.1038/nrd1718. [DOI] [PubMed] [Google Scholar]
- Kaundal RK, Shah KK, Sharma SS. Neuroprotective effects of NU1025, a PARP inhibitor in cerebral ischemia are mediated through reduction in NAD depletion and DNA fragmentation. Life Sci. 2006;79:2293–302. doi: 10.1016/j.lfs.2006.07.034. [DOI] [PubMed] [Google Scholar]
- Klaidman L, Morales M, Kem S, Yang J, Chang ML, Adams JD., Jr Nicotinamide offers multiple protective mechanisms in stroke as a precursor for NAD+, as a PARP inhibitor and by partial restoration of mitochondrial function. Pharmacology. 2003;69:150–7. doi: 10.1159/000072668. [DOI] [PubMed] [Google Scholar]
- Klein JA, Longo-Guess CM, Rossmann MP, Seburn KL, Hurd RE, Frankel WN, Bronson RT, Ackerman SL. The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature. 2002;419:367–74. doi: 10.1038/nature01034. [DOI] [PubMed] [Google Scholar]
- Koh DW, Lawler AM, Poitras MF, Sasaki M, Wattler S, Nehls MC, Stoger T, Poirier GG, Dawson VL, Dawson TM. Failure to degrade poly(ADP-ribose) causes increased sensitivity to cytotoxicity and early embryonic lethality. Proc Natl Acad Sci U S A. 2004;101:17699–704. doi: 10.1073/pnas.0406182101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampl Y, Boaz M, Gilad R, Lorberboym M, Dabby R, Rapoport A, Anca-Hershkowitz M, Sadeh M. Minocycline treatment in acute stroke: an open-label, evaluator-blinded study. Neurology. 2007;69:1404–10. doi: 10.1212/01.wnl.0000277487.04281.db. [DOI] [PubMed] [Google Scholar]
- Lang JT, McCullough LD. Pathways to ischemic neuronal cell death: are sex differences relevant? J Transl Med. 2008;6:33. doi: 10.1186/1479-5876-6-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansberg MG, Bluhmki E, Thijs VN. Efficacy and safety of tissue plasminogen activator 3 to 4.5 hours after acute ischemic stroke: a metaanalysis. Stroke. 2009;40:2438–41. doi: 10.1161/STROKEAHA.109.552547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Gharavi R, Pitta M, Gleichmann M, Mattson MP. Nicotinamide prevents NAD+ depletion and protects neurons against excitotoxicity and cerebral ischemia: NAD+ consumption by SIRT1 may endanger energetically compromised neurons. Neuromolecular Med. 2009a;11:28–42. doi: 10.1007/s12017-009-8058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Li Z, Li J, Siegel C, Yuan R, McCullough LD. Sex differences in caspase activation after stroke. Stroke. 2009b;40:1842–8. doi: 10.1161/STROKEAHA.108.538686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo VD. Linking sirtuins, IGF-I signaling, and starvation. Exp Gerontol. 2009;44:70–4. doi: 10.1016/j.exger.2008.06.005. [DOI] [PubMed] [Google Scholar]
- Love S, Barber R, Wilcock GK. Neuronal death in brain infarcts in man. Neuropathol Appl Neurobiol. 2000;26:55–66. doi: 10.1046/j.1365-2990.2000.00218.x. [DOI] [PubMed] [Google Scholar]
- MacDonald JF, Xiong ZG, Jackson MF. Paradox of Ca2+ signaling, cell death and stroke. Trends Neurosci. 2006;29:75–81. doi: 10.1016/j.tins.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Magistretti PJ, Pellerin L. Cellular mechanisms of brain energy metabolism and their relevance to functional brain imaging. Philos Trans R Soc Lond B Biol Sci. 1999;354:1155–63. doi: 10.1098/rstb.1999.0471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistretti PJ, Sorg O, Yu N, Martin JL, Pellerin L. Neurotransmitters regulate energy metabolism in astrocytes: implications for the metabolic trafficking between neural cells. Dev Neurosci. 1993;15:306–12. doi: 10.1159/000111349. [DOI] [PubMed] [Google Scholar]
- Magni G, Amici A, Emanuelli M, Orsomando G, Raffaelli N, Ruggieri S. Enzymology of NAD+ homeostasis in man. Cell Mol Life Sci. 2004;61:19–34. doi: 10.1007/s00018-003-3161-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough LD, Zeng Z, Blizzard KK, Debchoudhury I, Hurn PD. Ischemic nitric oxide and poly (ADP-ribose) polymerase-1 in cerebral ischemia: male toxicity, female protection. J Cereb Blood Flow Metab. 2005;25:502–12. doi: 10.1038/sj.jcbfm.9600059. [DOI] [PubMed] [Google Scholar]
- Pacher P, Szabo C. Role of the peroxynitrite-poly(ADP-ribose) polymerase pathway in human disease. Am J Pathol. 2008;173:2–13. doi: 10.2353/ajpath.2008.080019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci U S A. 1994;91:10625–9. doi: 10.1073/pnas.91.22.10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellerin L, Pellegri G, Bittar PG, Charnay Y, Bouras C, Martin JL, Stella N, Magistretti PJ. Evidence supporting the existence of an activity-dependent astrocyte-neuron lactate shuttle. Dev Neurosci. 1998;20:291–9. doi: 10.1159/000017324. [DOI] [PubMed] [Google Scholar]
- Puyal J, Vaslin A, Mottier V, Clarke PG. Postischemic treatment of neonatal cerebral ischemia should target autophagy. Ann Neurol. 2009;66:378–89. doi: 10.1002/ana.21714. [DOI] [PubMed] [Google Scholar]
- Raval AP, Dave KR, Perez-Pinzon MA. Resveratrol mimics ischemic preconditioning in the brain. J Cereb Blood Flow Metab. 2006;26:1141–7. doi: 10.1038/sj.jcbfm.9600262. [DOI] [PubMed] [Google Scholar]
- Renolleau S, Fau S, Goyenvalle C, Joly LM, Chauvier D, Jacotot E, Mariani J, Charriaut-Marlangue C. Specific caspase inhibitor Q-VD-OPh prevents neonatal stroke in P7 rat: a role for gender. J Neurochem. 2007;100:1062–71. doi: 10.1111/j.1471-4159.2006.04269.x. [DOI] [PubMed] [Google Scholar]
- Rosamond W, Flegal K, Furie K, Go A, Greenlund K, Haase N, Hailpern SM, Ho M, Howard V, Kissela B, Kittner S, Lloyd-Jones D, McDermott M, Meigs J, Moy C, Nichol G, et al. Heart disease and stroke statistics--2008 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2008;117:e25–146. doi: 10.1161/CIRCULATIONAHA.107.187998. [DOI] [PubMed] [Google Scholar]
- Sadanaga-Akiyoshi F, Yao H, Tanuma S, Nakahara T, Hong JS, Ibayashi S, Uchimura H, Fujishima M. Nicotinamide attenuates focal ischemic brain injury in rats: with special reference to changes in nicotinamide and NAD+ levels in ischemic core and penumbra. Neurochem Res. 2003;28:1227–34. doi: 10.1023/a:1024236614015. [DOI] [PubMed] [Google Scholar]
- Saunders LR, Verdin E. Sirtuins: critical regulators at the crossroads between cancer and aging. Oncogene. 2007;26:5489–504. doi: 10.1038/sj.onc.1210616. [DOI] [PubMed] [Google Scholar]
- Schulz JB, Weller M, Moskowitz MA. Caspases as treatment targets in stroke and neurodegenerative diseases. Ann Neurol. 1999;45:421–9. doi: 10.1002/1531-8249(199904)45:4<421::aid-ana2>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Siegel C, Turtzo C, McCullough LD. Sex differences in cerebral ischemia: possible molecular mechanisms. J Neurosci Res. 2010;88:2765–74. doi: 10.1002/jnr.22406. [DOI] [PubMed] [Google Scholar]
- Suh SW, Hamby AM, Swanson RA. Hypoglycemia, brain energetics, and hypoglycemic neuronal death. Glia. 2007;55:1280–6. doi: 10.1002/glia.20440. [DOI] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–6. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Pieper AA, Croul SE, Zhang J, Snyder SH, Greenberg JH. Post-treatment with an inhibitor of poly(ADP-ribose) polymerase attenuates cerebral damage in focal ischemia. Brain Res. 1999;829:46–54. doi: 10.1016/s0006-8993(99)01335-9. [DOI] [PubMed] [Google Scholar]
- Tang BL, Chua CE. SIRT1 and neuronal diseases. Mol Aspects Med. 2008;29:187–200. doi: 10.1016/j.mam.2007.02.001. [DOI] [PubMed] [Google Scholar]
- Tokime T, Nozaki K, Sugino T, Kikuchi H, Hashimoto N, Ueda K. Enhanced poly(ADP-ribosyl)ation after focal ischemia in rat brain. J Cereb Blood Flow Metab. 1998;18:991–7. doi: 10.1097/00004647-199809000-00008. [DOI] [PubMed] [Google Scholar]
- Tsacopoulos M, Magistretti PJ. Metabolic coupling between glia and neurons. J Neurosci. 1996;16:877–85. doi: 10.1523/JNEUROSCI.16-03-00877.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turtzo LC, McCullough LD. Sex differences in stroke. Cerebrovasc Dis. 2008;26:462–74. doi: 10.1159/000155983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungvari Z, Parrado-Fernandez C, Csiszar A, de Cabo R. Mechanisms underlying caloric restriction and lifespan regulation: implications for vascular aging. Circ Res. 2008;102:519–28. doi: 10.1161/CIRCRESAHA.107.168369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilchez D, Ros S, Cifuentes D, Pujadas L, Valles J, Garcia-Fojeda B, Criado-Garcia O, Fernandez-Sanchez E, Medrano-Fernandez I, Dominguez J, Garcia-Rocha M, Soriano E, Rodriguez de Cordoba S, Guinovart JJ. Mechanism suppressing glycogen synthesis in neurons and its demise in progressive myoclonus epilepsy. Nat Neurosci. 2007;10:1407–13. doi: 10.1038/nn1998. [DOI] [PubMed] [Google Scholar]
- Virag L, Szabo C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev. 2002;54:375–429. doi: 10.1124/pr.54.3.375. [DOI] [PubMed] [Google Scholar]
- Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, Tatar M, Sinclair D. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature. 2004;430:686–9. doi: 10.1038/nature02789. [DOI] [PubMed] [Google Scholar]
- Xiong ZG, Pignataro G, Li M, Chang SY, Simon RP. Acid-sensing ion channels (ASICs) as pharmacological targets for neurodegenerative diseases. Curr Opin Pharmacol. 2008;8:25–32. doi: 10.1016/j.coph.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Sauve AA. NAD metabolism and sirtuins: metabolic regulation of protein deacetylation in stress and toxicity. AAPS J. 2006;8:E632–43. doi: 10.1208/aapsj080472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying W, Alano CC, Garnier P, Swanson RA. NAD+ as a metabolic link between DNA damage and cell death. J Neurosci Res. 2005;79:216–23. doi: 10.1002/jnr.20289. [DOI] [PubMed] [Google Scholar]
- Ying W, Garnier P, Swanson RA. NAD+ repletion prevents PARP-1-induced glycolytic blockade and cell death in cultured mouse astrocytes. Biochem Biophys Res Commun. 2003;308:809–13. doi: 10.1016/s0006-291x(03)01483-9. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Limmroth V, Irikura K, Moskowitz MA. The NOS inhibitor, 7-nitroindazole, decreases focal infarct volume but not the response to topical acetylcholine in pial vessels. J Cereb Blood Flow Metab. 1994;14:924–9. doi: 10.1038/jcbfm.1994.123. [DOI] [PubMed] [Google Scholar]
- Yu SW, Andrabi SA, Wang H, Kim NS, Poirier GG, Dawson TM, Dawson VL. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc Natl Acad Sci U S A. 2006;103:18314–9. doi: 10.1073/pnas.0606528103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu SW, Wang H, Dawson TM, Dawson VL. Poly(ADP-ribose) polymerase-1 and apoptosis inducing factor in neurotoxicity. Neurobiol Dis. 2003;14:303–17. doi: 10.1016/j.nbd.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Yuan M, Siegel C, Zeng Z, Li J, Liu F, McCullough LD. Sex differences in the response to activation of the poly (ADP-ribose) polymerase pathway after experimental stroke. Exp Neurol. 2009;217:210–8. doi: 10.1016/j.expneurol.2009.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Xu F, Wang X, Shibata M, Uchiyama Y, Blomgren K, Hagberg H. Different apoptotic mechanisms are activated in male and female brains after neonatal hypoxia-ischaemia. J Neurochem. 2006;96:1016–27. doi: 10.1111/j.1471-4159.2005.03639.x. [DOI] [PubMed] [Google Scholar]
- Zhu K, Swanson RA, Ying W. NADH can enter into astrocytes and block poly(ADP-ribose) polymerase-1-mediated astrocyte death. Neuroreport. 2005;16:1209–12. doi: 10.1097/00001756-200508010-00015. [DOI] [PubMed] [Google Scholar]