

Abstract
Pd(II)-catalyzed β-C(sp3)–H carbonylation of N-arylamides under CO (1 atm) has been achieved. Following amide-directed C(sp3)–H cleavage and insertion of CO into the resulting [Pd(II)–C(sp3)] bond, intramolecular reductive amination gave the corresponding succinimides, which could be readily converted to 1,4-dicarbonyl compounds. This method was found to be effective with substrates containing α-hydrogen atoms and could be applied to effect methylene C(sp3)–H carbonylation of cyclopropanes.
The development of diverse transformations using Pd catalysis to functionalize unactivated β-C(sp3)–H bonds in aliphatic acids and their amide derivatives could offer a new strategy for synthesizing molecules with complex tertiary and quaternary carbon centers.1–3 To broaden the scope of this approach, we have reported a variety of novel C–C and C–heteroatom bond–forming reactions.1,3b We envisioned that a reaction to effect β-carbonylation of C(sp3)–H bonds would be a highly desirable addition to our collection of transformations because replacement of C–H bonds by a highly oxidized carbonyl group installs a versatile handle for further structural elaboration. For example, β-carbonylation of aliphatic acids would provide a novel route to synthetically important 1,4-dicarbonyl compounds4,5 that are wide spread in biologically important natural products (Figure 1).6
Figure 1.
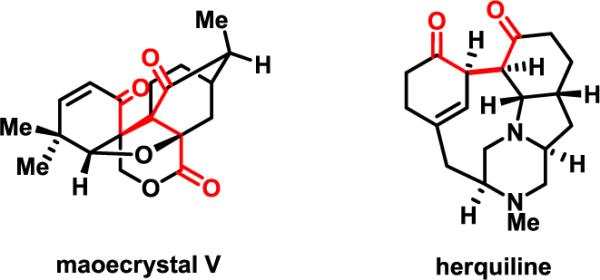
Natural products that contain 1,4-dicarbonyl moieties.
Despite landmark developments in Pd(0)-catalyzed carbonylation of aryl halides, triflates and tosylates7 and recent progress on Pd(II)-catalyzed carbonylation of aryl C(sp2)–H bonds (eq 1),8–11 Pd(II)-catalyzed C(sp3)–H carbonylation reactions that use the substrate as limiting reagent have yet to be reported. Herein, we disclose a protocol for Pd(II)-catalyzed β-C(sp3)–H carbonylation of aliphatic amides to give succinimides which are readily hydrolyzed to afford broadly useful 1,4-dicarbonyl compounds (eq 2). The succinimide intermediates are also well-established synthons for preparation of syn-disubstituted 1,4-dicarbonyl compounds through regioselective and diastereoselective enolate chemistry (eq 2).12 TEMPO was found to be a crucial co-coxidant for efficient reoxidation of Pd(0) to Pd(II) in the presence of CO.
 |
(1) |
 |
(2) |
Early reports by Fujiwara described a Pd-catalyzed C(sp3)–H carbonylation reaction of gaseous alkanes (used in excess) under CO (20 to 50 atm) using K2S2O8 as the reoxidant in TFA at 80 °C, which gave regioisomeric mixture of the corresponding carboxylic acid products.13 The use of excess substrate and the lack of regioselectivity limited the usefulness of this chemistry for synthetic applications. Although regioselective C(sp2)–H carbonylation using 1 equiv of substrate under CO (1 atm) has been accomplished8–11 through the use of proximate directing groups, extending this approach to C(sp3)–H carbonylation represents a significant challenge. One major obstacle is that excess CO inhibits the activation of the inert C(sp3)–H bonds by competitively occupying coordination sites on the Pd(II) center, thereby preventing the requisite C–H agostic interaction. In addition, both the insertion of CO into a [Pd(II)–C(sp3)] cyclopalladated intermediate and reductive elimination from Pd involving a C(sp3)–CO moiety have limited number of precedents.14
With these considerations in mind, we initiated our investigation of Pd-catalyzed C(sp3)–H carbonylation by first identifying a highly efficient directing group for facile C(sp3)–H activation. Recently, our group established that acidic amides are superior directing group for promoting C–H activation reactions with both Pd(0)/PR3 and Pd(II) catalysts.1c,1d We therefore focused our efforts on using acidic amide substrates for our exploratory studies (Table 1).
Table 1.
Optimization of Reaction Conditions for Pd-Catalyzed C(sp3)–H Carbonylationa
| entry | additive | solvent | yield (%)b |
|---|---|---|---|
| 1 | none | DMF | <1 |
| 2 | none | toluene | 7 |
| 3 | none | C6F6 | 8 |
| 4 | none | n-hexane | 30 |
| 5 | BQ | n-hexane | 13 |
| 6 | Cu(OAc)2 | n-hexane | 4 |
| 7 | DMF | n-hexane | 54 |
| 8 | PivOH | n-hexane | 50 |
| 9 | TEMPOc | n-hexane | 80 |
| 10 | TEMPO | n-hexane | 95 |
Reaction conditions: 1a (0.1 mmol), Pd(OAc)2 (10 mol %), AgOAc (2.0 equiv), KH2PO4 (2.0 equiv), additive (2.0 equiv), solvent (1 mL), CO (1 atm), 130 °C, 18 h.
The yield was determined by 1H NMR analysis of the crude product using 1,1,2,2-tetrachloroethane as an internal standard.
TEMPO (0.2 equiv).
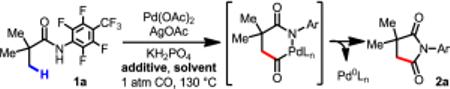
Gratifyingly, we observed that 1a could be transformed into the desired carbonylation product 2a in 30% yield (based on 1H NMR), using Pd(OAc)2 (10 mol %) as the catalyst, AgOAc (2 equiv) as the oxidant, and KH2PO4 as the base, under CO (1 atm) in n-hexane at 130 °C (Table 1, entry 4). Following 1,1-migratory insertion to forge the C(sp3)–CO bond, the intermediate undergoes Pd-mediated reductive amination to give the corresponding succinimide products.
After surveying a wide array of organic solvents, we observed that n-hexane gave the best reactivity. This finding was unexpected, as n-hexane has rarely been an effective solvent in our previous C–H activation reactions.1 Several additives that are known to positively influence Pd-catalyzed C–H functionalization reactions (e.g., 1,4-benzoquinone and Cu(OAc)2) were tested; however, they proved to be incompatible with the reaction conditions (entries 5 and 6). Among the other additives tested, DMF (2 equiv) increased conversion to 54% (entry 7), and pivalic acid improved the yield to 50% (entry 8). Further screening of the additives revealed that the addition of a catalytic amount of TEMPO (2,2,6,6-Tetramethylpiperidine-1-oxyl) (0.2 equiv) as a co-oxidant with AgOAc dramatically improved the conversion to over 80% (entry 9). Although the precise role of TEMPO remains to be elucidated, one plausible explanation is that oxoammonium salt15 (the oxidized form of TEMPO) reoxidizes Pd(0) to Pd(II) more efficiently than solely AgOAc. Stoichiometric amount of both TEMPO and AgOAc is required to achieve full conversion; using 2.0 equiv of TEMPO and AgOAc increased yield to 95% (entry 10), while using 2.0 equiv of TEMPO gave only 12 % conversion (see Supporting Information).
Notably, other directing groups that have previously been utilized in our laboratory for C(sp3)–H activation, such as carboxylic acids, hydroxamic acids, oxazolines, and pyridines, were unreactive under these optimized conditions. An analogous acidic directing group N-toluenesulfonyl amide (CONHTs), however, gives corresponding product in lower yield (63%, see SI).
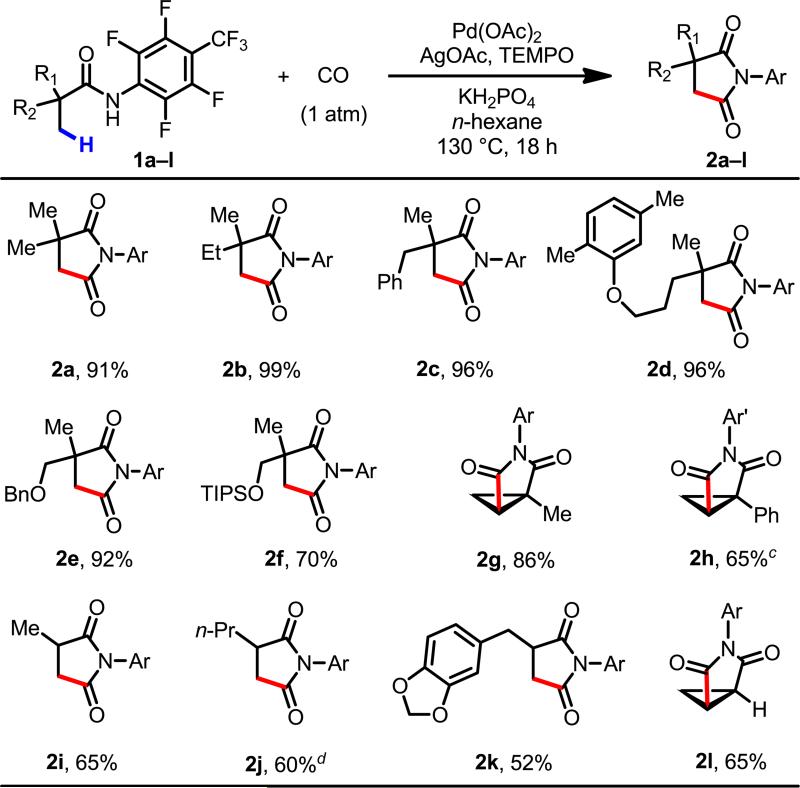
With the optimized conditions in hand, we converted a range of commercial aliphatic acids into the corresponding CONHAr amides to examine the scope of this carbonylation protocol. Substrates with a quaternary α-carbon atom gave good to excellent yields of the succinimide products (2a–h). Products containing ether groups (2d–f and 2k) could also be obtained in good yields. The benzyl moiety proved to be a better protecting group than TIPS group for β-hydroxyl substrates (1e and 1f), while TBS-protected substrates gave none of the desired product. Notably, this method was also effective for the carbonylation of methylene C–H bonds in cyclopropane substrates (2g, 2h and 2l). Intriguingly, cyclopropyl C(sp3)–H bonds could be selectively carbonylated over a methyl C(sp3)–H bond (in 1g) and an ortho aryl C(sp2)–H bond (in 1h).
In our early efforts to develop C(sp3)–H functionalization reactions with aliphatic carboxylic acids and their derivatives, substrates containing α-hydrogen atoms were often unreactive, restricting the substrate scope to those containing quaternary α-carbon atoms. We were pleased to find that, in the present study, carbonylated products 2i–l could be obtained in acceptable yields from substrates containing α-hydrogen atoms. These types of products are highly valuable synthons for preparation of 1,4-dicarbonyl compounds.4–6
To demonstrate the synthetic utility of this reaction, succinimide product 2a was subjected to different ring-opening conditions to obtain either 1,4-dicarboxylic acid 3 or 1,4-dicarbonyl molecule 4 (Scheme 1). Upon treatment of 2a with TFA/AcOH under reflux, hydrolysis occurs to give 2,2-dimethylsuccinic acid 3 in 93% isolated yield. Similarly, treatment of 2a with NaOMe in methanol at room temperature gave 80% of the ester product 4 without concomitant hydrolysis of the amide moiety; thus this group could potentially be used to direct further elaboration of the gem-dimethyl unit via Pd-catalyzed C–H functionalization.
Scheme 1.
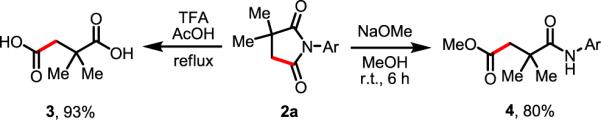
Ring Opening of Succinimides
In summary, we have developed a novel protocol to effect carbonylation of C(sp3)–H bonds under CO (1 atm). Studies to expand the scope of the reaction to simple carboxylic acid substrates and to develop an enantioselective variant for substrates containing gem-dimethyl or cyclopropyl groups are currently underway in our laboratory.
Supplementary Material
Table 2.
|
Reaction conditions: amide substrate 1 (0.1 mmol), Pd(OAc)2 (10 mol %), TEMPO (2.0 equiv), AgOAc (2.0 equiv), KH2PO4 (2.0 equiv), n-hexane (1 mL), CO (1 atm), 130 °C, 18 h.
Isolated yield.
Ar' = C6F5.
Pd(OAc)2 (20 mol %).
Acknowledgement
We gratefully acknowledge The Scripps Research Institute, the National Institutes of Health (NIGMS, 1 R01 GM084019-02), the NSF (NSF CHE-1011898), Amgen and Eli Lilly for financial support. This work was supported by a National Research Foundation of Korea Grant funded by the Korean Government (NRF-2009-352-C00077). We also thank Bristol Myers Squibb for a predoctoral fellowship (M. W.).
References
- (1).(a) Giri R, Maugel N, Li J-J, Wang D-H, Breazzano SP, Saunders LB, Yu J-Q. J. Am. Chem. Soc. 2007;129:3510. doi: 10.1021/ja0701614. [DOI] [PubMed] [Google Scholar]; (b) Wang D-H, Wasa M, Giri R, Yu J-Q. J. Am. Chem. Soc. 2008;130:7190. doi: 10.1021/ja801355s. [DOI] [PubMed] [Google Scholar]; (c) Wasa M, Engle KM, Yu J-Q. J. Am. Chem. Soc. 2009;131:9886. doi: 10.1021/ja903573p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Wasa M, Engle KM, Yu J-Q. J. Am. Chem. Soc. 2010;132:3680. doi: 10.1021/ja1010866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Renaudat A, Jean-Gerard L, Jazzar R, Kefalidis CE, Clot E, Baudoin O. Angew. Chem., Int. Ed. 2010 doi: 10.1002/anie.201003544. DOI: 10.1002/anie.201003544. [DOI] [PubMed] [Google Scholar]
- (3).For early studies on β-C(sp3)–H functionalization reactions of carboxylic acids using strongly coordinating auxiliaries, see: Zaitsev VG, Shabashov D, Daugulis O. J. Am. Chem. Soc. 2005;127:13154. doi: 10.1021/ja054549f. Giri R, Chen X, Yu J-Q. Angew. Chem., Int. Ed. 2005;44:2112. doi: 10.1002/anie.200462884.
- (4).For examples of a classic disconnection involving conjugate addition of acyl anions, see: Corey EJ, Hegedus LS. J. Am. Chem. Soc. 1969;91:4926. Stetter H, Schreckenberg M. Angew. Chem., Int. Ed. 1973;12:81. Groebel B-T, Seebach D. Synthesis. 1977:357. Kubota Y, Nemoto H, Yamamoto Y. J. Org. Chem. 1991;56:7195.
- (5).For recent reports describing the synthesis of 1,4-dicarbonyl compounds, see: Jang H-Y, Hong J-B, MacMillan DWC. J. Am. Chem. Soc. 2007;129:7004. doi: 10.1021/ja0719428. DeMartino MP, Chen K, Baran PS. J. Am. Chem. Soc. 2008;130:11546. doi: 10.1021/ja804159y. Liu Q, Perreault S, Rovis T. J. Am. Chem. Soc. 2008;130:14066. doi: 10.1021/ja805680z.
- (6).(a) Li S-H, Wang J, Niu X-M, Shen Y-H, Zhang H-J, Sun H-D, Li M-L, Tian Q-E, Lu Y, Cao P, Zheng Q-T. Org. Lett. 2004;6:4327. doi: 10.1021/ol0481535. [DOI] [PubMed] [Google Scholar]; (b) Omura S, Hirano A, Iwai Y, Masuma R. J. Antibiot. 1979;32:786. doi: 10.7164/antibiotics.32.786. [DOI] [PubMed] [Google Scholar]; (c) Furusaki A, Matsumoto T, Ogura H, Takayanagi H, Hirano A, Omura S. J. Chem. Soc., Chem. Commun. 1980:698. [Google Scholar]
- (7).(a) Negishi E, de Meijere A, editors. Handbook of Organopalladium Chemistry for Organic Synthesis. Vol. 2. Wiley; New York: 2002. p. 2309. [Google Scholar]; (b) Munday RH, Martinelli JR, Buchwald SL. J. Am. Chem. Soc. 2008;130:2754. doi: 10.1021/ja711449e. [DOI] [PubMed] [Google Scholar]; (c) Barnard CFJ. Organometallics. 2008;27:5402. [Google Scholar]; (d) Brennführer A, Neumann H, Beller M. Angew. Chem., Int. Ed. 2009;48:4114. doi: 10.1002/anie.200900013. [DOI] [PubMed] [Google Scholar]
- (8).For lactam formation via C–H carbonylation, see: Orito K, Horibata A, Nakamura T, Ushito H, Nagasaki H, Yuguchi M, Yamashita S, Tokuda M. J. Am. Chem. Soc. 2004;126:14342. doi: 10.1021/ja045342+.
- (9).For the use of diethyl azodicarboxylate for C–H carbonylation, see: Yu W-Y, Sit WN, Lai K-M, Zhou Z, Chan ASC. J. Am. Chem. Soc. 2008;130:3304. doi: 10.1021/ja710555g.
- (10).For Pd-catalyzed C–H carbonlylation with CO, see: Giri R, Yu J-Q. J. Am. Chem. Soc. 2008;130:14082. doi: 10.1021/ja8063827. Houlden CE, Hutchby M, Bailey CD, Ford JG, Tyler SNG, Gagne MR, Lloyd-Jones GC, Booker-Milburn KI. Angew. Chem., Int. Ed. 2009;48:1830. doi: 10.1002/anie.200805842. Giri R, Lam JK, Yu J-Q. J. Am. Chem. Soc. 2010;132:686. doi: 10.1021/ja9077705. Wu X-F, Anbarasan P, Neumann H, Beller M. Angew. Chem., Int. Ed. 2010 doi: 10.1002/anie.201003895. DOI: 10.1002/anie.201003895.
- (11).For carbonylation reactions of C(sp2)–H bonds using other metals, see: Guan Z-H, Ren Z-H, Spinella SM, Yu S, Liang Y-M, Zhang X. J. Am. Chem. Soc. 2009;131:729. doi: 10.1021/ja807167y. Inoue S, Shiota H, Fukumoto Y, Chatani N. J. Am. Chem. Soc. 2009;131:6898. doi: 10.1021/ja900046z.
- (12).(a) Bennett DJ, Pickering PL, Simpkins NS. Chem. Comm. 2004:1392. doi: 10.1039/b403193h. [DOI] [PubMed] [Google Scholar]; (b) Pohlmannm J, Lampe T, Shimada N, Nell PG, Pernerstorfer J, Svenstrup N, Brunner NA, Schiffer G, Freiberg C. Bioorg. Med. Chem. Lett. 2005;15:1189. doi: 10.1016/j.bmcl.2004.12.002. [DOI] [PubMed] [Google Scholar]
- (13).(a) Nishiguchi T, Nakata K, Takaki K, Fujiwara Y. Chem. Lett. 1992;21:1141. [Google Scholar]; (b) Miyata T, Nakata K, Yamaoka Y, Taniguchi Y, Takaki K, Fujiwara Y. Chem. Lett. 1993;22:1005. [Google Scholar]; (c) Nakata K, Yamaoka Y, Miyata T, Taniguchi Y, Takaki K, Fujiwara Y. J. Organomet. Chem. 1994;473:329. [Google Scholar]
- (14).(a) Balavoine G, Clinet JC. J. Organomet. Chem. 1990;390:C84. [Google Scholar]; (b) Dangel BD, Godula K, Youn SW, Sezen B, Sames D. J. Am. Chem. Soc. 2002;124:11856. doi: 10.1021/ja027311p. [DOI] [PubMed] [Google Scholar]; (c) Bloome KS, Alexanian EJ. J. Am. Chem. Soc. 2010;132:12823. doi: 10.1021/ja1053913. [DOI] [PubMed] [Google Scholar]
- (15).(a) Semmelhack MF, Schmid CR, Cortés DA, Chou CS. J. Am. Chem. Soc. 1984;106:3374. [Google Scholar]; (b) Kochkar H, Lassalle L, Morawietz M, Hölderich WF. J. Catal. 2000;194:343. [Google Scholar]; (c) Kirchberg S, Vogler T, Studer A. Synlett. 2008:2841. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.