

Glutamate dehydrogenase (GDH) has been extensively studied for more than 50 years. Of particular interest is the fact that, while considered by most to be a ‘housekeeping’ enzyme, the animal form of GDH is heavily regulated by a wide array of allosteric effectors and exhibits extensive inter-subunit communication. While the chemical mechanism for GDH has remained unchanged through epochs of evolution, it was not clear how or why animals needed to evolve such a finely tuned form of this enzyme. As reviewed here, recent studies have begun to elucidate these issues. Allosteric regulation first appears in the Ciliates and may have arisen to accommodate evolutionary changes in organelle function. The occurrence of allosteric regulation appears to be coincident with the formation of an ‘antenna’ like feature rising off the tops of the subunits that may be necessary to facilitate regulation. In animals, this regulation further evolved as GDH became integrated into a number of other regulatory pathways. In particular, mutations in GDH that abrogate GTP inhibition result in dangerously high serum levels of insulin and ammonium. Therefore, allosteric regulation of GDH plays an important role in insulin homeostasis. Finally, several compounds have been identified that block GDH-mediated insulin secretion that may be to not only find use in treating these insulin disorders but to kill tumors that require glutamine metabolism for cellular energy.
Homotropic and heterotropic regulation of GDH
Glutamate dehydrogenase (GDH) is found in nearly all living organisms and catalyzes the reversible oxidative deamination of L-glutamate to 2-oxoglutarate using NAD(P)+ as coenzyme (Hudson and Daniel, 1993). This homohexameric enzyme has subunits comprised of ~450 and ~500 amino acids in bacteria and animals, respectively. In eukaryotic organisms, GDH resides within the inner mitochondrial matrix where it catabolizes glutamate to feed 2-oxoglutarate to the Krebs cycle. Although there is some debate as to the directionality of the reaction, the high Km for ammonium in the reductive amination reaction seems to prohibit the reverse reaction under normal conditions in most organisms (Smith et al., 1975). Even in plants, recent 15N incorporation studies in the presence of excess ammonium have shown that GDH functions in the oxidative deamination reaction (Aubert et al., 2001). However, some bacteria may use GDH rather than the normal glutamine synthetase-glutamate synthase (GS-GOGAT) pathway to fix nitrogen under high ammonia conditions (Kanamori et al., 1987). Under most in-vitro conditions, coenzyme release is the rate-limiting step, particularly at higher substrate concentrations (diFranco, 1974). Since the major allosteric regulators noted below all seem to act by either promoting or inhibiting product release, it seems highly likely this is the rate-limiting step in vivo as well.
GDH from animals, but not other kingdoms (Frieden, 1965), is allosterically regulated by a wide array of ligands (Bailey et al., 1982; Dieter et al., 1981; Frieden, 1959a, 1965; Sener and Malaisse, 1980; Tomkins et al., 1962; Yielding and Tomkins, 1961). GTP (Dieter et al., 1981; Iwatsubo and Pantaloni, 1967; Koberstein and Sund, 1973) is a potent inhibitor of the reaction and acts by increasing the binding affinity for the product, thereby decreasing enzymatic turnover (Koberstein and Sund, 1973). ATP is also an inhibitor of the enzyme, albeit with ~100-fold lower affinity than GTP (Frieden, 1965). In both the reductive amination and the oxidative deamination reactions, the enzyme is inhibited by high substrate concentrations in a pH dependent manner (Bailey et al., 1982). In the case of the oxidative deamination reaction, this is due to the product, 2-oxoglutarate, being replaced by glutamate before the reduced coenzyme is able to dissociate from the active site. Reduced coenzyme binds very tightly in this abortive complex (GDH·NAD(P)H·Glu) and must dissociate before catalytic turnover can continue. ADP is an activator of GDH (Bailey et al., 1982; Frieden, 1965; Iwatsubo and Pantaloni, 1967; Koberstein and Sund, 1973; Markau et al., 1972) that likely acts by helping to resolve these abortive complexes by decreasing the affinity of the coenzyme and substrate to the active site (Bailey et al., 1982). This is in direct contrast to the effects of GTP that enhances substrate and coenzyme binding to the active site. The fact that ADP decreases substrate affinity for the enzyme leads to rather complex regulation. ADP affects GDH activity in a continuum ranging from activation under conditions where abortive complexes tend to form, to inhibition at conditions where substrate and coenzyme bind poorly (Bailey et al., 1982). ATP is similarly complex in that it at low concentrations it activates like ADP but at high concentrations it apparently binds to the GTP site and inhibits the reaction (e.g. (Banerjee et al., 2003)). There is very strong mutual antagonism between ADP and GTP and it seems likely that, in vivo, GTP and ADP largely act to finely regulate GDH activity via this competition. Leucine is a poor substrate for GDH and an allosteric activator for the enzyme (Yielding and Tomkins, 1961). Its activation is akin to ADP but acts at site distinct from ADP (Prough et al., 1973). As discussed below, leucine activation of GDH may play a larger role in GDH regulation than implied by its rather high ED50. Palmitoyl CoA (Fahien and Kmiotek, 1981), steroid hormones (Yielding et al., 1960), and diethylstilbestrol (Tomkins et al., 1962) (DES) are also potent inhibitors. GDH also has a second binding site for NADH that has been suggested to inhibit the enzyme and bind synergistically with GTP (Frieden, 1958, 1959a, b). However, NADH inhibition alone only occurs at high, non-physiological concentrations. Therefore, it may be that the binding synergism between GTP and NADH is more important in vivo than NADH inhibition alone.
Mammalian GDH also exhibits unusual homotropic regulation with regard to coenzyme, but with an unclear physiological purpose. Negative cooperativity is observed as ‘breaks’ in Lineweaver-Burk plots with NAD(P)+ varied (Engel and Dalziel, 1969). Subsequent studies demonstrated that coenzyme (NAD(P)(H)) binding to the initial subunits weakens the affinity to subsequent subunits (Bell and Dalziel, 1973; Melzi-D’eril and Dalziel, 1973). This process involves the substrate since negative cooperativity has been shown to be dependent upon the substituent at the α-carbon of the substrate backbone (Bell et al., 1985). Negative cooperativity, in general, is thought to help the enzyme maintain a uniform catalytic rate as coenzyme or substrate concentrations vary in vivo (Koshland, 1996). However, it is also possible that negative cooperativity is a consequence of inter-subunit communication that is used for other purposes rather than regulation in its own right. This seems to be more likely since the ratios of the various forms of coenzyme do not vary to a very large degree in vivo. With GDH, this communication is made evident by abrupt and striking changes in the circular dichroism and fluorescence spectra when GDH is half saturated (Bell and Dalziel, 1973) and from chemical modification studies demonstrating that the loss of enzymatic activity is extremely disproportional to the number of subunits modified (Piszkiewicz and Smith, 1971; Rasool et al., 1976; Syed and Engel, 1984). Therefore, it is clear that there is extensive inter-subunit communication in animal GDH and, as shown below, such communication is essential for catalysis and regulation.
Atomic structure of animal GDH
Mammalian GDH (Figure 1) is arranged as two trimers stacked directly on top of each other (Banerjee et al., 2003; Peterson and Smith, 1999; Smith et al., 2001; Smith et al., 2002). Each subunit is composed of three domains (Banerjee et al., 2003; Peterson and Smith, 1999; Smith et al., 2001; Smith et al., 2002). The first domain makes extensive contacts with the subunit from the other trimer (e.g. the blue and light blue subunits in Figure 1). Resting on top of this domain is the ‘NAD binding domain’ that has the conserved nucleotide-binding motif. Rising above these two domains is a long protrusion, ‘antenna’, that is a helix-loop-helix composed of ~50 residues. From sequence alignments, this feature is not found in bacteria, plants, fungi, and the vast majority of protists. The antenna from each subunit lies immediately behind the adjacent, counter-clockwise neighbor within the trimer. That these intertwined antennae are only found in the forms of GDH allosterically regulated by numerous ligands, leads to the obvious suggestion that it plays a unique and major role in animal GDH regulation.
Figure 1.

Atomic structure of animal glutamate dehydrogenase. This is a ribbon diagram of bovine glutamate dehydrogenase complexed with glutamate (yellow), NADH, and the inhibitor GTP (brown). There are two molecules of NADH bound; one at the active site (grey) and one at the allosteric inhibitory site (cyan). ADP also binds at this second NADH site and activates the enzyme. The ribbons are colored according to three pairs of dimers with shades of blue, red, and green.
From the structures of a number of GDH/ligand complexes, we developed a model whereby GDH performs a complex network of motions during catalytic turnover (Banerjee et al., 2003; Li et al., 2009; Peterson and Smith, 1999; Smith et al., 2001; Smith et al., 2002; Smith and Stanley, 2008). Substrate binds to the deep recesses of the cleft between the NAD binding domain and the lower domain. Coenzyme binds along the NAD binding domain surface of the cleft. Upon binding, the NAD binding domain rotates by ~18° to firmly close down upon the substrate and coenzyme. As the catalytic cleft closes, the base of each of the long ascending helices in the antenna appears to rotate out in a counter-clockwise manner to push against the ‘pivot’ helix of the adjacent subunit. There is a short helix in the descending loop of the antenna that becomes distended as the mouth closes in a manner akin to an extending spring. The ‘pivot helix’ rotates in a counter clockwise manner along the helical axes as well as rotating counter clockwise around the trimer 3-fold axis. Finally, the entire hexamer seems to ‘exhale’, or compress, as the mouth closes. This compression is where the three stacked dimers draw closer to each other, drawing the 2-fold related subunits closer and compressing the inner core. Therefore, it is clear that the conformational changes associated with, and necessary for, catalysis involve the entire hexamer. This not only might explain the complex kinetic behavior such as negative cooperativity, but also creates a number of potential binding sites for allosteric regulators.
The importance of these complex conformational changes has been borne out by the structures of various GDH-ligand complexes (Figures 1 and 2) that demonstrate that allosteric regulators bind to key junctions in this network of structural transitions. GTP binds to the side of the ‘jaw’ of each subunit. As the NAD binding domain closes, a space opens between the pivot helix and the back of the NAD domain. This affords enough space for GTP to bind mainly via interactions with its triphosphate moiety (Peterson and Smith, 1999; Smith et al., 2001). In contrast, ADP binds behind the NAD binding domain immediately under the pivot helix (Banerjee et al., 2003). We have shown that R463 on the pivot helix is essential to ADP activation in that the R463A mutation abrogates ADP-mediated activation without affecting ADP binding (Banerjee et al., 2003). We propose that ADP may activate by helping the catalytic cleft to open by interacting with the pivot helix as it rotates during the opening and closing of the catalytic cleft. In essence, ADP and GTP cause opposite effects on the dynamics of the enzyme upon binding to their respective sites. GTP ‘waits’ for the catalytic cleft to close, binds to its allosteric site, and inhibits the enzyme by making it harder for it to release the products of the reaction. In contrast, we propose that ADP binds to the back of the pivot helix and, via interactions with residues such as R463, decreases the energy required to open the catalytic cleft and release product. This is consistent with the binding and regulatory antagonism observed between these two allosteric regulators. In terms of in-vivo activity, these two regulators essentially act as energy switches for the enzyme. When the mitochondria are at a high-energy state and rich in triphosphates, GDH is inhibited by GTP and, to a lesser extent, ATP. When the mitochondria are low in energy, the elevated ADP levels activate GDH to catabolize glutamate and feed the Krebs cycle with 2-oxoglutarate. Therefore, in vivo, GDH is kept in a tense state where activity is finely tuned depending upon subtle changes in the ADP/GTP ratios. The binding locations and modes of action of other regulators such as leucine and palmitoyl CoA are not clear, but, as detailed below, may play a role in insulin homeostasis.
Figure 2.
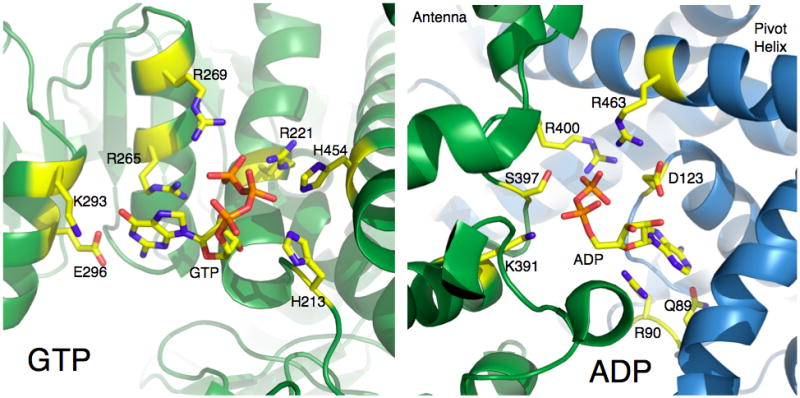
Atomic details of the ADP and GTP allosteric regulatory sites on animal GDH. In these figures, the residue numbering of human GDH is used. The side chains that form hydrogen bonds with the ligands are represented by sticks and colored according to atom type.
ADP/second NADH site paradox
Perhaps one of the most confusing regulator sites is the ADP activation site. What makes this site complex is that NADH also binds to this allosteric site but causes inhibition. In spite of having atomic details as to the interaction of these ligands with the enzyme, it is not at all clear how these regulators can cause opposite effects upon binding to the same site.
The existence of a second NADH binding site per subunit was demonstrated both kinetically and by binding analysis (Frieden, 1959a, b; Shafer et al., 1972). It was observed that NADH alone binds with a stoichiometry of 7–8 molecules per hexamer. In the presence of glutamate, NADH binds more tightly and the stoichiometry increases to 12 per hexamer (Shafer et al., 1972). Similarly, GTP also increases the affinity and binding stoichiometry (Koberstein and Sund, 1973). This second coenzyme site strongly favors NADH over NADPH with Kd’s of 57μM and 700μM, respectively. In the case of oxidized coenzyme, NAD+, two binding sites were also observed. While the recent structures of the various complexes have demonstrated that ADP and NAD(H) bind to the same site (Banerjee et al., 2003; Smith et al., 2001), this was first suggested by ADP binding competition with NAD+ (Limuti, 1983) and NADH (Dieter et al., 1981). Further, these binding studies provided direct evidence that GTP and glutamate enhance binding of NADH to a second site and ADP blocks binding of both NAD+ and NADH to a second site. A number of chemical reagents affect NADH inhibition by binding to disparate sites of the enzyme; the antenna (TNBS (Goldin and Frieden, 1971), FSBA {Pal, 1975 #548; Schmidt, 1984 #2658}), the core of the hexamer (FSBAzA (Dombrowski et al., 1992), FSBA (Pal et al., 1975; Schmidt and Colman, 1984)), and the outer portion of the NAD binding domain (6-BDB-TADP (Batra and Colman, 1986)). These results demonstrate that a number of regions distal to the NADH binding site are involved in NADH inhibition. Since, in general terms, NADPH is involved in anabolic reactions in the cell while NADH is important for catabolic processes, it is possible that this regulation offers a feedback mechanism to curtail glutamate oxidation when catabolic reductive potentials (NADH) are high.
In nearly every way, ADP acts in a manner opposite to NADH binding to this site. While GTP and glutamate bind synergistically with NADH to inhibit GDH, ADP activates the reaction by decreasing the affinity of the enzyme for coenzyme at the active site. It should be noted that substrate (2-oxoglutarate) inhibition in the reductive amination reaction is only observed using NADH as coenzyme. This was suggested to be due to the fact that NADH, not NADPH, binds to the second, inhibitory coenzyme site. Further, it was suggested that ADP activation under these conditions was due to ADP displacement of NADH from the second allosteric site (Frieden, 1965).
The structures of GDH complexed with NADH, NADPH, and NAD have all been determined (Smith et al., 2001). Because NADH (but not NADPH) has been suggested to be an inhibitor of the reaction, it is somewhat surprising that it binds to the ADP activation site (Banerjee et al., 2003). The adenosine-ribose moiety location exactly matched that of ADP. The electron density of the ribose-nicotinamide moiety was much weaker and was initially build in two alternative conformations. However, the stronger density for this portion of NADH suggests that it points down into the interface between adjacent subunits. As predicted from the binding studies reviewed above, NADPH was found bound to the active site but not the second, allosteric site. From the preferred orientation, this is likely due to the fact that there is not enough room to accommodate the additional phosphate on the ribose ring that is buried at the subunit interface.
NAD+ was found to bind in a manner essentially identical to NADH (Smith et al., 2001). From steady state kinetic analysis, it was initially thought that NAD+ binding to this second site causes activation of the enzyme (Frieden, 1959a), even though NADH causes apparent inhibition. However, subsequent studies demonstrated that this apparent activation was due to negatively cooperative binding with respect to coenzyme (Dalziel and Engel, 1968). Therefore, it is not clear what difference there might be, if any, between NAD+ and NADH binding to GDH at this location. It is interesting to note that modification of the ADP site with an ADP analog did not eliminate NADH inhibition (Wrzeszczynski and Colman, 1994). Perhaps this is due to the nicotinamide moiety still binding to the pocket between the subunits in spite of AMPSBDB being bound to R459. As will be detailed below, recent studies on new GDH inhibitors have shown that compounds binding to subunit interfaces can be potent inhibitors of the enzyme. Perhaps the ribose-nicotinamide moiety is acting in a similar manner.
The physiological role of ADP activation is easily understood; when the energy level of the mitochondria is low and ADP levels are high, the catabolism of glutamate is facilitated for energy production. However, the possible in-vivo role of NADH inhibition is less clear. In mammalian mitochondria, assuming a matrix volume of 1μl/mg of protein, the concentrations of NAD(H) and NADP(H) are approximately 0.5–2.0mM (Lenartowicz, 1990). However, activity of the transhydrogenase transfers much of the reductive power of NADH to NADPH. Using metabolite indicators, the mitochondrial NADH/NAD+ ratio was estimated to be ~0.2 and the NADPH/NADP+ ratio was ~200 (Hoek and Rydström, 1988). In experiments on submitochondrial particles, the energy-linked transhydrogenase was found to maintain NADP up to 500 times more reduced than NAD (Rydström et al., 1970). These results suggest that the range of NADH concentration is ~0.083–0.33mM. NADH inhibition is observed at concentrations above 0.2mM (e.g. see (Batra and Colman, 1986)), but only reaches ~50% inhibition at 1mM NADH. Therefore, if NADH inhibition is physiologically relevant, it seems more likely that its purpose is to synergistically enhance GTP inhibition; under conditions of high reductive potential, NADH acts with GTP to keep GDH in a tonic state.
At an atomic level, there is a very clear delineation between ligands binding to the open and closed conformations. NADH alone only binds to the active site. When glutamate is added, the catalytic cleft closes and NADH is able to bind to the second, allosteric site. Further, the GTP binding site collapses when the catalytic cleft opens and therefore GTP also favors the closed conformation. Therefore, the synergism between NADH and GTP is likely due to both ligands binding to, and stabilizing the closed conformation. Again, this supports the contention that NADH inhibition alone may not have a significant physiological role, but rather its main function is the enhancement of GTP inhibition.
Role of GDH regulation in vivo
While GDH has been studied for more than 50 years, it was not at all clear why a ‘house keeping’ enzyme needed such complex allosteric regulation in animals. More recent studies have shown that GDH sits at a crucial intersection for several metabolic pathways and therefore requires fine-tuned regulation. In particular, it is clear that GDH is intimately involved in insulin homeostasis. The connection between GDH and insulin regulation was initially found with a nonmetabolizable analog of leucine (Sener and Malaisse, 1980; Sener et al., 1981), BCH (β-2-aminobicycle (2.2.1)-heptane-2-carboxylic acid). These studies demonstrated that activation of GDH was tightly correlated with increased glutaminolysis and release of insulin. In addition, it has also been noted that factors that regulate GDH also affect insulin secretion (Fahien et al., 1988). Subsequently, it was postulated that glutamine could also play a secondary messenger role and that GDH plays a role in its regulation (Li et al., 2004; Li et al., 2003; Stanley, 2000). The in-vivo importance of GDH in glucose homeostasis was demonstrated by the finding that a genetic hypoglycemic disorder, the hyperinsulinemia/hyperammonemia (HHS) syndrome, is caused by loss of GTP regulation (MacMullen et al., 2001; Stanley et al., 2000; Stanley et al., 1998). Children with HHS have increased β-cell responsiveness to leucine and susceptibility to hypoglycemia following high protein meals (Hsu et al., 2001). This is likely due to uncontrolled catabolism of amino acids yielding high ATP levels that stimulate insulin secretion as well as high serum ammonium levels.
The loss of GTP inhibition, observed in HHS, results in profound effects on several major organs (Figure 3). As noted above, dysregulated GDH causes pancreatic tissue to secrete more insulin than is needed, thereby decreasing blood glucose levels to dangerous levels. In the liver and/or the kidneys, the hyperactive GDH diminishes the pool of glutamate, thereby producing excessive ammonium. This hyperammonemia is further exacerbated since the lower glutamate concentrations also decreases the production of N-acetyl-glutamate that is a necessary activator for carbamoylphosphate synthetase (CPS) that is in the ureagenesis pathway. Together, these effects elevate serum ammonium concentrations to extremely high levels. While the glucose and ammonium levels in patients with HHS are alone sufficient to potentially cause damage to the CNS, recent studies have suggested a high correlation between HHS and childhood-onset epilepsy, learning disabilities, and seizures (Bahi-Buisson et al., 2008). Some of these pathologies have been shown to be unrelated to serum glucose and ammonium levels. This is not entirely surprising considering the importance of glutamate and its derivative, γ-aminobutyric acid, as neurotransmitters. The current treatment for HHS is to pharmaceutically control insulin secretion (e.g. diazoxide, a potassium channel activator) but this does not address the liver and CNS problems. Therefore, there is a significant need to develop a new inhibitor of GDH that acts independently of the GTP affinity site.
Figure 3.

Overview of the multi-organ effects of HHS. The top figure shows that the loss of GTP inhibition increases the flux of glutamate through GDH. The resulting stimulation of Krebs cycle activity results in higher a ATP:ADP ratio and causes degranulation of the β-cells. The middle figure shows that in the liver and/or the kidneys, the increased activity of GDH diminishes the glutamate pool that results in lower N-acetylglutamate levels followed by a loss of carbamoyl phosphate synthetase activation. This leads to higher serum ammonium levels not only due to the deamination of glutamate but also the decrease in urea synthesis. The bottom panel shows the large number of glutamate dependent ion channels involved in neuronal synapsis and the role that glial cells play in glutamate recycling. Unregulated GDH will very likely affect glutamate homeostasis.
Other forms of GDH regulation
There is evidence that GDH activity in vivo might be modulated by covalent modifications in the mitochondria. In recent studies, it has been suggested that sirtuin 4 (SIRT4) may inhibit GDH activity by ADP-ribosylation (Haigis et al., 2006). SIRT4 has been suggested to be a mitochondrial protein (Michishita et al., 2005) and does not display NAD-dependent deacetylase activity but instead uses NAD to ADP-ribosylate GDH. Typically, this NAD-dependent acetylase can occur at arginine, cysteine, and histidine residues. In SIRT4 deficient insulinoma cells, GDH is activated, and, akin to HHS, causes hypersensitivity to amino acid stimulated insulin secretion. A similar effect is seen in wild-type mice that are on a calorie-restricted diet, where repression of GDH by ADP-ribosylation is removed and the β cells are hypersensitive to glutamine and leucine stimulation. This is entirely consistent with the pattern of GDH allosteric regulation where it appears that GDH is kept very tightly controlled in vivo and repression is only relieved when energy sources other than amino acids are depleted. In this case, the NAD+:NADH ratio may play a role in this regulation.
In recent work, it has also been suggested that GDH activity may also be modulated by protein-protein associations within the mitochondria (Li et al., 2010). Several cases of hyperinsulinism have been described that were associated with a deficiency of a mitochondrial fatty acid β-oxidation enzyme, short-chain 3-hydroxyacyl-CoA dehydrogenase (SCHAD). SCHAD catalyzes the third step in the β-oxidation cycle for medium and short chain 3-hydroxy fatty acyl-CoAs. Other genetic disorders of mitochondrial fatty acid oxidation do not cause hyperinsulinism (Stanley et al., 2006). Interestingly, insulin dysregulation in SCHAD deficiency has shown to be associated with sensitivity to protein-induced hypoglycemia much like HHS (Kapoor et al., 2009). The mechanism of this form of insulin dysregulation was examined in mice with knockout of the hadh gene (hadh−/−). Similar to the SCHAD deficient children, the hadh−/− mice had reduced levels of plasma glucose and elevated plasma insulin levels and were hypersensitive to orally administered amino acid with decrease of glucose level and elevation of insulin. Specificially, leucine, glutamine and alanine are responsible for amino acid hypersensitivity in islets. hadh−/− islets have lower intracellular glutamate and aspartate levels that can be prevented by high glucose. hadh−/− islets also have increased [U-14C]glutamine oxidation. In contrast, hadh−/− mice have similar glucose tolerance and insulin sensitivity compared to controls. Perifused hadh−/− islets showed no differences from controls in response to glucose-stimulated insulin secretion, even with addition of either a medium-chain fatty acid (octanoate) or a long-chain fatty acid (palmitate). Pull-down experiments with SCHAD, anti-SCHAD, or anti-GDH antibodies showed protein-protein interactions between SCHAD and GDH. GDH enzyme kinetics of hadh−/− islets showed an increase in GDH affinity for its substrate, alpha-ketoglutarate. Together, these studies indicate that SCHAD deficiency causes hyperinsulinism by release of SCHAD inhibition of GDH likely due to the loss of protein-protein interactions.
Role and evolution of the antenna domain
There are two general locations for the GDH HHS mutants that abrogate GTP inhibition (Figure 4); immediately adjacent to the GTP binding site and in the antenna region. The effects on GTP inhibition due to the first group of mutations likely have direct effects on GTP binding. Most of the mutations either eliminate the basic charge character in the GTP binding site or place bulky side chains that sterically interfere with GTP binding. The second group of mutations (underlined labels in Figure 4) is harder to understand in that none of the mutated residues interact with bound GTP. Most of mutations lie in the short helix in the descending strand of the antenna. From our structural studies, this helix unwinds like a stretched spring when the catalytic cleft is closed and ‘rewinds’ upon cleft opening (Smith et al., 2002). It is possible that these mutations affect those conformational transitions. Since this section is immediately attached to the pivot helix, it is also possible that these mutations block the opening of the cleft between the pivot helix and the coenzyme-binding domain that is necessary for GTP binding. However, the remaining two mutations in this group (N410 and L413) lie on the major, ascending helix in the antenna and point towards the center of the antenna. This suggests that the effect of these antenna mutations may be more complex that just indirectly affecting the GTP binding site within a particular subunit. The ascending helixes of the antennae within the trimers of the hexamer wrap around each other and rotate back and forth as the active sites open and close. These mutations may be affecting that particular ‘washing machine like’ motion. It is therefore possible that antennae interactions between the subunits within the trimers are necessary to effectively communicate GTP inhibition. Indeed, it may be these types of inter-subunit communications that give rise to negative cooperativity reviewed above.
Figure 4.
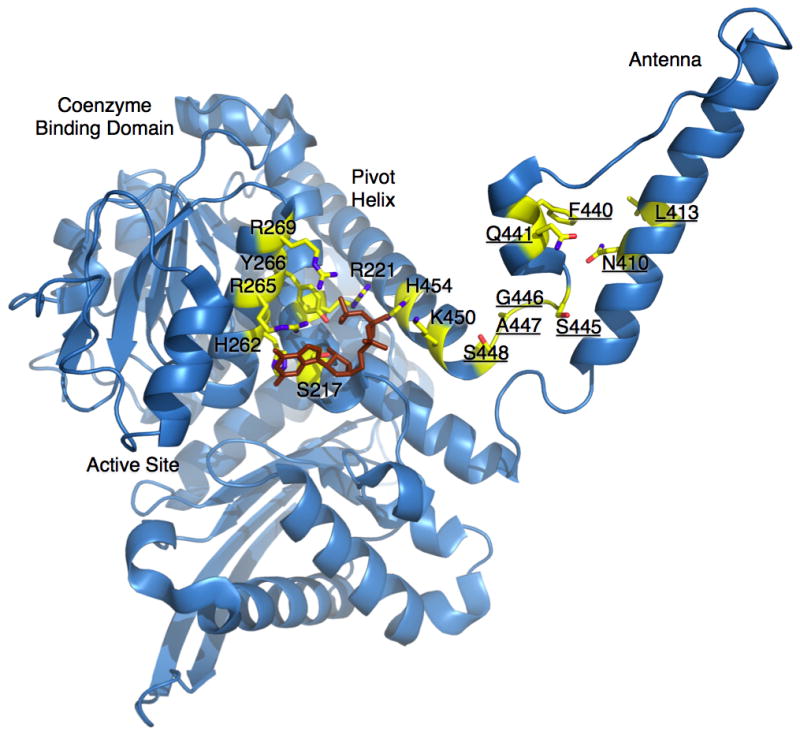
Locations of the HHS mutations. Shown here is a ribbon diagram of one GDH subunit. The side chains of the known HHS mutations are represented as stick figures colored according to the atom type. The numbering used here correlates to the human GDH amino acid sequence. The structure of bound GTP is shown as a brown stick figure. The residues with underlined labels are those that are not in contact with the bound GTP and are clustered around the antenna region.
It is clear that the antenna is a feature essential for allosteric regulation. However, the next question is when and why did it evolve. Genomics studies demonstrated that, in fact, the antenna first appeared in the Ciliates within the Protista. From sequence alignments, it is apparent that these organisms have an antenna that is slightly smaller than that found in the Animalia kingdom. Notably, other members of the Protista, such as trypanosomes, have GDH nearly identical to bacterial forms (Allen et al., 2004). Therefore, the Ciliates are an unexpected ‘missing link’ between animals and the other kingdoms with regard to the evolution of GDH allostery.
To better understand the connection between the antenna and GDH regulation, the allosteric behavior of several forms of GDH was examined (Allen et al., 2004). In this regard, Tetrahymena GDH is indeed between the Protista and Animalia kingdoms (Table 1); it is activated by ADP, inhibited by palmitoyl CoA, but unaffected by GTP and leucine. Interestingly, the lack of leucine activation of Tetrahymena GDH suggests, but does not prove, that there could indeed be a binding site for leucine other than just the active site since the catalytic site is highly conserved amongst the GDH’s from all sources. To directly ascertain which allosteric regulators require the antenna, the antenna was removed from human GDH and replaced with the short loop found in bacterial GDH. This should not directly affect GTP and ADP binding because none of the contact residues for these regulators reside on the antenna. However, the ‘antenna-less’ form of human GDH lost all forms of regulation except leucine activation. When the antenna from Tetrahymena GDH was spliced onto the main body of human GDH, this hybrid GDH exhibited all of the allosteric regulation found in human GDH. This demonstrates that, if the allosteric regulator sites exist on the main body of the enzyme, the Tetrahymena antenna is capable of communicating the associated regulation among the subunits. This is fairly strong evidence that the antenna evolved for inter-subunit communication that is necessary for effective allosteric regulation.
Table 1.
Allosteric properties of four different forms of GDH; human GDH (hGDH), human GDH with the antenna replaced by a loop found in bacterial GDH (Antenna-less hGDH), Tetrahymena GDH (tGDH), and Tetrahymena GDH with the antenna replaced with that found in human GDH (tGDH with hGDH antenna).
| hGDH | Antenna-less hGDH | tGDH | tGDH with hGDH antenna | |
|---|---|---|---|---|
| GTP inhibition | + | − | − | + |
| PalmCoA inhibition | + | − | + | + |
| ADP activation | + | − | + | + |
| Leucine activation | + | + | − | + |
While this better explains the function of the antenna, it does not explain why the Ciliates were the only early eukaryotes to require such regulation. One explanation may be that GDH allostery evolved in response to the changing functions of the cellular organelles (Allen et al., 2004). In the other eukaryotes, all fatty acid oxidation occurs in the peroxisomes (Erdmann et al., 1997; Gerhardt, 1992). In the Ciliates, fatty acid oxidation is shared between the peroxisomes and the mitochondria (Blum, 1973; Muller et al., 1968) and eventually in animals all of the medium and long chain fatty oxidation occurs in the mitochondria (Hashimoto, 1999; Reddy and Mannaerts, 1994). From the pattern of regulation in Ciliates, it seems that if the mitochondria have sufficient levels of fatty acids, then the catabolism of amino acids is blocked. Only when the mitochondria are in a low energy state (high ADP), will amino acids be catabolized.
The causal link between the loss of GTP inhibition and the hypersecretion of insulin suggests that animals further evolved the allostery created in the Ciliates to link GDH-mediated amino acid oxidation with insulin homeostasis. Thus, GDH is activated when amino acids (protein) are ingested to promote insulin secretion and appropriate anabolic effects on peripheral tissues; in the glucose-fed state, GDH is inhibited in pancreas perhaps to redirect amino acids into glutamine synthesis in order to amplify insulin release. Similarly, adjustment of hepatic GDH allows amino acid degradation to be suppressed when other fuels, such as fatty acid are available, but to be increased when protein (amino acids) are ingested and surplus amino acids can be oxidized. To this end, mammals developed leucine activation and GTP inhibition while using the antenna architecture created by the Ciliates. The choice of leucine as a regulator is likely not an accident, because leucine is the most abundant amino acid in protein (10%) and provides a good measure of protein abundance. Similarly, the marked sensitivity of GDH for GTP over ATP is also with good purpose. Most of the ATP in the mitochondria is produced from oxidative phosphorylation that is driven by the potential across the mitochondrial membrane created by NADH oxidation. Therefore, the number of ATP molecules generated from one turn of the TCA cycle can vary between 1 and 29. In contrast, one GTP is generated per turn of the TCA cycle and therefore, with the slow mitocondria/cytoplasm exchange rate, the GTP/GDP ratio is much better metric of TCA cycle activity than the ATP/ADP ratio. Indeed, recent results have demonstrated that mitochondrial GTP, but not ATP, regulates glucose-stimulated insulin secretion (Kibbey et al., 2007). This is also consistent with the HHS disorder in that without GTP inhibition of GDH, glutamate will be catabolized in an uncontrolled manner, the TCA cycle will generate more GTP, and more insulin will be released. Therefore, the addition of GTP and leucine regulation to GDH makes it acutely sensitive to amino acid and glucose catabolism with obvious implications for insulin homeostasis.
Development of novel GDH inhibitors
As discussed above, the HHS is a multi-organ disorder that needs to be treated with compounds that directly target the dysregulated form of GDH rather than just the insulin hypersecretion effects in the pancreas. To this end, we have been working to create agents that can directly and systemically control the dysregulated HHS form of GDH to alleviate all HHS-related pathologies. Along the way, studies on these compounds are elucidating the molecular details of GDH allostery and the role that GDH regulation plays in vivo.
We used two approaches to find new inhibitors for GDH. From some very old publications (Konayagi and Minowada, 1933), it was suggested that the polyphenols from green tea might be efficacious in treating diabetes and therefore the effects of this class of compounds were tested on GDH (Li et al., 281). Subsequently, a more systematic screen was performed using ~30,000 compounds (Li et al., 2007). For these latter studies very high glutamate and coenzyme concentrations were used to search for allosteric inhibitors that were not substrate or coenzyme analogs. In this way, it was hoped that whatever compounds were identified, they would not interfere with other metabolic processes that use these common substrates and cofactors. A number of compounds were found to have sub-micromolar ED50’s. Importantly, the active green tea compounds we found previously (EGCG and ECG) also appeared in the screen while the inactive compounds (e.g. EC) were similarly inactive in this screen.
Shown in Figure 5 is a summary of our most active compounds thus far. It should be noted that, as with all screening results, there was concern as to the specificity of these compounds. For example, hits from screens could aggregate the enzyme or cause irreversible denaturation in a non-specific manner. In the case of aggregation, none of these compounds caused changes in the aggregation state as per dynamic light scattering measurements. Further, GDH crystallized in the presence of high concentrations of all of the active compounds shown in Figure 5 and we have determined the structures of GDH complexed with hexachlorophene, bithionol, GW5074, and EGC. In the case of possible denaturation, the inhibitory effects of these compounds are completely reversed by either dialysis or dilution during activity measurements. Therefore, none of our active compounds are false positives or non-specific inhibitors, as is commonly found in high throughput screening.
Figure 5.
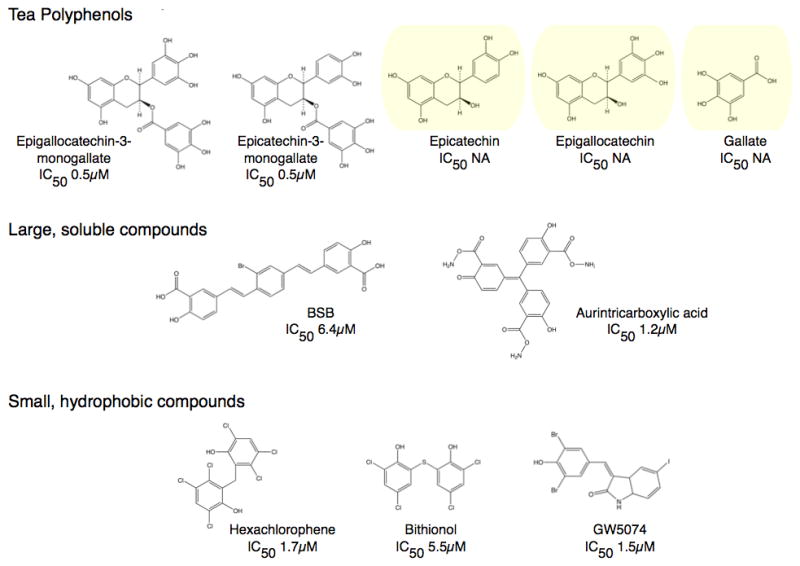
Structures of some of the new and novel inhibitors for GDH. The compounds fall into three general classes; the tea polyphenols, large soluble compounds, and small hydrophobic molecules. The three polyphenols highlighted in yellow do not inhibit GDH activity even at high concentrations even though they have essentially the same chemical properties.
Structures of GDH complexed with hexachlorophene, biothionol, and GW5074
Six molecules of hexachlorophene (HCP) form a ring in the inner cavity of the hexamer. These six drug molecules alternate between two different conformations around the ring (figure 5). One conformer is relatively flat and tucks into a pocket at the interface between diagonal subunits. Essentially, the symmetrical HCP binds at the interface between two-fold related subunits with one ring interacting with one subunit and the other ring interacting with the other. The majority of the interactions between HCP and GDH are hydrophobic, but there is also an almost ‘chain link’ of aromatic stacking interactions. The two rings of HCP in this first orientation approximately stack against two Y190 sidechains from diagonally adjacent subunits rather than the two subunits stacked on top of each other. The interactions between the other HCP conformer and GDH are also symmetrical with each HCP ring making the same hydrophobic interactions with the two diagonal subunits. The hydrophobic pocket for this orientation is mainly comprised of M150, I187, Y190 and the methylene side chain atoms from T186 and K154. There are also approximate stacking interactions among the bound HCP molecules. As shown in figure 5, the top ring of one conformer interacts with the bottom ring of the other.
Bithionol and GW5074 do not bind to the same site as HCP. The electron densities of the drugs agreed well with the structures of the compounds; bithionol has a marked kink between the two rings while GW5074 has a planar structure. While HCP binds to the inner core, these two drugs bind halfway between the core and the exterior of the hexamer. Unlike HCP, each of the six drug molecules are associated with separate subunits rather than one molecule contacting two symmetrical sites simultaneously. Instead, two drug molecules form pairs that are related by the hexameric two-fold axes. The binding environments of the two drugs are nearly identical. Residues 138–155 of the glutamate-binding domain form an α-helix that makes most of the contact between diagonal subunits and draw closer together when the catalytic cleft is closed. These two drugs stack against each other and interact with hydrophobic residues and the aliphatic portions of the polar and charged side chains of residues K143, R146, R147, and M150. These drugs, therefore, appear to directly bind to the area that compresses during mouth closure.
There are some differences, however, in the interactions between GW5074 and bithionol and GDH. The acidic brominated phenol group in GW5074 points towards R147 while the planar heteroaromatic fused ring stacks against the planar guanidinium head group of R146. The latter aromatic ring – guanidinium head group interaction is commonly observed in proteins (Flocco and Mowbray, 1994). In the case of the halogenated, biphenolic compound, bithionol, one of the two aromatic rings is approximately planar to the guanidinium head group of R146, but does not stack nearly to the degree as GW5074. The two hydroxyl groups of the acidic phenol rings point toward the head group of R147 in a manner similar, but not identical to that of GW5074. Nevertheless, it is interesting that both of these phenolic compounds bind within this cluster of lysine and arginine residues.
As discussed above, the core of GDH expands and contracts as the catalytic mouth opens and closes, respectively. The stacked dimers effectively act like a single rigid body because of the extensive β-sheet interactions at the dimer interface. During mouth opening and closing, the distance between these dimers change by up to 4% while the distance between the subunits within the dimer change by ~0.1%. The binding locations for these drugs are right in the middle of these conformational changes in that GW5074 and bithionol bind between two pairs of helices that form the dimer-dimer interface and HCP forms dimer-dimer interactions in the core of the enzyme. Therefore, it seems highly likely that these compounds are inhibiting the enzyme by ‘wedging’ into these junction points and keeping the stacked dimers from being able to freely move. This would explain how these compounds are able to inhibit catalytic turnover while being bound distal to the active and regulatory sites.
Effects of polyphenols on GDH activity in-vitro and in-situ
Green tea is a significant source of a type of flavonoids called catechins; epigallocatechin gallate (EGCG), epigallocatechin (EGC), epicatechin gallate (ECG), and epicatechin (EC). One 200 ml cup of green tea supplies 140, 65, 28, and 17 mg of these polyphenols, respectively (Yang and Wang, 1993). Of the four major catechins found in green tea, only two showed inhibitory activity against GDH; ECG and EGCG. Essentially, activity of this family of compounds is dependent upon the presence of the third ring structure, the gallate, on the flavonoid moiety. EGCG and ECG allosterically inhibit purified animal GDH in-vitro with a nanomolar ED50. Since EC or EGC were not active against GDH, but have the same anti-oxidant activity as ECG and EGCG, the anti-oxidant property of these catechins cannot be relevant to GDH inhibition. As mentioned above, neither ECG nor EGCG aggregate or denature the enzyme. EGCG inhibition is non-competitive and allosteric since leucine, BCH, and ADP can all abrogate this inhibition. As reviewed above, the antenna is necessary for GTP inhibition and ADP activation (Allen et al., 2004). Similarly, EGCG does not inhibit the ‘antenna-less’ form of GDH, thus is further evidence that EGCG is a specific and allosteric inhibitor. Most importantly, EGCG inhibits HHS GDH mutants as effectively as wild type (Li et al., 281), making it a possible therapeutic lead compound.
The next step was to ascertain whether EGCG was active in tissue. Studies have demonstrated that GDH plays a major role in leucine stimulated insulin secretion (LSIS) by controlling glutaminolysis (Li et al., 2004; Li et al., 2003). Therefore, EGCG was tested on pancreatic β-cells using the perifusion assay (Li et al., 281). Importantly, EGCG, but not EGC, blocked the GDH-mediated stimulation of insulin secretion by the β-cells but did not affect insulin secretion, glucose oxidation, or cellular respiration during glucose stimulation where GDH is known to not play a major role in the regulation of insulin secretion. Therefore, EGCG is indeed a specific inhibitor of GDH both in-vitro and in-situ and ongoing studies are evaluating whether it will be similarly active in vivo.
Other uses for GDH inhibitors
More recent studies have demonstrated that pharmaceutically inhibiting GDH activity is not only needed in controlling HHS but may be useful in treating other diseases as well. Studies on glioblastoma cells have demonstrated that EGCG inhibition of GDH may have a role in antitumor therapy. Increased glucose and glutamine utilization are hallmarks of tumor metabolism (DeBerardinis et al., 2008; Kim and Dang, 2006). The phosphatidylinositol 3′-kinase/Akt pathway is enhanced in many human tumors and up-regulates glucose uptake and utilization (Bauer et al., 2005; Elstrom et al., 2004). c-Myc, on the other hand, up-regulates glutamine utilization by increasing cell surface transporters and enzymes (Gao et al., 2009; Wise et al., 2008). At least in-vitro, the enhanced utilization of one of these carbon sources also makes the cells sensitive to its withdrawal (Buzzai et al., 2005; Wise et al., 2008). Extending upon all of these results and from our demonstration that EGCG inhibits GDH activity in tissue, the DeBerardinis laboratory tested the effects of this catechin on glioblastoma cells. They demonstrated that EGCG sensitizes glioblastoma cells to glucose withdrawal and to inhibitors of Akt signaling and glycolysis (Yang et al., 2009). Indeed, the addition of EGCG mirrored the effects of knocking out GDH in the tissue. Therefore, these results suggest that anti-cancer therapy that combines GDH inhibitors with those that inhibit glucose utilization could be very effective in treating tumors.
Work from the Blenis group (Choo et al., 2010) also demonstrated EGCG inhibition of GDH activity may be useful in treating the tuberous sclerosis complex (TSC) disorder. The TSC disorder is characterized by benign tumors due to the loss of either TSC1 or TSC2 and the subsequent hyperactivation of the mammalian target of rapamycin (mTOR). The deregulation of mTOR is associated with a number of disorders including neurological dysfunction, cancer, and inflammation (Shaw, 2006). Inhibitors of mTOR can decrease the size of TSC associated tumors, but only in a cytostatic manner. Similar to the findings of the DeBerardinis group and the glioblastoma cells, the TSC1/2 −/− cells are hypersensitive to glucose deprivation and highly dependent on GDH mediated catabolism of glutamine. Nearly all of the TSC1/2 −/− cells that were deprived of glucose and given rapamycin died upon administration of EGCG. As expected, this cytoxic effect of EGCG was reversed if GDH mediated oxidation of glutamate was circumvented by the addition of 2-oxoglutarate, pyruvate, or aminooxyacetate. As noted by the authors, attempts to make glutamine analogs to inhibit glutamate oxidation have only yielded toxic compounds, presumably because of its effects on numerous pathways. Therefore, our new allosteric GDH inhibitors are more likely to produce families of safer compounds.
Research Highlights.
Animal glutamate dehydrogenase is a homohexamer.
Only animal GDH has a large antenna protruding from both ends.
Loss of allosteric regulation leads to insulin dysregulation
Compounds from green tea (EGCG and ECG) block GDH activity
Regulation is likely mediated by changes in protein dynamics.
Figure 6.

Locations of the binding sites for the small hydrophobic compounds. On the left, the orange and mauve molecules at the GDH two-fold axes represent the pair of drugs bound at the expansion point between the dimers of GDH subunits. The figure on the right is a top-down view of the core of the enzyme showing the relative locations of bithionol and hexachlorophene. Hexachlorophene also binds as three pairs of molecules that are represented by cyan and black molecules.
Figure 7.

Effects of polyphenols on GDH. A) Using steady-state kinetic assays on purified enzyme, this figure shows that only EGCG and ECG inhibit the enzyme with nanomolar ED50’s while the chemically similar compounds, EC and EGC, had no effect on GDH activity. B) This figure shows that EGCG inhibits five different GDH mutants that lead to the HHS syndrome. Also shown here is the fact that Tetrahymena GDH, that has an antenna and activated by ADP, is also affected by EGCG. However, if the antenna is removed from human GDH and replaced by the short loop found in bacterial sources, EGCG has no effect on the enzyme. C) Using β-cell islet perifusion assays, EGCG clearly blocks GDH-mediated, BCH stimulation of insulin secretion in a dose-dependent manner. Importantly, EGC, that was shown to be in active in figure (B), has no effect on insulin secretion.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allen A, Kwagh J, Fang J, Stanley CA, Smith TJ. Evolution of glutamate dehydrogenase regulation of insulin homeostasis is an example of molecular exaptation. Biochemistry. 2004;43:14431–14443. doi: 10.1021/bi048817i. [DOI] [PubMed] [Google Scholar]
- Aubert S, Bligny R, Douce R, Ratcliffe RG, Roberts JKM. Contribution of glutamate dehydrogenase to mitochondrial metabolism studied by 13C and 31P nuclear magnetic resonance. J Exp Bot. 2001;52:37–45. [PubMed] [Google Scholar]
- Bahi-Buisson N, Roze E, Dionisi C, Escande F, Valayannopoulos V, Feillet F, Heinrichs C. Neurological aspects of hyperinsulinism-hyperammonaemia syndrome. Dev Med & Child Neurol. 2008;50:945–949. doi: 10.1111/j.1469-8749.2008.03114.x. [DOI] [PubMed] [Google Scholar]
- Bailey JS, Bell ET, Bell JE. Regulation of bovine glutamate dehydrogenase. J Biol Chem. 1982;257:5579–5583. [PubMed] [Google Scholar]
- Banerjee S, Schmidt T, Fang J, Stanley CA, Smith TJ. Structural studies on ADP activation of mammalian glutamate dehydrogenase and the evolution of regulation. Biochemistry. 2003;42:3446–3456. doi: 10.1021/bi0206917. [DOI] [PubMed] [Google Scholar]
- Batra SP, Colman RF. Isolation and identification of cysteinyl peptide labeled by 6-[(4-bromo-2,3-dioxobutyl)thio]-6-deaminoadenosine 5′-diphosphate in the reduced diphosphopyridine nucleotide inhibitory site of glutamate dehydrogenase. Biochemistry. 1986;25:3508–3515. doi: 10.1021/bi00360a005. [DOI] [PubMed] [Google Scholar]
- Bauer DE, Hatzivassiliou G, Zhao F, Andreadis C, Thompson CB. ATP citrate lyase is an important component of cell growth and transformation. Oncogene. 2005;24:6314–6322. doi: 10.1038/sj.onc.1208773. [DOI] [PubMed] [Google Scholar]
- Bell ET, LiMuti C, Renz CL, Bell JE. Negative co-operativity in glutamate dehydrogenase. Involvement of the 2-position in the induction of conformational changes. Biochem J. 1985;225:209–217. doi: 10.1042/bj2250209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell JE, Dalziel K. A conformational transition of the oligomer of glutamate dehydrogenase induced by half-saturation with NAD+ or NADP+ Biochim Biophys Acta. 1973;309:237–242. doi: 10.1016/0005-2744(73)90336-7. [DOI] [PubMed] [Google Scholar]
- Blum JJ. Localization of some enzymes of β-oxidation of fatty acids in the peroxisomes of Tetrahymena. J Protozool. 1973;20:688–692. doi: 10.1111/j.1550-7408.1973.tb03600.x. [DOI] [PubMed] [Google Scholar]
- Buzzai M, Bauer DE, Jones RG, Deberardinis RJ, Hatzivassiliou G, Elstrom RL, Thompson CB. The glucose dependence of Akt-transformed cells can be reversed by pharmacologic activation of fatty acid β-oxidation. Oncogene. 2005;24:4165–4173. doi: 10.1038/sj.onc.1208622. [DOI] [PubMed] [Google Scholar]
- Choo AY, Kim SG, Vander Heiden MG, Mahoney SJ, Vu H, Yoon SO, Cantley LC, Blenis J. Glucose addiction of TSC null cells Is caused by failed mTORC1-dependent balancing of metabolic demand with supply. Molecular Cell. 2010;38:487–499. doi: 10.1016/j.molcel.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalziel K, Engel PC. Antagonistic homotropic interactions as a possible explanation of coenzyme activation of glutamate dehydrogenase. FEBS Lett. 1968;1:349–352. doi: 10.1016/0014-5793(68)80153-x. [DOI] [PubMed] [Google Scholar]
- DeBerardinis RJ, Sayed N, Ditsworth D, Thompson CB. Brick by brick: metabolism and tumor cell growth. Curr Opin Gent Dev. 2008;18:54–61. doi: 10.1016/j.gde.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieter H, Koberstein R, Sund H. Studies of glutamate dehydrogenase. The interaction of ADP, GTP, and NADPH in complexes with glutamate dehydrogenase. Eur J Biochem. 1981;115:217–226. [PubMed] [Google Scholar]
- diFranco A. Reaction mechanism of glutamate dehydrogenase. Transient complexes in the oxidative deamination of L-glutamate catalyzed by N~D(P)-dependent L-glutamate dehydrogenase. Eur J Biochem. 1974;45:407–424. doi: 10.1111/j.1432-1033.1974.tb03565.x. [DOI] [PubMed] [Google Scholar]
- Dombrowski KE, Huang YC, Colman RF. Identification of amino acids modified by the bifunctional affinity label 5′-(p-fluorosulfonyl)benzoyl)-8-azidoadenosine in the reduced coenzyme regulatory site of bovine liver glutamate dehydrogenase. Biochemistry. 1992;31:3785–3793. doi: 10.1021/bi00130a008. [DOI] [PubMed] [Google Scholar]
- Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM, Thompson CB. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004;64:3892–3899. doi: 10.1158/0008-5472.CAN-03-2904. [DOI] [PubMed] [Google Scholar]
- Engel P, Dalziel K. Kinetic studies of glutamate dehydrogenase with glutamate and norvaline as substrates. Biochem J. 1969;115:621–631. doi: 10.1042/bj1150621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdmann R, Veenhuis M, Kunau WH. Peroxisomes: organelles at the crossroads. Trends Cell Biol. 1997;7:400–407. doi: 10.1016/S0962-8924(97)01126-4. [DOI] [PubMed] [Google Scholar]
- Fahien LA, Kmiotek E. Regulation of glutamate dehydrogenase by palmitoyl-coenzyme A. Arch Biochem Biophys. 1981;212:247–253. doi: 10.1016/0003-9861(81)90364-7. [DOI] [PubMed] [Google Scholar]
- Fahien LA, MacDonald MJ, Kmiotek EH, Mertz RJ, Fahien CM. Regulation of insulin release by factors that also modify glutamate dehydrogenase. J Biol Chem. 1988;263:13610–13614. [PubMed] [Google Scholar]
- Flocco MM, Mowbray SL. Planar stacking interactions of arginine and aromatic side-chains in proteins. J Mol Biol. 1994;235:709–717. doi: 10.1006/jmbi.1994.1022. [DOI] [PubMed] [Google Scholar]
- Frieden C. The dissociation of glutamate dehydrogenase by reduced diphosphopyridine nucleotide (DPNH) Biochim et Biophys Acta. 1958;27:431–432. doi: 10.1016/0006-3002(58)90364-0. [DOI] [PubMed] [Google Scholar]
- Frieden C. Glutamic dehydrogenase I. The effect of coenzyme on the sedimentation velocity and kinetic mechanism. J Biol Chem. 1959a;234:809–814. [PubMed] [Google Scholar]
- Frieden C. Glutamic dehydrogenase II The effect of various nucleotides on the association-disassociation and kinetic properties. J Biol Chem. 1959b;234:815–819. [PubMed] [Google Scholar]
- Frieden C. Glutamate dehydrogenase VI. Survey of purine nucleotides and other effects on the enzyme from various sources. J Biol Chem. 1965;240:2028–2037. [PubMed] [Google Scholar]
- Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT, Dang CV. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458:762–765. doi: 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhardt B. Fatty acid degradation in plants. Prog Lipid Res. 1992;31:417–446. doi: 10.1016/0163-7827(92)90004-3. [DOI] [PubMed] [Google Scholar]
- Goldin BR, Frieden C. Effect of trinitrophenylation of specific lysyl residues on the catalytic, regulatory, and molecular properties of bovine liver glutamate dehydrogenase. Biochemistry. 1971;10:3527–3534. doi: 10.1021/bi00795a006. [DOI] [PubMed] [Google Scholar]
- Haigis MC, Mostoslavsky R, Haigis KM, Fahie K, Christodoulou DC, Murphy AJ, Valenzuela DM, Yancopoulos GD, Karow M, Blander G, Wolberger C, Prolla TA, Weindruch R, Alt FW, Guarente L. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell. 2006;126:941–954. doi: 10.1016/j.cell.2006.06.057. [DOI] [PubMed] [Google Scholar]
- Hashimoto T. Peroxisomal beta-oxidation enzymes. Neurochem Res. 1999;24:551–563. doi: 10.1023/a:1022540030918. [DOI] [PubMed] [Google Scholar]
- Hoek JB, Rydström J. Physiological roles of nicotinamide nucleotide transhydrogenase. Biochem J. 1988;254:1–10. doi: 10.1042/bj2540001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu BY, Kelly A, Thornton PS, Greenberg CR, Dilling LA, Stanley CA. Protein-sensitive and fasting hypoglycemia in children with the hyperinsulinism/hyperammonemia syndrome. J Pediatr. 2001;138:383–389. doi: 10.1067/mpd.2001.111818. [DOI] [PubMed] [Google Scholar]
- Hudson RC, Daniel RM. L-Glutamate dehydrogenases: distribution, properties and mechanism. Comp Biochem Physiol. 1993;106B:767–792. doi: 10.1016/0305-0491(93)90031-y. [DOI] [PubMed] [Google Scholar]
- Iwatsubo M, Pantaloni D. Regulation De L’ Activite’ De La glutamate dehydrogenase par les effecteurs GTP et ADP: ETUDE par “stopped flow”. Bull Soc Chem Biol. 1967;49:1563–1572. [PubMed] [Google Scholar]
- Kanamori K, Weiss RL, Roberts JD. Role of glutamate dehydrogenase in ammonia assimilation in nitrogen-fixing Bacillus macerans. J Bacteriol. 1987;169:4692–4695. doi: 10.1128/jb.169.10.4692-4695.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor RR, James C, Flanagan SE, Ellard S, Eaton S, Hussain K. 3-Hydroxyacyl-coenzyme A dehydrogenase deficiency and hyperinsulinemic hypoglycemia: characterization of a novel mutation and severe dietary protein sensitivity. Journal of Clinical Endocrinology and Metabolism. 2009;94:2221–2225. doi: 10.1210/jc.2009-0423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kibbey RG, Pongratz RL, Romanelli AJ, Wollheim CB, Cline GW, Shulman GI. Mitochondrial GTP regulates glucose-stimulated insulin secretion. Cell Metabolism. 2007;5:253–264. doi: 10.1016/j.cmet.2007.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Dang CV. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 2006;66:8927–8930. doi: 10.1158/0008-5472.CAN-06-1501. [DOI] [PubMed] [Google Scholar]
- Koberstein R, Sund H. The influence of ADP, GTP and L-glutamate on the binding of the reduced coenzyme to beef-liver glutamate dehydrogenase. Eur J Biochem. 1973;36:545–552. doi: 10.1111/j.1432-1033.1973.tb02942.x. [DOI] [PubMed] [Google Scholar]
- Konayagi S, Minowada M. On the effect of green tea for diabettes mellitus. Study of Physiology; Kyoto University. 1933;10:449–454. [Google Scholar]
- Koshland DEJ. The structural basis of negative cooperativity: receptors and enzymes. Cur Opin Struc Biol. 1996;6:757–761. doi: 10.1016/s0959-440x(96)80004-2. [DOI] [PubMed] [Google Scholar]
- Lenartowicz E. A complex effet of arsenite on the formation of a-ketoglutarate in rate liver mitochondria. Arch Biochem Biophys. 1990;283:388–396. doi: 10.1016/0003-9861(90)90659-m. [DOI] [PubMed] [Google Scholar]
- Li C, Allen A, Kwagh K, Doliba NM, Qin W, Najafi H, Collins HW, Matschinsky FM, Stanley CA, Smith TJ. 281. Green Tea Polyphenols Modulate Insulin Secretion by Inhibiting Glutamate Dehydrogenase. J Biol Chem. 2006:10214–10221. doi: 10.1074/jbc.M512792200. [DOI] [PubMed] [Google Scholar]
- Li C, Buettger C, Kwagh J, Matter A, Daihkin Y, Nissiam I, Collins HW, Yudkoff M, Stanley CA, Matschinsky FM. A signaling role of glutamine in insulin secretion. J Biol Chem. 2004;279:13393–13401. doi: 10.1074/jbc.M311502200. [DOI] [PubMed] [Google Scholar]
- Li C, Chen P, Palladino A, Narayan S, Russell LK, Sayed S, Xiong G, Chen J, Stokes D, Butt YM, Jones PM, Collins HW, Cohen NA, Cohen AS, Nissim I, Smith TJ, Strauss AW, Matschinsky FM, Bennett MJ, Stanley CA. Mechanism of hyperinsulinism in short-chain 3-hydroxyacyl-CoA dehydrogenase deficiency involves activation of glutamate dehydrogenase. J Biol Chem. 2010 doi: 10.1074/jbc.M110.123638. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Najafi H, Daikhin Y, Nissim I, Collins HW, Yudkoff M, Matschinsky FM, Stanley CA. Regulation of leucine stimulated insulin secretion and glutamine metabolism in isolated rat islets. J Biol Chem. 2003;278:2853–2858. doi: 10.1074/jbc.M210577200. [DOI] [PubMed] [Google Scholar]
- Li M, Allen A, Smith TJ. High throughput screening reveals several new classes of glutamate dehydrogenase inhibitors. Biochemistry. 2007;46:15089–15102. doi: 10.1021/bi7018783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Smith CJ, Walker MT, Smith TJ. Novel inhibitors complexed with glutamate dehydrogenase: allosteric regulation by control of protein dynamics. J Biol Chem. 2009;284:22988–23000. doi: 10.1074/jbc.M109.020222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limuti CM. Glutamate dehydrogenase: Equilibrium and kinetic studies. Department of Biochemistry, University of Rochester; Rochester, New York: 1983. [Google Scholar]
- MacMullen C, Fang J, Hsu BYL, Kelly A, deLonlay-Debeney P, Saudubray JM, Ganguly A, Smith TJ, Stanley CA. The Hyperinsulinism/hyperammonemia Contributing Investigators. Hyperinsulinism/hyperammonemia syndrome in children with regulatory mutations in the inhibitory guanosine triphosphate-binding domain of glutamate dehydrogenase. J Clin Endocrinol Metab. 2001;86:1782–1787. doi: 10.1210/jcem.86.4.7414. [DOI] [PubMed] [Google Scholar]
- Markau K, Schneider J, Sund H. Kinetic studies on the mechanism of the action of ADP on the glutamate dehydrogenase reaction. FEBS Lett. 1972;24:32–36. doi: 10.1016/0014-5793(72)80819-6. [DOI] [PubMed] [Google Scholar]
- Melzi-D’eril G, Dalziel K. Negative cooperativity in glutamate dehydrogenase. Coenzyme binding studies. Biochem J. 1973;130:3P. doi: 10.1042/bj1300003pa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michishita E, Park JY, Burneskis JM, Barrett JC, Horikawa I. Evolutionarily conserved an nonconserved cellular localizaitons and functions of human SIRT proteins. Mol Biol Cell. 2005;16:4623–4635. doi: 10.1091/mbc.E05-01-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller M, Hogg JF, De Duve C. Distribution of tricarboxylic acid cycle enzymes and glyoxylate cycle enzymes between mitochondria and peroxisomes in Tetrahymena pyriformis. J Biol Chem. 1968;243:5385–5395. [PubMed] [Google Scholar]
- Pal PK, Wechter WJ, Colman RF. Affinity labeling of a regulatory site of bovine liver glutamate dehydrogenase. Biochemistry. 1975;14:707–715. doi: 10.1021/bi00675a010. [DOI] [PubMed] [Google Scholar]
- Peterson PE, Smith TJ. The structure of bovine glutamate dehydrogenase provides insights into the mechanism of allostery. Structure Fold Des. 1999;7:769–782. doi: 10.1016/s0969-2126(99)80101-4. [DOI] [PubMed] [Google Scholar]
- Piszkiewicz D, Smith EL. Bovine liver glutamate dehydrogenase. Equilibria and kinetics of inactivation by pyridoxal. Biochemistry. 1971;10:4538–4544. doi: 10.1021/bi00800a030. [DOI] [PubMed] [Google Scholar]
- Prough RA, Culver JM, Fisher HF. The mechanism of activation of glutamate dehydrogenase-catalyzed reactions by two different, cooperatively bound activators. J Biol Chem. 1973;248:8528–8533. [PubMed] [Google Scholar]
- Rasool CG, Nicolaidis S, Akhtar M. The asymmetric distribution of enzymic activity between the six subunits of bovine liver glutamate dehydrogenase. Use of D- and L-glutamyl alpha-chloromethyl ketones (4-amino-6-chloro-5-oxohexanoic acid. Biochem J. 1976;157:675–686. doi: 10.1042/bj1570675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy JK, Mannaerts GP. Peroxisomal lipid metabolism. Ann Rev Nutrition. 1994;14:343–370. doi: 10.1146/annurev.nu.14.070194.002015. [DOI] [PubMed] [Google Scholar]
- Rydström J, Teixeira da Cruz A, Ernster L. Factors governing the kinetics and steady state of the mitochondrial nicotinamide nucleotide transhydrogenase system. Eur J Biochem. 1970;17:56–62. doi: 10.1111/j.1432-1033.1970.tb01133.x. [DOI] [PubMed] [Google Scholar]
- Schmidt JA, Colman RF. Identification of the lysine and tyrosine peptides labeled by 5′-p-fluorosulfonylbenzoyladenosine in the NADH inhibitory site of glutamate dehydrogenase. J Biol Chem. 1984;259:14515–14519. [PubMed] [Google Scholar]
- Sener A, Malaisse WJ. L-leucine and a nonmetabolized analogue activate pancreatic islet glutamate dehydrogenase. Nature. 1980;288:187–189. doi: 10.1038/288187a0. [DOI] [PubMed] [Google Scholar]
- Sener A, Malaisse-Lagae F, Malaisse WJ. Stimulation of pancreatic islet metabolism and insulin release by a nonmetabolizable amino acid. Proc Natl Acad Sci USA. 1981;78:5460–5464. doi: 10.1073/pnas.78.9.5460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafer JA, Chiancone E, Vittorelli LM, Spagnuolo C, Machler B, Antonini E. Binding of reduced cofactor to glutamate dehydrogenase. Eur J Biochem. 1972;31:166–171. doi: 10.1111/j.1432-1033.1972.tb02515.x. [DOI] [PubMed] [Google Scholar]
- Shaw RJ. Glucose metabolism and cancer. Curr Opin Cell Biol. 2006;18:598–608. doi: 10.1016/j.ceb.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Smith EL, Austen BM, Blumenthal KM, Nyc JF. Glutamate dehydrogenases. In: Boyer PD, editor. The Enzymes. Vol. 11. Academic Press; New York: 1975. pp. 293–367. [Google Scholar]
- Smith TJ, Peterson PE, Schmidt T, Fang J, Stanley C. Structures of bovine glutamate dehydrogenase complexes elucidate the mechanism of purine regulation. J Mol Biol. 2001;307:707–720. doi: 10.1006/jmbi.2001.4499. [DOI] [PubMed] [Google Scholar]
- Smith TJ, Schmidt T, Fang J, Wu J, Siuzdak G, Stanley CA. The structure of apo human glutamate dehydrogenase details subunit communication and allostery. J Mol Biol. 2002;318:765–777. doi: 10.1016/S0022-2836(02)00161-4. [DOI] [PubMed] [Google Scholar]
- Smith TJ, Stanley CA. Untangling the glutamate dehydrogenase allosteric nightmare. Trends in Biological Chemistry. 2008;33:557–564. doi: 10.1016/j.tibs.2008.07.007. [DOI] [PubMed] [Google Scholar]
- Stanley C, Bennett MJ, Mayatepek E. Disorders of Mitochondrial Fatty Acid Oxidation and Related Metabolic Pathways. In: Fernandes J, Saudubray J-M, van den Berg G, Walter JH, editors. Inborn metabolic diseases. 4. Springer; Heidelberg: 2006. pp. 177–188. [Google Scholar]
- Stanley CA. The hyperinsulinism-hyperammonemia syndrome: gain-of-function mutations of glutamate dehydrogenase. In: O’Rahilly S, Dunger DB, editors. Genetic Insights in Paediatric Endocrinology and Metabolism. Bio Scientifica, Ltd; Bristol: 2000. pp. 23–30. [Google Scholar]
- Stanley CA, Fang J, Kutyna K, Hsu BYL, Ming JE, Glaser B, Poncz M. Molecular basis and characterization of the hyperinsulinism/hyperammonemia syndrome of the glutamate dehydrogenase gene. Diabetes. 2000;49:667–673. doi: 10.2337/diabetes.49.4.667. [DOI] [PubMed] [Google Scholar]
- Stanley CA, Lieu YK, Hsu BY, Burlina AB, Greenberg CR, Hopwood NJ, Perlman K, Rich BH, Zammarchi E, Poncz M. Hyperinsulinism and hyperammonemia in infants with regulatory mutations of the glutamate dehydrogenase gene. New England Journal of Medicine. 1998;338:1352–1357. doi: 10.1056/NEJM199805073381904. [DOI] [PubMed] [Google Scholar]
- Syed SEH, Engel PC. Ox liver glutamate dehydrogenase. The use of chemical modification to study the rlationship between catalytic sites for differeent amino acid substrates andthe question of kinetic non-equivalence of the subunits. Biochem J. 1984;222:621–626. doi: 10.1042/bj2220621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomkins GM, Yielding KL, Curran JF. The influence of diethylstilbestrol and adenosine diphosphate on pyridine nucleotide coenzyme binding by glutamic dehydrogenase. J Biol Chem. 1962;237:1704–1708. [PubMed] [Google Scholar]
- Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB, Thompson CB. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A. 2008;105:18782–18787. doi: 10.1073/pnas.0810199105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrzeszczynski KO, Colman RF. Activation of bovine liver glutamate dehydrogenase by covalent reaction of adenosine 5′-O-[S-(4-bromo-2,3-dioxobutyl)thiophosphate] with arginine-459 at an ADP regulatory site. Biochemistry. 1994;33:11544–11553. doi: 10.1021/bi00204a017. [DOI] [PubMed] [Google Scholar]
- Yang C, Sudderth J, Dang T, Bachoo RG, McDonald JG, DeBerardinis RJ. Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or Akt signaling. Cancer Res. 2009;69:7986–7993. doi: 10.1158/0008-5472.CAN-09-2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CS, Wang ZY. Tea and cancer. J National Cancer Inst. 1993;85:1038–1049. doi: 10.1093/jnci/85.13.1038. [DOI] [PubMed] [Google Scholar]
- Yielding KL, Tomkins GM. An effect of L-leucine and other essential amino acids on the structure and activity of glutamate dehydrogenase. Proc Natl Acad Sci. 1961;47:983. doi: 10.1073/pnas.47.7.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yielding KL, Tomkins GM, Munday JS, Curran JF. The effects of steroid hormones on the glutamic dehydrogenase reaction. Biochem Biophys Res Comm. 1960;2:303–306. [Google Scholar]