

Abstract
Native chemical ligation (NCL) is widely applicable for building proteins in the laboratory. Since the discovery of this method, many strategies have been developed to enhance its capability and efficiency. Because of the poor reactivity of proline thioesters, ligation at a C-terminal proline site is not readily accomplished. Here, we demonstrate that ligation at an N-terminal protein is feasible using the combined logic of NCL and metal free dethiylation (MFD).
The groundbreaking discovery of native chemical ligation (NCL) by Kent and colleagues was a seminal event in the chemical synthesis of protein-based biopolymers with precisely defined structures.1,2 Typically, NCL involves the reaction between an unprotected peptidyl C-terminal thioester with another peptide bearing an N-terminal cysteine. Following rate determining trans thioacylation, a fast S→N acyl transfer presumably passing through a five membered tetrahedral intermediate leads to product (Scheme 1, eq 1, Kent et al.).3
Scheme 1.

NCL, desulfurization, and extended NCL.a
a P1 and P2 are peptide fragments. R can be alkyl or aryl. R1 can be varied according to the amino acid structure.
While the original discovery of NCL was based on a peptidyl N-terminal cysteine which appeared at the ligation site, it was soon extended to alanine ligation, via desulfurization of the thiol function by Raney Nickel reduction,4 to phenylalanine ligation, using nickel boride as reductant,5 and to serine ligation, through cyanobromide-mediated cleavage of the methylcysteine precursor.6 The finding that high yielding metal free de-thiylation is feasible in aqueous medium and specific to SH groups constituted a major expansion of NCL (Scheme 1, eq 2). Not only did it enable alanine ligation in the presence of existing ACM protected cysteines (as well as methionines), it opened up an entirely new set of possibilities for ligation. One could now incorporate a thiol containing unnatural amino acid at the N-terminus.7,8 The combination of thiol and amino groups allows for simulations of the qualitative mechanism of cysteine based NCL. Metal free dethiylation (MFD) generates either a proteogenic or unnatural amino acid at the ligation site (Scheme 1, eq 3).7a
Using this MFD enabled logic, we and others have demonstrated valine,9,10 leucine,11,12 threonine,13 and lysine ligations.14,15 Thus, MFD has brought with it a major expansion in the synthesis of reasonable sized proteins, using the powerful underlying logic of NCL.
The research described herein was directed to a remaining but important problematic issue – i.e. that of proline ligation.16 It turns out that for reasons that have been elegantly interpreted, C-terminal proline thioesters are not particularly effective acyl donors, even in the context of cysteine based NCL.17–19 While the corresponding C-terminal proline p-nitrophenyl esters are more effective,10 they too carry a serious liability – i.e. susceptibility to hydrolysis in a ligation context. We were thus led to ask whether, in light of the considerations discussed above, a proline–like moiety at the N-terminus of a peptide might serve as a ligation site. This led us to consider the possibility of utilizing an N-terminal thioproline at the ligation site. In so doing, we would be extending the mechanistic concepts of NCL in two major respects. First, the S→N acyl migration would have to occur even at a secondary amine in a ring context. Furthermore, the transfer would be occurring in the context of a bridged ring tetrahedral intermediate. To probe the feasibility of the idea, we took recourse to the commercially available diastereomeric 4-thioproline building blocks 1 and 2.
We began by synthesizing peptides 4 and 7 incorporating N-terminal γ-thioprolines [(2S,4R)-Mpt] and [(2S,4S)-Mpt], respectively, by the solid-phase peptide synthesis (SPPS) method using the Fmoc chemistry. We went on to evaluate the ligation of both 4 and 7 with peptide 3, presenting a C-terminal glycine residue. For this purpose, we took recourse to the surrogate acyl donor system bearing a 2-(ethyldithiophenyl)ester, previously invented in our laboratory as a more durable thioester equivalent.20,21 The required thioester is generated in situ, as previously described, via unidirectional O→S acyl transfer.21 The reaction between 4 and 3 was initiated by the addition of the standard phosphate ligation buffer, and was monitored by ultra performance liquid chromatography (UPLC). As shown in Scheme 2, under standard ligation conditions, peptide 4 readily underwent transthioesterification with 3 to afford 5 (90% conversion),22 within 10 minutes at room temperature.23 At a considerably slower rate, the thioester intermediate 5 underwent the S→N acyl transfer reaction to afford the desired peptide product in 85% yield within 2.5h. By contrast, under identical reaction conditions, peptide 7, incorporating the diastereomeric γ-thioproline, [(2S,4S)-Mpt], demonstrated very low proclivity toward ligation with peptide 3. Although the trans thioesterification could be achieved as efficiently as in the case of 5, progression to peptide product 9 essentially failed. After 28 h, less than 5% conversion was observed. Instead, about 90% of thioester 8 was hydrolyzed to peptide 7.
Scheme 2.

Proline ligation with two Pro(SH) diastereomers.a
a Key: (a) 6 M Gn•HCl, 100 mM NaH2PO4, 50 mM TCEP, pH 7.5; (b) TCEP, VA-044, tBuSH, MeCN/H2O, 37 °C, 10 min, 88%. P1: ALLVNSS–; P2: –WEPLN; Ar = 2-(ethyldithio)phenyl.
This finding of a large difference is readily interpreted. Thus, in the case of the productive trans series, in the presumed tetrahedral S→N acyl transfer intermediate, the large COP2 substructure is exo. By contrast, in the unproductive cis series, based on 2, the large COP2 moiety would have to be contained in a highly hindered endo disposition.
Having established an efficient and practical γ-thioproline ligation protocol, albeit only with a terminal glycine, we sought to probe the versatility of this protocol in the context of varying the nature of the C-terminal amino acid residue. Toward this end, we prepared several peptide esters, which would allow us to evaluate the effects of steric hindrance on peptide bond formation. In order to avoid any potential complications associated with residues not involved in amide bond formation, these peptide esters only differ by a side chain at the C-terminus. In accordance with previous NCL studies, the rate and efficiency of ligation was found to be dependent on the steric hindrance of the C-terminal residue.9,10,12,16 Under standard conditions, peptide 10 and 12 underwent rapid transthioesterification with the peptide 4 to generate the corresponding thioester intermediates. Although more time was required to complete the S→N acyl transfer, good yields were obtained (entries 1 and 2). As expected, the coupling reactions at the valine and proline sites were significantly less efficient (entries 3 and 4). Interestingly, the factors that caused the low conversion in these two reactions are different. In the case of valine, the trans-thioesterification step was fast and apparently efficient. However, the resulting thioester intermediate was insufficiently reactive to allow for reasonable rates of acyl transfer. By contrast, the proline peptide ester 16 was found to exhibit remarkably diminished levels of reactivity in the transthioacylation step. Only about 5% of 16 was converted to the thioester intermediate, which was rapidly hydrolyzed under attempted ligation conditions to peptide 4.
Next, in order to probe the origin of the observed low reactivity of the proline peptide ester 16, an additional control ligation was performed between peptide 4 and proline ester 18. Under the standard conditions, 4 and 18 underwent rapid transthioesterification and acyl transfer to furnish adduct 19 in 73% yield within 3h (entry 5). This result agreed well with the observations of Kent et al, suggesting that the presence of a peptidyl group on the α-amine of the C-terminal proline is the dominant factor affecting the reactivity of the proline peptide ester in NCL.17
Furthermore, the versatility of the proline ligation protocol was demonstrated in the synthesis of the proline-rich peptide 21. In the event, peptide 20, containing 50% proline, was coupled with the glutamine peptide ester 10. The result is shown in Table 1, entry 6. Within 10h, a 66% isolated yield of 21 was obtained.
Table 1.
Substrate scope of proline ligation.a
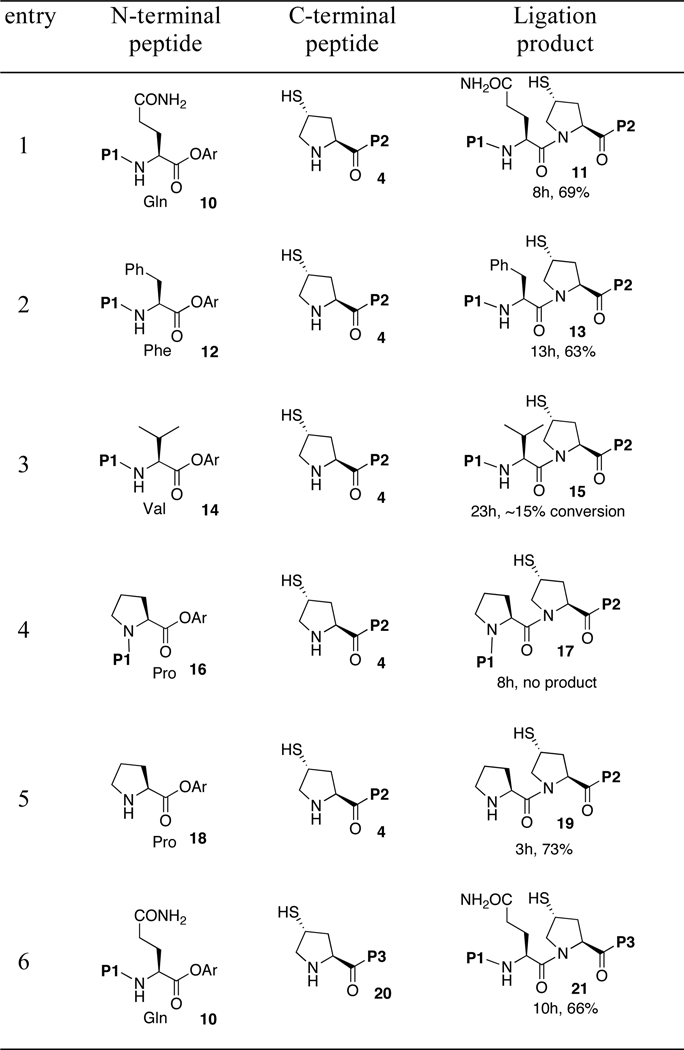 |
Reaction conditions: 6 M Gn•HCl, 100 mM NaH2PO4, 50 mM TCEP, pH 7.5. P1 ALLVNSS–; P2 –WEPLN; P3 –PPWEPLN; Ar = 2-(ethyldithio)phenyl.
Finally, we demonstrated the ability to efficiently convert the γ-thioproline to the natural amino acid proline in the context of a complex peptide. Upon exposure to our standard metal-free desulfurization conditions, the γ-thioproline containing peptides 11, 13, 19, and 21 were readily converted to the target peptide products in less than 10 minutes.7a,10,12,24
Since both native chemical ligation and direct aminolysis are possible options for the synthesis of large peptides and small proteins, we also directly compared these two methods for synthesizing desulfurized 11.25,26 The direct aminolysis reaction was conducted in DMSO using modified Blake-Aimoto conditions (SI, Figure S4).27 At room temperature, it proceeded well to give coupling product in a slightly higher yield. However, because of the difficulties related to the preparation and purification of partially protected peptide fragments28 and the risk of epimerization of the residue on the C-terminal side of the condensation site,27 in many cases, proline ligation would be a better choice for coupling two synthetic fragments at the proline site.
In summary, an efficient and broadly useful two-stage proline ligation protocol has been developed. The likely basis for the different ability of the peptide esters to undergo proline ligation has been rationalized. The proline ligation approach provides a significant advance in the field of protein chemical synthesis, and will likely find broad application in the study of proline-rich proteins and glycoproteins. It is of note that proline-rich proteins frequently participate in diverse signaling pathways and serve many crucial biological functions.29,30 The unique conformational properties of proline inserts provide the molecular basis for highly discriminatory recognition during the formation of multiprotein signaling complexes. Understanding how these interactions contribute to the stability of the complex should prove valuable in the rational design of peptide mimics that could potentially be used to disrupt the interfaces between proline-rich proteins and their binding partners.31 The methodology developed herein is expected to facilitate the preparation of proline-rich polypeptide probe structures, thus enhancing opportunities for detailed analysis of protein-protein interactions.
Supplementary Material
ACKNOWLEDGMENT
Support for this research was provide by the National Institute of Health (CA28824 to SJD). We thank Rebecca Lambert for valuable discussions. We also thank Dr. George Sukenick, Hui Fang, and Sylvi Rusli of SKI’s NMR core facility for mass spectral and NMR assistance and Laura Wilson and Jason Chan for assistance with the preparation of the manuscript.
Footnotes
SUPPORTING INFORMATION. Experimental procedures and characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Dawson PE, Muir TW, Clark-Lewis I, Kent SB. Science. 1994;266:776. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- 2.Tam JP, Lu YA, Liu CF, Shao J. Proc. Natl. Acad. Sci. USA. 1995;92:12485. doi: 10.1073/pnas.92.26.12485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnson EC, Kent SB. J. Am. Chem. Soc. 2006;128:6640. doi: 10.1021/ja058344i. [DOI] [PubMed] [Google Scholar]
- 4.Yan LZ, Dawson PE. J. Am. Chem. Soc. 2001;123:526. doi: 10.1021/ja003265m. [DOI] [PubMed] [Google Scholar]
- 5.Crich D, Banerjee A. J. Am. Chem. Soc. 2007;129:10064. doi: 10.1021/ja072804l. [DOI] [PubMed] [Google Scholar]
- 6.Okamoto R, Kajihara Y. Angew. Chem. Int. Ed. 2008;47:5402. doi: 10.1002/anie.200801097. [DOI] [PubMed] [Google Scholar]
- 7. Wan Q, Danishefsky S. J. Angew. Chem. Int. Ed. 2007;46:9248. doi: 10.1002/anie.200704195.; For earlier work on phosphorus derivatives-mediated radical desulfurization, see: Walling C, Rabinowitz R. J. Am. Chem. Soc. 1957;79:5326. Walling C, Basedow OH, Savas ES. J. Am. Chem. Soc. 1960;82:2181.
- 8.A futuristic statement of this concept can be found in Botti P, Tchertchian S. Patent No. WO2006133962. 2006
- 9.Haase C, Rohde H, Seitz O. Angew. Chem. Int. Ed. 2008;47:6807. doi: 10.1002/anie.200801590. [DOI] [PubMed] [Google Scholar]
- 10.Chen J, Wan Q, Yuan Y, Zhu J, Danishefsky SJ. Angew. Chem. Int. Ed. 2008;47:8521. doi: 10.1002/anie.200803523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harpaz Z, Siman P, Kumar KS, Brik A. Chembiochem. 2010;11:1232. doi: 10.1002/cbic.201000168. [DOI] [PubMed] [Google Scholar]
- 12.Tan Z, Shang S, Danishefsky SJ. Angew. Chem. Int. Ed. 2010;49:9500. doi: 10.1002/anie.201005513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen J, Wang P, Zhu JL, Wan Q, Danishefsky SJ. Tetrahedron. 2010;66:2277. doi: 10.1016/j.tet.2010.01.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang R, Pasunooti KK, Li F, Liu XW, Liu CF. J. Am. Chem. Soc. 2009;131:13592. doi: 10.1021/ja905491p. [DOI] [PubMed] [Google Scholar]
- 15.Kumar KSA, Haj-Yahya M, Olschewski D, Lashuel HA, Brik A. Angew. Chem. Int. Ed. 2009;48:8090. doi: 10.1002/anie.200902936. [DOI] [PubMed] [Google Scholar]
- 16.Hackeng TM, Griffin JH, Dawson PE. Proc. Natl. Acad. Sci. USA. 1999;96:10068. doi: 10.1073/pnas.96.18.10068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pollock SB, Kent SB. Chem. Commun. 2011;47:2342. doi: 10.1039/c0cc04120c. [DOI] [PubMed] [Google Scholar]
- 18.Hinderaker MP, Raines RT. Protein Sci. 2003;12:1188. doi: 10.1110/ps.0241903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choudhary A, Raines RT. Protein Sci. 2011;20:1077. doi: 10.1002/pro.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Warren JD, Miller JS, Keding SJ, Danishefsky SJ. J. Am. Chem. Soc. 2004;126:6576. doi: 10.1021/ja0491836. [DOI] [PubMed] [Google Scholar]
- 21.Chen G, Warren JD, Chen J, Wu B, Wan Q, Danishefsky SJ. J. Am. Chem. Soc. 2006;128:7460. doi: 10.1021/ja061588y. [DOI] [PubMed] [Google Scholar]
- 22.Thioester intermediate 5 could be observed by LC-MS analysis, and was differentiated from the N-linked product 6 by the addition of MESNa, where disappearance of 5 would be observed.
- 23.Canne LE, Bark SJ, Kent SBH. J. Am. Chem. Soc. 1996;118:5891. [Google Scholar]
- 24.Shang S, Tan Z, Danishefsky SJ. Proc. Natl. Acad. Sci. USA. 2011;108:5986. doi: 10.1073/pnas.1103118108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen G, Wan Q, Tan Z, Kan C, Hua Z, Ranganathan K, Danishefsky SJ. Angew. Chem. Int. Ed. 2007;46:7383. doi: 10.1002/anie.200702865. [DOI] [PubMed] [Google Scholar]
- 26.Tan Z, Shang S, Halkina T, Yuan Y, Danishefsky SJ. J. Am. Chem. Soc. 2009;131:5424. doi: 10.1021/ja808704m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aimoto S. Curr. Org. Chem. 2001;5:45. [Google Scholar]
- 28.Tan Z, Shang S, Danishefsky SJ. Proc. Natl. Acad. Sci. USA. 2011;108:4297. doi: 10.1073/pnas.1100195108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vanhoof G, Goossens F, Demeester I, Hendriks D, Scharpe S. FASEB J. 1995;9:736. [PubMed] [Google Scholar]
- 30.Ball LJ, Kuhne R, Schneider-Mergener J, Oschkinat H. Angew. Chem. Int. Ed. 2005;44:2852. doi: 10.1002/anie.200400618. [DOI] [PubMed] [Google Scholar]
- 31.Kuriyan J, Cowburn D. Annu. Rev. Biophys. Biomol. Struct. 1997;26:259. doi: 10.1146/annurev.biophys.26.1.259. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.