

Abstract
Background
Ewing sarcoma can arise in either bone or soft tissue locations. We sought to investigate if patient characteristics, treatment strategies, and outcomes differ between skeletal Ewing sarcoma and extraskeletal Ewing sarcoma (EES).
Procedure
Patients < 40 years of age with Ewing sarcoma or peripheral primitive neuroectodermal tumor (PNET) reported to the US SEER database from 1973 to 2007 were evaluated based on skeletal (n=1519) vs. extraskeletal (n=683) site of origin. Patient characteristics were compared using Fisher exact tests. Overall survival was estimated by Kaplan-Meier methods and compared using log-rank tests and Cox models.
Results
Patients with EES had a higher mean age (19.5 vs. 16.3 years; p < 0.001) and were less likely to be male (53.4% vs. 63.3%; p < 0.001) or white (84.8% vs. 92.5%; p < 0.001) compared to patients with skeletal tumors. Extraskeletal tumors were more likely to arise in axial locations (72.9% vs. 54.2%; p = 0.001), though less likely to arise specifically in the pelvis (19.8% vs. 26.6%; p < 0.001). Metastatic status or tumor size did not differ by group. Five-year overall survival was superior for localized EES compared to localized skeletal tumors (69.7% vs. 62.6%; p = 0.02). The hazard ratio for death in patients with localized skeletal tumors compared to localized EES was 2.36 (95% CI 1.61-3.44) beyond 24 months from initial diagnosis.
Conclusions
Patient characteristics and outcomes differ among patients with EES compared to patients with skeletal Ewing sarcoma. These findings may have important implications for patient care.
Keywords: Ewing sarcoma, extraskeletal, extraosseous, PNET, SEER
Introduction
Ewing sarcoma is the second most common childhood primary bone cancer, though a substantial proportion of these tumors arise from extraskeletal sites.1 Several studies have demonstrated the prognostic impact of tumor stage, size, site, and age at diagnosis, with most studies focused on Ewing sarcoma of the bone.2-9 Few studies have directly compared skeletal Ewing sarcoma to extraskeletal Ewing sarcoma (EES). Smaller studies have evaluated this population and suggest that outcomes for patients with EES are at least similar to that of Ewing sarcoma of the bone.10-16 This finding has also been observed in two recently reported multicenter clinical trials.1, 17 However, these studies have focused on relatively small numbers of patients and have not focused on all age ranges. Moreover, small sample sizes have precluded more detailed analysis of the prognostic impact of skeletal vs. extraskeletal origin of these tumors.
Based on the above evidence, the extent to which the clinical characteristics and outcomes for skeletal Ewing sarcoma and EES are similar or different is uncertain. Developing a better understanding of this relationship is important for the development of future clinical trials. We hypothesized that EES has different tumor characteristics, treatment approaches, and outcomes compared to skeletal Ewing sarcoma. Using data from the Surveillance, Epidemiology and End Results Program (SEER), we evaluated this hypothesis in a cohort of patients diagnosed with either EES or skeletal Ewing sarcoma.
Methods
Patients and Variables
We gathered patient information from the US National Cancer Institute's SEER database, which included data from 1973-2007. The SEER system is used in regions that represent ~26% of the US population. Beginning January 1, 1973, the SEER program initiated the collection and publication of cancer incidence, patient demographics, primary tumor site, tumor morphology, stage at diagnosis, limited treatment data, and survival data.
Patients with histologically confirmed Ewing sarcoma, Askin tumor, or peripheral primitive neuroectodermal tumor (PNET) between 0 and 39 years of age were eligible for the study. This age range was chosen to capture the majority of typical cases of Ewing sarcoma. Patients with PNET were included as these tumors are now believed to be belong to the family of Ewing sarcoma.18 From 1973-2007, the 17 population based SEER registries reported 2,221 cases of Ewing sarcoma or PNET in patients < 40 years of age. One patient with melanotic neuroectodermal tumor was excluded from analysis, as this is a distinct tumor from Ewing sarcoma.19 We also excluded patients with unknown skeletal vs. extraskeletal status (n=18; see below), as this was the primary predictor variable of interest. The remaining 2,202 patients formed the analytic cohort for this study.
Ewing sarcoma or PNET were classified in the SEER database according to the International Classification of Childhood Cancer and/or the International Classification of Disease for Oncology, third revision (ICD-O-3). Anatomic site codes were classified according to the ICD-O coding scheme. Patients were classified as EES or skeletal Ewing sarcoma based on ICD-10-CM coding used by the SEER database. These codes are specific for bone vs. soft tissue sites. Patients with the following ambiguous tumor locations were excluded (n = 18): unknown; thorax NOS; orbit NOS; and palate NOS. Patient characteristics and clinical presentation were evaluated according to classification as EES or skeletal Ewing sarcoma. Variables of interest included: sex; stage (metastatic vs. localized); year of diagnosis (in sequential 5-year blocks); race (white vs. non-white); histology (PNET vs. Ewing sarcoma); primary tumor site (axial vs. appendicular and also pelvic vs. non-pelvic); and tumor size (< 10 vs. ≥ 10 cm in maximal dimension).
Data on treatment received were also collected. Data regarding the use of surgery as local control was inconsistent and not felt to be reliable for further analysis. Radiation therapy was dichotomized as not given or given if delivered to any tumor site (primary and/or metastatic) at any time point during treatment (including radioactive implants and radioisotopes). For analysis related to use of radiation, patients were excluded if they had missing data for radiation (n=19). Other treatment details, including the use of chemotherapy, were not available.
Statistical Methods
Patient, tumor, and treatment categorical characteristics were evaluated for differences between EES and skeletal Ewing sarcoma using the Fisher exact test, with the exception of year of diagnosis for which a chi square test was used. A two-sided unpaired t-test was used to compare patient age between groups.
A logistic regression model was constructed to evaluate differences in the frequency of radiation use between EES and skeletal Ewing sarcoma while controlling for year of diagnosis, tumor location, age, and metastatic status. This was further confirmed with a sensitivity analysis that also controlled for size and included the 50.3% of patients for whom size data were available.
Overall survival from the time of diagnosis was estimated by Kaplan-Meier methods and potential differences between EES and skeletal Ewing sarcoma were evaluated using the log-rank test. Overall survival was expressed as Kaplan-Meier estimates with 95% confidence interval (CI). The median follow-up time for the analyzed cohort was 86 months.
Cox proportional hazard models were used to assess the effect of EES vs. skeletal origin on overall survival while controlling for known prognostic factors. Visual inspection of the univariate Kaplan-Meyer curve comparing EES to skeletal Ewing sarcoma showed that the survival functions crossed at approximately 24 months. This suggested that standard proportional hazards modeling might not be appropriate. A formal test of the proportional hazard assumption was performed using time-varying covariates and confirmed violation of this assumption20. We therefore constructed a model that allowed the hazards to differ before and after 24 months from initial diagnosis. A sensitivity analysis was performed that also controlled for tumor size.
The SEER database was accessed using SEER*Stat version 6.6.2. All statistical analyses were performed using SAS, version 9 and STATA, version 10.
Results
Patient Characteristics
Of the 2,202 patients in the analytic cohort, 683 (31%) had EES and 1519 (69%) had skeletal tumors. The clinical characteristics of patients in these two groups are shown in Table 1.
Table 1.
Patient characteristics by extraskeletal vs. skeletal tumor origin in 2202 patients with Ewing sarcoma.
| Characteristic | Extraskeletal Ewing Sarcoma n = 683 (31.0%) | Skeletal Ewing Sarcoma n = 1519 (69.0%) | p-value |
|---|---|---|---|
| Mean age | 19.5 years | 16.3 years | <0.001 |
| Range | 0-39 years | 0-39 yeas | |
| Male | 53.4% | 63.3% | <0.001 |
| Race | |||
| White | 84.8% | 92.5% | |
| Non-White | 15.2% | 7.5% | <0.001 |
| Stage | |||
| Metastatic | 30.1% | 29.9% | |
| Localized | 69.9% | 70.1% | 0.92 |
| Histology | |||
| PNET | 58.3% | 6.4% | |
| Ewing sarcoma | 41.7% | 93.6% | <0.001 |
| Primary Site | |||
| Axial | 72.9% | 54.2% | |
| Non-axial | 27.1% | 45.8% | <0.001 |
| Pelvic | 19.8% | 26.6% | |
| Non-pelvic | 80.2% | 73.4% | 0.001 |
| Percent of axial tumors in pelvis | 28.0% | 49.1% | <0.001 |
| Size | |||
| ≥ 10cm | 40.7% | 35.9% | 0.12 |
| Year of Diagnosis | |||
| 1973-1977 | 0.29% | 7.4% | <0.001* |
| 1978-1982 | 3.1% | 10.1% | |
| 1983-1987 | 1.2% | 10.5% | |
| 1988-1992 | 6.3% | 9.4% | |
| 1993-1997 | 20.6% | 13.8% | |
| 1998-2002 | 31.6% | 22.5% | |
| 2003-2007 | 36.9% | 26.3% | |
Calculated using chi square analysis
Significant differences in clinical presentation were noted between EES and skeletal Ewing sarcoma. Patients with EES had a higher mean age compared to patients with bone tumors (19.5 vs. 16.3 years; p < 0.001). In order to explore this difference further, we also evaluated age in 5-year categories. Compared to patients with skeletal Ewing sarcoma, patients with EES were more likely to be < 5 (9.7% vs. 4.9%; p < 0.001) or ≥ 35 (10.7% vs. 3.4%; p < 0.001) years old at initial diagnosis. Patients with skeletal Ewing sarcoma were more likely to be male (63.3% vs. 53.4%; p < 0.001) or white (92.5% vs. 84.8%; p < 0.001) compared to patients with extraskeletal tumors. Extraskeletal tumors were more likely to arise in axial locations (72.9% vs. 54.2%; p = 0.001), though less likely to arise specifically in the pelvis (19.8% vs. 26.6%; p < 0.001). EES was also more likely to have a histological classification of PNET vs. Ewing sarcoma (p < 0.001) and be diagnosed in more recent years (p < 0.001). There were no differences in tumor size or metastatic status between EES and bone tumors.
Use of Radiotherapy
Radiation therapy was used in 50.1% of all patients. Patients with EES were less likely to receive radiation compared to patients with skeletal tumors (42.3% vs. 54.1%; p < 0.001). Given that the choice to utilize radiation treatment may be confounded by other patient and tumor characteristics, we constructed a logistic regression model to control for these potential confounders. After controlling for age, tumor site, metastatic status, and year of diagnosis, bone tumors continued to demonstrate higher radiation rates compared to EES. The odds ratio for receiving radiation for bone tumors was 1.48 (95% CI 1.20-1.82; p < 0.001). A sensitivity analysis that also controlled for tumor size was performed and yielded similar results.
Patient Outcomes
Among patients with localized disease, overall survival was superior for patients with EES compared to patients with skeletal tumors (p = 0.02; Figure 1). Five- and ten-year Kaplan-Meier estimates of overall survival for localized EES compared to localized skeletal tumors were 69.7% (95% CI 64.7-74.2%) vs. 62.6% (95% CI 59.1-65.9%) and 65.2% (95% CI 59.3-70.5%) vs. 55.3% (95% CI 51.5-58.8%), respectively. Among all patients with either metastatic or localized tumors, overall survival did not differ between EES and skeletal Ewing sarcoma. Similarly, overall survival did not differ between patients with metastatic EES and metastatic skeletal tumors.
Figure 1.
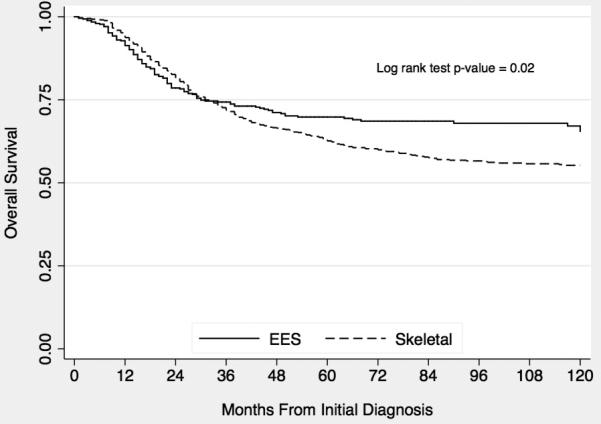
Kaplan-Meier estimates of overall survival from time of diagnosis according to extraskeletal (EES) vs. skeletal origin in patients with localized Ewing sarcoma.
In order to evaluate this survival difference further, we constructed a Cox proportional hazards model that also controlled for age, year of diagnosis (in five year blocks), tumor site, and race in patients with localized tumors. Since the survival hazard for EES vs. bone origin differed over time, we incorporated a time-varying covariate to allow the hazard ratio to differ before and after 24 months from initial diagnosis. This time point was chosen based on inspection of the Kaplan-Meier overall survival curve, indicating more favorable early outcomes for patients with bone tumors and more favorable late outcomes for patients with EES. From 0 – 24 months after initial diagnosis, the hazard ratio for death for patients with bone tumors was 0.71 (95% CI 0.53-0.94) compared to EES. After 24 months, the hazard ratio for patients with bone tumors was 2.36 (95% CI 1.61-3.44) compared to EES. Other time points evaluated in the range of 12 – 30 months yielded similar findings. A sensitivity analysis that also controlled for tumor size was performed and yielded similar results.
Discussion
In this study, we found significant differences in patient demographics and other important clinical features between skeletal Ewing sarcoma and EES. We also found that patients with EES were more likely to be treated with radiotherapy and that their overall survival was worse in the first 24 months but better after 24 months compared to patients with skeletal Ewing sarcoma. The differences in tumor characteristics and clinical behavior between EES and skeletal Ewing sarcoma suggest that they may have subtle biologic differences.21 Some of these differences may arise from differences in the tumor microenvironment between Ewing sarcoma arising from the bone vs. an extraskeletal location. Our study represents a significant advance from previous studies because it is the largest and most comprehensive analysis of patient characteristics, treatment strategies, and outcomes in EES compared to skeletal Ewing sarcoma.
We clearly demonstrated that patient characteristics differ between EES and skeletal tumors. Patients with EES have a higher mean age, but also a bimodal distribution with EES more commonly found in those older than 35 and less than 5 years compared with skeletal tumors. This finding is consistent with prior reports.11 However, our study also noted other important differences between EES and skeletal tumors. Patients with EES were less likely to be male, white or have pelvic primary tumors, though more likely to have tumors arising in other axial locations. These extraskeletal tumors were more likely to have a histological classification of PNET and to be diagnosed more recently. The increased proportion of extraskeletal tumors classified as PNET may reflect changing nomenclature, as a common misconception is that soft tissue origin is synonymous with PNET histology.22 PNET histology has been described in bone tumors,23 a finding we observed in 6.4% of osseous tumors in the current study. Similarly, the increased number of cases reported in more recent years likely reflects the more recent recognition and reporting of EES as opposed to a true increase in the incidence of EES. In particular, improved cytogenetic and molecular diagnostic methods now allow for more reliable detection of genetic translocations characteristic of this disease.
The use of radiation therapy for Ewing sarcoma has evolved over the past several decades.24 Radiation has generally become less common, though still plays an important role.25 We found that radiation therapy is more common in skeletal Ewing sarcoma. This is somewhat surprising as more of these tumors were in the extremities, a site which would typically be more amenable to surgical resection. Recognizing that the use of radiation has diminished over time, we also controlled for year of diagnosis with similar results. Therefore, the reasons for this observation are unclear and require further study.
Previous smaller prior studies have suggested that outcomes for EES and skeletal Ewing sarcoma are similar.9, 12, 15, 26 Two prospective treatment studies have also shown similar outcomes for EES when treated with Ewing sarcoma protocols.1, 17 In contrast, our findings suggest that outcome differences for patients with EES compared to skeletal Ewing sarcoma are more complex. We found that patients with localized EES have an unfavorable prognosis prior to two years from initial diagnosis, but then the outcomes for EES are significantly better. These results were confirmed even after controlling for other known prognostic factors. Of note, the magnitude of this difference was much greater after 24 months, suggesting that extraskeletal site has a greater impact on long-term rather than short-term outcomes. It is possible that patients with EES are diagnosed later in their disease course compared to skeletal tumors, causing a shift in the survival curve. This hypothesis can not be tested using data available in the SEER database and will require further study. There may also be other biologic or treatment differences that account for the observed increased early mortality seen in EES. Unfortunately, additional biological and treatment data are not available from the SEER database and these possibilities can not be further elucidated by this study. Finally, we must be cautious in the conclusions we draw from this finding because our study involved patients over several decades and information about how these patients were treated is limited. Nevertheless, these outcome differences highlight the importance of further research in this area.
A main strength of this study is the large number of EES cases analyzed by using the SEER database. These results are more representative of the general population, as the database reflects many areas of the United States and many different modalities of treatment. This strategy enabled us to evaluate the largest group of EES patients published to date to our knowledge. However, there are several limitations to analyzing data from SEER, which are similar to any study using a tumor registry. We were limited to the available data in the registry, which does not provide information on cause of death, time to tumor recurrence, or chemotherapy utilization. In addition, tumor histology could not be independently confirmed. Most importantly, site of origin in either bone or soft tissue could not be confirmed. The definition of EES is not standardized. Indeed, utilizing the reported site of origin data from SEER revealed regional differences in the incidence of EES (data not shown). Nevertheless, the proportion of cases with EES (31%) out of all reported cases of Ewing sarcoma is similar to prior reports.1
In order to develop the largest cohort possible, we included patients diagnosed in the 1970s. Both the role of chemotherapy and surgical treatments evolved significantly over the past decades which could have influenced our results. It is also possible that cases of EES were misclassified in the past as it has only been recognized as a distinct entity more recently. In order to account for these changes over time, we controlled for year of diagnosis in our regression models and were able to confirm our univariate results. Another limitation is the paucity of data for tumor size in the SEER database. Tumor size is a recognized prognostic indicator in patients with Ewing sarcoma.3, 27, 28 In the SEER database, tumor size was unavailable for approximately half of the analyzed population. To address this, we performed sensitivity analyses using size data among the patients for whom these data were available and obtained similar results to regression models not including size.
Based on our findings, we conclude that there are significant differences in clinical presentation, treatment strategy, and outcomes for EES compared to skeletal Ewing sarcoma. Our findings support the idea that EES is an important subtype of Ewing sarcoma that may require different treatment strategies. Future studies should investigate the biologic basis for these differences. Additional efforts should be directed at determining optimal treatment strategies to maximize outcomes in these tumors.
Acknowledgments
Support: Supported by the Campini Foundation and NIH/NCRR/OD UCSF-CTSI Grant Number KL2 RR024130. The contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Footnotes
Disclosures: None
References
- 1.Granowetter L, Womer R, Devidas M, et al. Dose-intensified compared with standard chemotherapy for nonmetastatic Ewing sarcoma family of tumors: A Children's Oncology Group Study. J Clin Oncol. 2009;27:2536–2541. doi: 10.1200/JCO.2008.19.1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cotterill SJ, Ahrens S, Paulussen M, et al. Prognostic factors in Ewing's tumor of bone: Analysis of 975 patients from the European Intergroup Cooperative Ewing's Sarcoma Study Group. J Clin Oncol. 2000;18:3108–3114. doi: 10.1200/JCO.2000.18.17.3108. [DOI] [PubMed] [Google Scholar]
- 3.Hense HW, Ahrens S, Paulussen M, Lehnert M, Jurgens H. Factors associated with tumor volume and primary metastases in Ewing tumors: Results from the (EI)CESS studies. Ann Oncol. 1999;10:1073–1077. doi: 10.1023/a:1008357018737. [DOI] [PubMed] [Google Scholar]
- 4.Jurgens H, Exner U, Gadner H, et al. Multidisciplinary treatment of primary Ewing's sarcoma of bone. A 6-year experience of a European Cooperative Trial. Cancer. 1988;61:23–32. doi: 10.1002/1097-0142(19880101)61:1<23::aid-cncr2820610106>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 5.Bacci G, Ferrari S, Bertoni F, et al. Prognostic factors in nonmetastatic Ewing's sarcoma of bone treated with adjuvant chemotherapy: Analysis of 359 patients at the Istituto Ortopedico Rizzoli. J Clin Oncol. 2000;18:4–11. doi: 10.1200/JCO.2000.18.1.4. [DOI] [PubMed] [Google Scholar]
- 6.Nesbit ME, Jr, Gehan EA, Burgert EO, Jr, et al. Multimodal therapy for the management of primary, nonmetastatic Ewing's sarcoma of bone: A long-term follow-up of the First Intergroup study. J Clin Oncol. 1990;8:1664–1674. doi: 10.1200/JCO.1990.8.10.1664. [DOI] [PubMed] [Google Scholar]
- 7.Craft A, Cotterill S, Malcolm A, et al. Ifosfamide-containing chemotherapy in Ewing's sarcoma: The Second United Kingdom Children's Cancer Study Group and the Medical Research Council Ewing's Tumor Study. J Clin Oncol. 1998;16:3628–3633. doi: 10.1200/JCO.1998.16.11.3628. [DOI] [PubMed] [Google Scholar]
- 8.Rosito P, Mancini AF, Rondelli R, et al. Italian cooperative study for the treatment of children and young adults with localized Ewing sarcoma of bone: A preliminary report of 6 years of experience. Cancer. 1999;86:421–428. doi: 10.1002/(sici)1097-0142(19990801)86:3<421::aid-cncr10>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 9.Lee J, Hoang BH, Ziogas A, Zell JA. Analysis of prognostic factors in Ewing sarcoma using a population-based cancer registry. Cancer. 2010;116:1964–1973. doi: 10.1002/cncr.24937. [DOI] [PubMed] [Google Scholar]
- 10.Raney RB, Asmar L, Newton WA, Jr, et al. Ewing's sarcoma of soft tissues in childhood: A report from the Intergroup Rhabdomyosarcoma Study, 1972 to 1991. J Clin Oncol. 1997;15:574–582. doi: 10.1200/JCO.1997.15.2.574. [DOI] [PubMed] [Google Scholar]
- 11.Lee JA, Kim DH, Lim JS, et al. Soft-tissue Ewing sarcoma in a low-incidence population: Comparison to skeletal Ewing sarcoma for clinical characteristics and treatment outcome. Jpn J Clin Oncol. 2010 doi: 10.1093/jjco/hyq080. [DOI] [PubMed] [Google Scholar]
- 12.El Weshi A, Allam A, Ajarim D, et al. Extraskeletal Ewing's sarcoma family of tumours in adults: Analysis of 57 patients from a single institution. Clin Oncol (R Coll Radiol) 2010;22:374–381. doi: 10.1016/j.clon.2010.02.010. [DOI] [PubMed] [Google Scholar]
- 13.Xie CF, Liu MZ, Xi M. Extraskeletal Ewing's sarcoma: A report of 18 cases and literature review. Chin J Cancer. 2010;29:420–424. doi: 10.5732/cjc.009.10402. [DOI] [PubMed] [Google Scholar]
- 14.Marina NM, Etcubanas E, Parham DM, Bowman LC, Green A. Peripheral primitive neuroectodermal tumor (peripheral neuroepithelioma) in children. A review of the St. Jude experience and controversies in diagnosis and management. Cancer. 1989;64:1952–1960. doi: 10.1002/1097-0142(19891101)64:9<1952::aid-cncr2820640931>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 15.van den Berg H, Heinen RC, van der Pal HJ, Merks JH. Extra-osseous Ewing sarcoma. Pediatr Hematol Oncol. 2009;26:175–185. doi: 10.1080/08880010902855581. [DOI] [PubMed] [Google Scholar]
- 16.Ahmad R, Mayol BR, Davis M, Rougraff BT. Extraskeletal Ewing's sarcoma. Cancer. 1999;85:725–731. [PubMed] [Google Scholar]
- 17.Castex MP, Rubie H, Stevens MC, et al. Extraosseous localized Ewing tumors: Improved outcome with anthracyclines--the French society of pediatric oncology and International society of pediatric oncology. J Clin Oncol. 2007;25:1176–1182. doi: 10.1200/JCO.2005.05.0559. [DOI] [PubMed] [Google Scholar]
- 18.Sorensen PH, Liu XF, Delattre O, et al. Reverse transcriptase PCR amplification of EWS/FLI-1 fusion transcripts as a diagnostic test for peripheral primitive neuroectodermal tumors of childhood. Diagn Mol Pathol. 1993;2:147–157. [PubMed] [Google Scholar]
- 19.Kapadia SB, Frisman DM, Hitchcock CL, Ellis GL, Popek EJ. Melanotic neuroectodermal tumor of infancy. clinicopathological, immunohistochemical, and flow cytometric study. Am J Surg Pathol. 1993;17:566–573. doi: 10.1097/00000478-199306000-00004. [DOI] [PubMed] [Google Scholar]
- 20.Cortese G, Scheike TH, Martinussen T. Flexible survival regression modelling. Stat Methods Med Res. 2010;19:5–28. doi: 10.1177/0962280209105022. [DOI] [PubMed] [Google Scholar]
- 21.Bovee JV, Hogendoorn PC. Molecular pathology of sarcomas: Concepts and clinical implications. Virchows Arch. 2010;456:193–199. doi: 10.1007/s00428-009-0828-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grier HE. The Ewing family of tumors. Ewing's sarcoma and primitive neuroectodermal tumors. Pediatr Clin North Am. 1997;44:991–1004. doi: 10.1016/s0031-3955(05)70541-1. [DOI] [PubMed] [Google Scholar]
- 23.Dehner LP. Primitive neuroectodermal tumor and Ewing's sarcoma. Am J Surg Pathol. 1993;17:1–13. doi: 10.1097/00000478-199301000-00001. [DOI] [PubMed] [Google Scholar]
- 24.Donaldson SS. Ewing sarcoma: Radiation dose and target volume. Pediatr Blood Cancer. 2004;42:471–476. doi: 10.1002/pbc.10472. [DOI] [PubMed] [Google Scholar]
- 25.Dunst J, Schuck A. Role of radiotherapy in Ewing tumors. Pediatr Blood Cancer. 2004;42:465–470. doi: 10.1002/pbc.10446. [DOI] [PubMed] [Google Scholar]
- 26.Gururangan S, Marina NM, Luo X, et al. Treatment of children with peripheral primitive neuroectodermal tumor or extraosseous Ewing's tumor with Ewing's-directed therapy. J Pediatr Hematol Oncol. 1998;20:55–61. doi: 10.1097/00043426-199801000-00009. [DOI] [PubMed] [Google Scholar]
- 27.Paulussen M, Ahrens S, Dunst J, et al. Localized Ewing tumor of bone: Final results of the cooperative Ewing's sarcoma study CESS 86. J Clin Oncol. 2001;19:1818–1829. doi: 10.1200/JCO.2001.19.6.1818. [DOI] [PubMed] [Google Scholar]
- 28.Grier HE, Krailo MD, Tarbell NJ, et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med. 2003;348:694–701. doi: 10.1056/NEJMoa020890. [DOI] [PubMed] [Google Scholar]