

Abstract
The identification of genetic causes for Mendelian disorders has been based on the collection of multi-incident families, linkage analysis, and sequencing of genes in candidate intervals. This study describes the application of next-generation sequencing technologies to a Swiss kindred presenting with autosomal-dominant, late-onset Parkinson disease (PD). The family has tremor-predominant dopa-responsive parkinsonism with a mean onset of 50.6 ± 7.3 years. Exome analysis suggests that an aspartic-acid-to-asparagine mutation within vacuolar protein sorting 35 (VPS35 c.1858G>A; p.Asp620Asn) is the genetic determinant of disease. VPS35 is a central component of the retromer cargo-recognition complex, is critical for endosome-trans-golgi trafficking and membrane-protein recycling, and is evolutionarily highly conserved. VPS35 c.1858G>A was found in all affected members of the Swiss kindred and in three more families and one patient with sporadic PD, but it was not observed in 3,309 controls. Further sequencing of familial affected probands revealed only one other missense variant, VPS35 c.946C>T; (p.Pro316Ser), in a pedigree with one unaffected and two affected carriers, and thus the pathogenicity of this mutation remains uncertain. Retromer-mediated sorting and transport is best characterized for acid hydrolase receptors. However, the complex has many types of cargo and is involved in a diverse array of biologic pathways from developmental Wnt signaling to lysosome biogenesis. Our study implicates disruption of VPS35 and retromer-mediated trans-membrane protein sorting, rescue, and recycling in the neurodegenerative process leading to PD.
Main Text
Parkinson disease (PD [MIM 168600]) affects 1% of individuals older than 65 years. Patients develop both motor and nonmotor symptoms and suffer an inexorable decline in function. Dopaminergic therapies provide temporary relief but are not without side-effects and do not slow disease progression.1 About 14% of patients with PD report one or more first-degree relatives with parkinsonism.2 Studies of familial parkinsonism have identified pathogenic mutations in several genes, and such identifications have provided mechanistic insight and novel targets for therapeutic intervention.3 These loci have been associated with idiopathic PD, emphasizing the importance of studying monogenic forms of disease (see the PDGene database in the Web Resources and Nalls et al.4).
All Mendelian forms of parkinsonism have been identified through classic linkage and Sanger sequence analysis.3 This study describes NGS applied to a multi-incident Swiss family with autosomal-dominant, late-onset PD (CH; Figure 1). The clinical features of this kindred, which includes eleven individuals with a diagnosis of PD, were previously described.5 DNA samples from six affected and 16 unaffected individuals excluded point mutations and copy-number variants (CNVs) in genes previously implicated in monogenic parkinsonism.5
Figure 1.
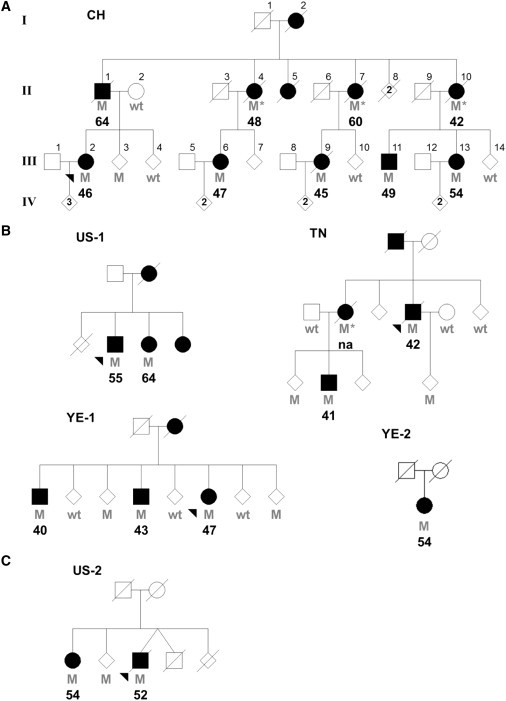
Pedigrees with VPS35 Mutations
Individual pedigrees are labeled and numbered according to their geographic origin (CH, Switzerland; US, United States; TN, Tunisia; YE, Yemenite Jews from Israel). An asterisk indicates an inferred mutation carrier. Filled symbols indicate affected individuals, and the corresponding age at disease onset is indicated; na, not available. Heterozygote mutation carriers (M) and wild-type (wt) genotypes are indicated.
(A) Original family used for exome sequencing and identification of VPS35 p.Asp620Asn.
(B) Additional kindreds presenting VPS35 p.Asp620Asn mutations.
(C) Pedigree presenting the p.Pro316Ser variant.
To identify the novel pathogenic mutation causing PD in this Swiss kindred, we performed exome sequencing analysis in an affected pair of first-degree cousins (Figure 1). The capture of exonic regions and the subsequent sequencing and characterization of variants was performed by Knome, Inc. NimbleGen Sequence Capture Arrays were used for isolating the genomic coding regions to be sequenced. The NimbleGen Arrays, which contain 2.1 million probes that capture ∼180,000 exons and ∼550 miRNA, were used for interrogation of genomic regions from CH III-2 and III-13. Sequencing was subsequently performed on an Illumina Genome Analyzer by 76 bp paired-end reads. The mean coverage per targeted base was 51.6-fold for III-2 and 43.8-fold for III-13, and 73.2% and 60.7% of bases, respectively, had ≥20-fold coverage. The percentage of targeted bases covered was 98.5% and 97.0% for III-2 and III-13, respectively. Adaptor sequences from reads were filtered via a local dynamic programming algorithm. Reads with more than 12 bp of overlap with adaptor sequences were deemed contaminated and removed; only those reads of 30 bp or longer were used for further analysis. Reads with more than six missing bases or 40 consecutive identical bases were omitted from further processing. The remaining sequences were aligned to a reference sequence (Hg18, build 36.1) with SOAPaligner (parameters -a -D -o -r 1 -t -c -f 4), followed by SOAPsnp so that consensus and call genotypes (parameters -i -d -o -r 0.00005 -e 0.0001 -M -t -u -L -s −2 -T) could be assembled. Validation of common SNP genotypes was conducted against dbSNP v129. Structural variations were determined by the paired-end method and clustering algorithm alignment against CNVs in the Database of Genomic Variants v6 (November 2008). This analysis identified a total of 34,754 and 29,952 variants for III-2 and III-13, respectively. Assuming these two family members have the same genetic cause of disease reduced the number of variants to 17,872 common to both individuals. On the basis of the genotypes of eight publicly available HapMap samples,6 additional polymorphisms were excluded, leaving 4,256 novel variants. After variants on the X chromosome and homozygous changes (autosomal-dominant inheritance of disease was assumed), noncoding variants, synonymous changes, and variants present in dbSNP v130 were excluded, only 69 variants remained.
We recognize that next-generation capture and sequencing methods are limited in coverage and that some bioinformatics results might be erroneous. Thus, we used Sanger sequencing of these novel variants in one sample used for NGS (III-2), two additional affected family members (III-6 and III-9), and one unaffected spouse (II-2) to assess the accuracy of NGS genotype calls and segregation with disease. Thirty-six variants were an artifact; they could not be confirmed by Sanger sequencing and appeared to be a consequence of misaligned repeat sequences. Thirty-three variants were validated, and six were present in family members with PD but not observed in the spouse (Table S1). Only two variants, c.3035C>T (p.Ala1012Val) in integrin alpha X (ITGAX; RefSeq accession number NM_000887.3 [MIM 151510]) and c.1858G>A (p.Asp620Asn) in vacuolar protein sorting 35 ortholog (VPS35; RefSeq accession number NM_018206.4 [MIM 601501]), were not observed in 184 randomly selected US control subjects of European descent. Both variants are on the same chromosomal region 16p12.1-q12.1 haplotype and were found in all affected family members and one unaffected family member (III-3; Figure 1). Additional support for variants in chromosomal region 16p12.1-q12.1 is provided by linkage data (Figure S1).
Both variants were subsequently genotyped in a multi-ethnic case-control series consisting of 4,326 patients and 3,309 controls. This series comprises eight independent PD series from the US, Canada, Norway, Poland, Ireland, Tunisia, Taiwan, and Israel (Yemenite Jews). The demographics for each series are presented in Table S2. All patients were examined and observed longitudinally by a movement-disorders neurologist and diagnosed with PD according to published criteria.7 Patients and unrelated controls of identical ethnicity but without evidence of neurological disease were consecutively selected. The ethical review boards at each institution approved the study, and all participants provided informed consent. A combination of Sequenom MassArray iPLEX (San Diego, CA) and TaqMan probes was used for genotyping, and all mutations were confirmed by direct sequencing. Four patients (three familial and one sporadic) and no controls were identified as having VPS35 p.Asp620Asn; in those three families, a total of nine affected individuals are present and carry the mutation (Figure 1). In contrast, the ITGAX p.Ala1012Val mutation was found in one control (61 years) and one familial case (42 years at onset), but no other samples were available to allow segregation to be examined. Carrier frequencies for both variants are provided in Table S3. This data indicate that VPS35 p.Asp620Asn is a pathogenic mutation, whereas ITGAX p.Ala1012Val is a rare variant. VPS35 is highly conserved from humans to yeast, supporting the pathogenicity of the p.Asp620Asn substitution (Figure 2), whereas ITGAX is not evolutionarily conserved. Additionally, p.Asp620Asn is an acidic-to-basic change and is predicted to be a deleterious substitution (SIFT score = 0), whereas p.Ala1012Val is not (SIFT score = 0.16).
Figure 2.

VPS35 Mutations and Cross-species Conservation
Protein orthologs were aligned via ClustalW. Amino acid positions for VPS35 p.Pro316Ser and p.Asp620Asn are highlighted in black. Protein orthologs with amino acid positions differing from those of the human sequence are indicated in gray. RefSeq accession numbers: Homo sapiens, NP_060676.2; Pan troglodytes, XP_001161439.1; Mus musculus, NP_075373.1; Rattus norvegicus, XP_214646.3; Bos taurus, NP_001039723.1; Canis familiaris, XP_532570.2; Gallus gallus, NP_001005842.1; Xenopus laevis, NP_001089981.1; Danio rerio, NP_001020688.2; Drosophila melanogaster, NP_726175.3; and Saccharomyces cerevisiae, NP_012381.1. An asterisk indicates that five aminoacids have been excluded from Drosophila melanogaster and Saccharomyces cerevisiae at this position.
Haplotype analysis of VPS35 p.Asp620Asn was performed with microsatellite markers between D16S401 and D16S3044 (Table 1). One primer of each pair was labeled with a fluorescent tag; sequences for novel markers are provided in Table S4. Polymerase chain reactions (PCRs) were performed under standard conditions, and products were run on an ABI 3730 genetic analyzer. Results were analyzed with GeneMapper 4.0 software (Applied Biosystems). Genotyping of VPS35 kindreds shows that the p.Asp620Asn substitution is a mutational hotspot; it has arisen independently from at least four separate mutational events (Table 1). Only two haplotypes (CH and YE-2) share alleles for an adjacent microsatellite marker (Chr16_45.333M). Interestingly, p.Asp620Asn in two families that share similar ethnicity does not appear to be identical by descent (YE-1 and YE-2).
Table 1.
16p12.1-q12.1 Haplotypes of Kindreds with the VPS35 p.Asp620Asn Mutation
| Positiona | cMb | CH | YE-2 | YE-1 | TN | US-1 | |
|---|---|---|---|---|---|---|---|
| D16S401 | 24,593,458 | 46.94 | 172 | 172 | 172 | 176 | 172/174 |
| D16S3068 | 25,468,102 | 48.53 | 139 | 141/147 | 143 | 147 | 141 |
| D16S753 | 31,180,950 | 57.79 | 260 | 264 | 272 | 264 | 264/268 |
| Chr16_45.023M | 45,023,561 | - | 295 | 295 | 295 | 293 | 295 |
| VPS35 p.Asp620Asn | 45,253,865 | - | A | A | A | A | A |
| Chr16_45.333M | 45,333,651 | - | 294 | 294 | 292 | 288 | 278 |
| D16S3105 | 45,349,601 | 58.46 | 187 | 187 | 187 | 193 | 191 |
| Chr16_45.615M | 45,615,739 | - | 147 | 147 | 149 | 149 | 149 |
| Chr16_45.806M | 45,806,285 | - | 246 | 242 | 246 | 246 | 246 |
| Chr16_45.835M | 45,835,953 | - | 235 | 243 | 237 | 237 | 235 |
| Chr16_45.855M | 45,855,831 | - | 212 | 206 | 210/212 | 210 | 210 |
| D16S3044 | 45,995,037 | 58.46 | 197 | 193 | 195 | 191 | 191/195 |
Microsatellite allele sizes are given in base pairs, consistent with Centre d'Étude du Polymorphisme Humain (CEPH) standards (Table S4). Haplotypes that are possibly shared between CH and YE-2 kindreds are italicized. For markers with an unknown phase, both alleles are given.
Markers are shown with their physical locations (NCBI Build 36.1).
Sex-averaged centimorgan positions are given from the Marshfield Comprehensive Human Genetic Map when available.
The average age at disease onset for the CH kindred is 50.6 ± 7.3 years (range 42–64 years) and is comparable to 48.3 ± 8.6 years (range 40–64 years), the mean age of onset for other families affected by VPS35 p.Asp620Asn. However, the mutation has incomplete, age-associated penetrance; among these pedigrees, there are four unaffected mutation carriers who were between 49 and 67 years at the time of exam (data not shown).
In an attempt to identify novel mutations, we sequenced all 17 VPS35 exons in 190 multiethnic familial probands from the US, Canada, Norway, Poland, Ireland, Tunisia, Taiwan, Israel (Yemenite Jews), France, Switzerland, and Sweden. The ethical review boards at each institution approved the study, and all participants provided informed consent. Primer pairs used for sequencing VPS35 exons and exon-intron boundaries by PCR are provided in Table S4. PCR products were purified from unincorporated nucleotides via Agencourt bead technology (Beverly, MA) with Biomek FX automation (Beckman Coulter, Fullerton, CA). Sequence analysis was performed as previously described.8 Sequencing of 106 US probands identified two novel variants, one silent (c.945A>T; p.Gly315Gly) and one missense (c.946C>T; p.Pro316Ser). The age at disease onset for the patients with these variants was 65 and 52 years, respectively. In 22 Tunisian probands, one additional variant (c.2210C>T; p.Ala737Val) was identified in one patient with disease onset at 60 years. Sequencing of VPS35 in ethnically matched controls (94 US, 47 Tunisian) identified p.Gly315Gly in one US control and p.Ala737Val in one Tunisian control. The presence of these two variants in their corresponding ethnically matched controls suggests they are nonpathogenic polymorphisms. No additional variants, including p.Pro316Ser, were identified in these control samples or in probands from other populations.
Two additional family members from the VPS35 p.Pro316Ser kindred were available (US-2, Figure 1), and both subjects were carriers; one was diagnosed with PD at 54 years, and the other displayed hand tremor and micrographia at 67 years but did not fulfill the clinical criteria for PD. To further discern the pathogenicity of this variant, we genotyped c.946C>T (p.Pro316Ser) in a multi-ethnic case-control series consisting of 4,326 patients and 3,309 controls (Table S2). One Norwegian control subject (86 years) with the mutation was identified. Thus, the genetic evidence for the pathogenicity of p.Pro316Ser is inconclusive. However, the high level of species conservation suggests an important functional role (Figure 2). Additional studies are required if we are to determine whether p.Pro316Ser is a pathogenic mutation, a risk factor, or a neutral polymorphism.
We also evaluated VPS35 CNVs as a cause of disease in the 190 familial probands previously described. We used two genomic probes against exon 6 and exon 15 of VPS35 and two endogenous control assays against exon 5 of PSEN2 [MIM 600759] and exon 5 of SNCA [MIM 162890] to identify whole-gene CNVs. All primers and probes were purchased from Applied Biosystems, and sequences are available on request. Quantitative PCR was carried out with the TaqMan expression chemistry protocol, 25 ng genomic DNA was amplified with 0.25 μl primer probe and 2.5 μl TaqMan 2× Universal PCR Master Mix (Applied Biosystems). The thermal cycle conditions inclued 50°C for 2 min, 95°C for 10 min, and then 40 cycles at 95°C for 15 s for denaturation and 60°C for 1 min for annealing and extension. All assays were performed in quadruplicate on the ABI 7900HT Fast Real-Time PCR System and analyzed with ABI SDS 2.2.2 Software (Applied Biosystems). This analysis did not identify CNVs in VPS35.
Genes linked to familial parkinsonism have highlighted loci associated with idiopathic PD. Using published genome-wide association study (GWAS) datasets,9–11 we examined evidence for disease association with VPS35 observed no such evidence (p values = 0.1–0.9; Table S5). Nevertheless, coverage of VPS35 was limited, and a lack of association should not preclude more detailed study in ethnically diverse populations.
VPS35 is a critical component of the retromer cargo-recognition complex, which is involved in the recycling of membrane proteins between endosomes and the trans-Golgi network.12,13 In mammals the pentameric assembly includes the recognition complex of VPS35, VPS29 [MIM 606932], and either VPS26A [MIM 605506] or VPS26B [MIM 610027], as well as sorting nexins (one of each of the following pairs: SNX1 [MIM 601272] or SNX2 [MIM 605929]; and SNX5 [MIM 605937] or SNX6 [MIM 606098]). Coimmunoprecipitation analyses of VPS35 p.Pro316Ser and p.Asp620Asn substitutions indicate that neither is likely to result in aberrant complex formation (Figure S2). However, VPS35 is known to directly associate with cargos, including soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins, SORL1 [MIM 602005] and SORT1 [MIM 602458], and MUL1 [MIM 612037].13–15 We hypothesize that mutations perturb cargo recognition and binding and that this perturbation results in deficient receptor recycling, but such effects are likely to be subtle given the late onset of disease. In parkinsonism, alpha-synuclein has been proposed to regulate SNARE proteins, which are involved in vesicle fusion and exocytosis,16 whereas leucine-rich repeat kinase 2 (LRRK2 [MIM 609007]) is a central regulator of neuritic outgrowth, morphology, and cellular homeostasis, in part through the regulation of endosomal trafficking.17
The identification of VPS35 pathogenic mutations in PD provides a compelling genetic link for the retromer in neurodegeneration. In Alzheimer disease [MIM 104300], retromer deficiency impairs Wnt signaling and amyloid precursor protein (APP [MIM 104760]) trafficking, and it is associated with an elevation in endogenous amyloid-beta peptide.18,19 Nevertheless, dementia is not prominent in patients with parkinsonism and VPS35 p.Asp620Asn. Cognitive deficits were observed in two family members with parkinsonism and VPS35 p.Pro316Ser mutations (US-2), but assessment of cognitive decline in VPS35 mutation carriers requires further evaluation. The recruitment of VPS26/29/35 complex to endosomes is initially catalyzed by RAB7 [MIM 602298].20 This dynamic interaction is perturbed by RAB7 mutations in Charcot-Marie-Tooth type 2B neuropathy (CMT2B [MIM 600882]).20,21 The human genome contains more than 60 RAB proteins that regulate vesicular transport between cellular compartments and may provide cell-type specificity. Of note, RAB7L1 [MIM 603949] is adjacent to rs947211 (PARK16), recently nominated in PD susceptibility through a GWAS.22 Interaction between dynactin (DCTN1 [MIM 601143]) and SNX6 is also required for endosome-trans-Golgi transport and provides another link between retromer function and parkinsonism in that mutations in DCTN1 have recently been implicated in Perry syndrome [MIM 168605].23,24
The retromer plays a central and intermediate role in recycling of membrane-associated proteins and endosomal-lysosomal trafficking. VPS35 mutations provide a mechanistic tool for exploring dominant-negative versus toxic gain of function and will allow the generation of in vitro and in vivo systems that address whether gene expression or transport of specific cargos explains the selective vulnerability of dopaminergic neurons. Endosomal trafficking appears to be a common pathway disrupted in neurodegenerative diseases. Screening of VPS35 and its interacting partners, not only in PD but also in other movement and cognitive disorders, is warranted if we are to fully understand the role of the retromer in disease development.
Acknowledgments
We are grateful to all individuals who generously participated in this study. We thank the Greenberg family for their 2011 AD/PD Conference Award generously donated by Evelyn Greenberg, in memory of Moshe Greenberg. C.V.-G. and exome sequencing were financed by the Parkinson Disease Foundation. Additional funding for clinical, genetic, and functional studies was provided by the National Institute of Neurological Disorders and Stroke, National Institutes of Health (NINDS/NIH) (Morris K. Udall Parkinson's Disease Research Center of Excellence; P50NS040256), the Swiss Parkinson's Disease Foundation, and the Michael J. Fox Foundation. Z.K.W. was partially supported by NINDS/NIH grant P50NS072187 and a gift from Carl Edward Bolch, Jr., and Susan Bass Bolch (MCF #90052031/PAU #90052). This research was undertaken, in part, thanks to funding from the Canada Excellence Research Chairs program (to M.J.F. and C.V.-G.). Leading Edge Endowment Funds provided by the Province of British Columbia, LifeLabs, and Genome BC support the Dr. Donald Rix BC Leadership Chair (M.J.F.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
PDGene, http://www.pdgene.org/
Database of Genomic Variants, http://projects.tcag.ca/variation/
Online Mendelian Inheritance in Man, http://www.omim.org/
SIFT, http://sift.jcvi.org/
Knome, http://www.knome.com/
References
- 1.Lang A.E., Lozano A.M. Parkinson's disease. Second of two parts. N. Engl. J. Med. 1998;339:1130–1143. doi: 10.1056/NEJM199810153391607. [DOI] [PubMed] [Google Scholar]
- 2.McDonnell S.K., Schaid D.J., Elbaz A., Strain K.J., Bower J.H., Ahlskog J.E., Maraganore D.M., Rocca W.A. Complex segregation analysis of Parkinson's disease: The Mayo Clinic Family Study. Ann. Neurol. 2006;59:788–795. doi: 10.1002/ana.20844. [DOI] [PubMed] [Google Scholar]
- 3.Farrer M.J. Genetics of Parkinson disease: Paradigm shifts and future prospects. Nat. Rev. Genet. 2006;7:306–318. doi: 10.1038/nrg1831. [DOI] [PubMed] [Google Scholar]
- 4.Nalls M.A., Plagnol V., Hernandez D.G., Sharma M., Sheerin U.M., Saad M., Simón-Sánchez J., Schulte C., Lesage S., Sveinbjörnsdóttir S., International Parkinson Disease Genomics Consortium Imputation of sequence variants for identification of genetic risks for Parkinson's disease: A meta-analysis of genome-wide association studies. Lancet. 2011;377:641–649. doi: 10.1016/S0140-6736(10)62345-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wider C., Skipper L., Solida A., Brown L., Farrer M., Dickson D., Wszolek Z.K., Vingerhoets F.J. Autosomal dominant dopa-responsive parkinsonism in a multigenerational Swiss family. Parkinsonism Relat. Disord. 2008;14:465–470. doi: 10.1016/j.parkreldis.2007.11.013. [DOI] [PubMed] [Google Scholar]
- 6.Ng S.B., Turner E.H., Robertson P.D., Flygare S.D., Bigham A.W., Lee C., Shaffer T., Wong M., Bhattacharjee A., Eichler E.E. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–276. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gelb D.J., Oliver E., Gilman S. Diagnostic criteria for Parkinson disease. Arch. Neurol. 1999;56:33–39. doi: 10.1001/archneur.56.1.33. [DOI] [PubMed] [Google Scholar]
- 8.Ross O.A., Soto A.I., Vilariño-Güell C., Heckman M.G., Diehl N.N., Hulihan M.M., Aasly J.O., Sando S., Gibson J.M., Lynch T. Genetic variation of Omi/HtrA2 and Parkinson's disease. Parkinsonism Relat. Disord. 2008;14:539–543. doi: 10.1016/j.parkreldis.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fung H.C., Scholz S., Matarin M., Simón-Sánchez J., Hernandez D., Britton A., Gibbs J.R., Langefeld C., Stiegert M.L., Schymick J. Genome-wide genotyping in Parkinson's disease and neurologically normal controls: first stage analysis and public release of data. Lancet Neurol. 2006;5:911–916. doi: 10.1016/S1474-4422(06)70578-6. [DOI] [PubMed] [Google Scholar]
- 10.Maraganore D.M., de Andrade M., Lesnick T.G., Strain K.J., Farrer M.J., Rocca W.A., Pant P.V., Frazer K.A., Cox D.R., Ballinger D.G. High-resolution whole-genome association study of Parkinson disease. Am. J. Hum. Genet. 2005;77:685–693. doi: 10.1086/496902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pankratz N., Wilk J.B., Latourelle J.C., DeStefano A.L., Halter C., Pugh E.W., Doheny K.F., Gusella J.F., Nichols W.C., Foroud T., Myers R.H., PSG-PROGENI and GenePD Investigators, Coordinators and Molecular Genetic Laboratories Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Hum. Genet. 2009;124:593–605. doi: 10.1007/s00439-008-0582-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hierro A., Rojas A.L., Rojas R., Murthy N., Effantin G., Kajava A.V., Steven A.C., Bonifacino J.S., Hurley J.H. Functional architecture of the retromer cargo-recognition complex. Nature. 2007;449:1063–1067. doi: 10.1038/nature06216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bonifacino J.S., Hurley J.H. Retromer. Curr. Opin. Cell Biol. 2008;20:427–436. doi: 10.1016/j.ceb.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arighi C.N., Hartnell L.M., Aguilar R.C., Haft C.R., Bonifacino J.S. Role of the mammalian retromer in sorting of the cation-independent mannose 6-phosphate receptor. J. Cell Biol. 2004;165:123–133. doi: 10.1083/jcb.200312055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Braschi E., Goyon V., Zunino R., Mohanty A., Xu L., McBride H.M. Vps35 mediates vesicle transport between the mitochondria and peroxisomes. Curr. Biol. 2010;20:1310–1315. doi: 10.1016/j.cub.2010.05.066. [DOI] [PubMed] [Google Scholar]
- 16.Burré J., Sharma M., Tsetsenis T., Buchman V., Etherton M.R., Südhof T.C. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329:1663–1667. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dächsel J.C., Behrouz B., Yue M., Beevers J.E., Melrose H.L., Farrer M.J. A comparative study of Lrrk2 function in primary neuronal cultures. Parkinsonism Relat. Disord. 2010;16:650–655. doi: 10.1016/j.parkreldis.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muhammad A., Flores I., Zhang H., Yu R., Staniszewski A., Planel E., Herman M., Ho L., Kreber R., Honig L.S. Retromer deficiency observed in Alzheimer's disease causes hippocampal dysfunction, neurodegeneration, and Abeta accumulation. Proc. Natl. Acad. Sci. USA. 2008;105:7327–7332. doi: 10.1073/pnas.0802545105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Small S.A., Kent K., Pierce A., Leung C., Kang M.S., Okada H., Honig L., Vonsattel J.P., Kim T.W. Model-guided microarray implicates the retromer complex in Alzheimer's disease. Ann. Neurol. 2005;58:909–919. doi: 10.1002/ana.20667. [DOI] [PubMed] [Google Scholar]
- 20.Seaman M.N., Harbour M.E., Tattersall D., Read E., Bright N. Membrane recruitment of the cargo-selective retromer subcomplex is catalysed by the small GTPase Rab7 and inhibited by the Rab-GAP TBC1D5. J. Cell Sci. 2009;122:2371–2382. doi: 10.1242/jcs.048686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Verhoeven K., De Jonghe P., Coen K., Verpoorten N., Auer-Grumbach M., Kwon J.M., FitzPatrick D., Schmedding E., De Vriendt E., Jacobs A. Mutations in the small GTP-ase late endosomal protein RAB7 cause Charcot-Marie-Tooth type 2B neuropathy. Am. J. Hum. Genet. 2003;72:722–727. doi: 10.1086/367847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Satake W., Nakabayashi Y., Mizuta I., Hirota Y., Ito C., Kubo M., Kawaguchi T., Tsunoda T., Watanabe M., Takeda A. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson's disease. Nat. Genet. 2009;41:1303–1307. doi: 10.1038/ng.485. [DOI] [PubMed] [Google Scholar]
- 23.Farrer M.J., Hulihan M.M., Kachergus J.M., Dächsel J.C., Stoessl A.J., Grantier L.L., Calne S., Calne D.B., Lechevalier B., Chapon F. DCTN1 mutations in Perry syndrome. Nat. Genet. 2009;41:163–165. doi: 10.1038/ng.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hong Z., Yang Y., Zhang C., Niu Y., Li K., Zhao X., Liu J.J. The retromer component SNX6 interacts with dynactin p150(Glued) and mediates endosome-to-TGN transport. Cell Res. 2009;19:1334–1349. doi: 10.1038/cr.2009.130. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.