

Abstract
Geleophysic (GD) and acromicric dysplasia (AD) belong to the acromelic dysplasia group and are both characterized by severe short stature, short extremities, and stiff joints. Although AD has an unknown molecular basis, we have previously identified ADAMTSL2 mutations in a subset of GD patients. After exome sequencing in GD and AD cases, we selected fibrillin 1 (FBN1) as a candidate gene, even though mutations in this gene have been described in Marfan syndrome, which is characterized by tall stature and arachnodactyly. We identified 16 heterozygous FBN1 mutations that are all located in exons 41 and 42 and encode TGFβ-binding protein-like domain 5 (TB5) of FBN1 in 29 GD and AD cases. Microfibrillar network disorganization and enhanced TGFβ signaling were consistent features in GD and AD fibroblasts. Importantly, a direct interaction between ADAMTSL2 and FBN1 was demonstrated, suggesting a disruption of this interaction as the underlying mechanism of GD and AD phenotypes. Although enhanced TGFβ signaling caused by FBN1 mutations can trigger either Marfan syndrome or GD and AD, our findings support the fact that TB5 mutations in FBN1 are responsible for short stature phenotypes.
Introduction
Geleophysic dysplasia (GD, [MIM 231050]) and acromicric dysplasia (AD, [MIM 102370]) belong to the acromelic dysplasia group and are both characterized by severe short stature (<−3 standard deviations [SD]), short hands and feet, joint limitations, and skin thickening.1 Radiological manifestations include delayed bone age, cone-shaped epiphyses, shortened long tubular bones, and ovoid vertebral bodies. GD is distinct from AD because it has an autosomal-recessive mode of inheritance, characteristic facial features—a “happy” face with full cheeks, a shortened nose, hypertelorism, a long and flat philtrum, and a thin upper lip—a progressive cardiac valvular thickening often leading to an early death, toe walking, tracheal stenosis, respiratory insufficiency, and lysosomal-like storage vacuoles in various tissues. AD has an autosomal-dominant mode of inheritance and is characterized by distinct facial features—a round face, well-defined eyebrows, long eyelashes, a bulbous nose with anteverted nostrils, a long and prominent philtrum, and thick lips with a small mouth—a hoarse voice, a pseudomuscular build, and distinct skeleton features, including an internal notch of the femoral head, an internal notch of the second metacarpal, and the external notch of the fifth metacarpal.2,3
We recently identified ADAMTSL2 [MIM 612277] mutations in GD and demonstrated a direct involvement of ADAMTSL2 in TGFβ bioavailability.4 However, the absence of ADAMTSL2 mutations in 19 out of 33 GD patients suggests genetic heterogeneity.5 The molecular etiology of AD has not been previously reported. The aim of our study was to identify (1) the gene mutated in AD and (2) an additional mutated gene in GD by studying those GD cases without ADAMTSL2 mutations.
Subjects and Methods
Patients
Nineteen GD cases were included in the study, and they all fulfilled the diagnostic criteria, namely short stature <−3 SD, short hands and feet, restricted joint mobility, characteristic facial features, and progressive cardiac involvement (Table 1, Figure 1). Ten AD cases, including two familial cases, were included in the study. They all fulfilled the diagnostic criteria for AD, namely severe short stature, short hands and feet, progressively stiff joints, and characteristic facial features (Table 1, Figure 1). We collected blood samples from affected individuals after obtaining written informed consent, in accordance with the ethical standards of our institutional review board on human experimentation.
Table 1.
Clinical Manifestations of GD and AD Patients
| Family | Origin | Diagnosis | Age (Years) | Height | Cardiac Involvement | Other |
|---|---|---|---|---|---|---|
| 1 | Belgium | GD | Death at 9 | <−6 SD (80 cm) | mitral stenosis and insufficiency | tracheotomy at 3 |
| 2 | France | GD | 18 | <−6 SD (112 cm) | mitral stenosis | HTAP, respiratory insufficiency, hepatomegaly, laryngeal stenosis |
| 3 | Russia | GD | 12 | <−6 SD (106 cm) | no | hepatomegaly |
| 4 | Switzerland | GD | 21 | <−6 SD (116 cm) | tricuspid stenosis, mild aortic insufficiency | |
| 5 | Russia | GD | 8 | −4 SD (103.5 cm) | no | |
| 6 | France | GD | 5.7 | −4 SD (97 cm) | no | laryngeal and respiratory insufficiency |
| 7 | U.K. | GD | Death at 3 | −5 SD (75 cm) | no | respiratory insufficiency, HTAP, Sleep apnea |
| 8 | Turkey | GD | 4.5 | −4 SD (85 cm) | mitral and tricuspide stenosis | respiratory insufficiency, hepatomegaly, spleep apnea |
| 9 | Algeria | GD | Death at 4 | <−6 SD (60 cm) | mitral and tricuspide stenosis | laryngeal and respiratory insufficiency, HTAP |
| 10 | Lebanon | GD | 14 | −3.5 SD (133 cm) | no | – |
| 11 | USA | GD | ? | ? | no | pyloric stenosis |
| 12 | Turkey | GD | 3 | −3 SD (85 cm) | no | |
| 13 | Russia | GD | 18 | −4 SD (134 cm) | mitral valve prolapse | |
| 14 | Iraq | GD | 11 | <−6 SD (92 cm) | yes | hepatomegaly |
| 15 | USA | GD | ? | ? | aortic stenosis, mitral and aortic valve insufficiencies | |
| 16 | Japan | GD | 9 | −6 SD (98 cm) | no | _ |
| 17 | Australia | GD | 3 | −4 SD (76 cm) | severe pulmonary hypertension | hepatomegaly |
| 18 | USA | GD | ? | ? | ||
| 19 | Korea/Japan | GD | 7 | −6 SD (88 cm) | mitral insufficiency | HTAP |
| 20 | France | AD | 10 | −6 SD (99 cm) | no | - |
| 21 | France | AD | 62 | −6 SD (125 cm) | no | broncho-pulmonary infection |
| 22-a | France | AD | 10 | −3 SD (121 cm) | no | |
| 22-b | France | AD | 13 | −3.5 SD (128 cm) | no | |
| 22-c | France | AD | 40 | −6 SD (128 cm) | no | mother of 22 a and 22b |
| 23 | Belgium | AD | 14 | <−6 SD (111 cm) | no | broncho-pulmonary infection |
| 24 | Netherlands | AD | 36 | <−6 SD (119cm) | no | carpal tunnel syndrome, laminectomy C1-C3 for cervical spine stenosis |
| 25 | France | AD | 13 | <−6 SD (104cm) | no | |
| 26 | Italy | AD | 43 | <−6 SD (129cm) | no | |
| 27-a | China | AD | 10 | −4 SD (117 cm) | no | |
| 27-b | China | AD | 35 | −5 SD (130 cm) | no | mother of 27a |
| 28 | France | AD | 54 | <−6 SD 125 cm | no | carpal tunnel syndrome |
| 29 | France | AD | 33 | −6 SD (125 cm) | no | asthma |
Figure 1.
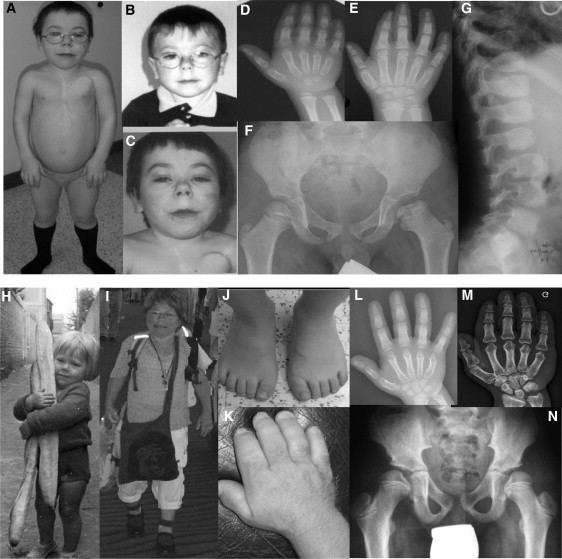
Clinical and Radiological Features in GD and AD
(B, D, F, and G) GD patient 1 at 5 years and (A, C, and E) 15 years; note the “happy” face with full cheeks, a shortened nose, and a long and flat philtrum with a thin upper lip. (D and E) Note the delayed bone age and cone-shaped epiphyses, (F) shortened long tubular bones, epiphyseal dysplasia, and (G) ovoid vertebral bodies.
(H and I) AD patient 21 at 3 years and 62 years. Note the round face, bulbous nose, pseudomuscular build, (J, K, L, and M) very short hands and feet with a delayed bone age, and (N) the internal notch of the femoral head.
Exome Sequencing
Enrichment was performed by hybridization of shotgun fragment libraries to Agilent SureSelect in solution capture assays. Using the Solid3.5 (Life Technologies), we generated an average of 5.1 Gb of mappable sequence data per sample to achieve more than 40× median coverage of the targeted exome (38 Mb, ∼18,000 genes). We first focused our exome analyses on nonsynonymous variants. We also defined variants as previously unidentified if they were absent from control populations and all datasets, including dbSNP129, the 1000 Genomes Project, and in-house exome data.
Mutation Detection
We designed a series of 66 intronic primers to amplify the 65 coding exons of FBN1 (MIM 134797; NM_000138.4). We purified the amplicons and sequenced them by using the fluorescent dideoxy-terminator method on an automatic sequencer (ABI 3100).
Immunofluorescence
Skin fibroblasts from affected individuals and controls were obtained after written informed consent, in accordance with the ethical standards of our institutional review board on human experimentation and were grown in cell-culture chambers and fixed in 4% paraformaldehyde (PFA). After being blocked with 3% bovine serum albumin (BSA), cells were incubated with FBN1 antibody (Millipore) overnight at 4°C, and followed by incubation with fluorescein-isothyocianate-conjugated secondary antibodies at room temperature.
Immunoblotting Analysis
For pSMAD2 immunoblotting, cell lysates were obtained from skin fibroblasts (controls and GD and AD patients) and actin (Invitrogen) and pSMAD2 (Cell signaling technology) antibodies were used. Respective protein species were quantified by densitometry (Kodak 1D image analysis software).
ELISA Assays for Active and Total TGF-β1
TGF-β1 present in 100 μl culture medium of confluent fibroblasts from two affected individuals and unaffected controls was quantitated with the TGF-β1 EMax Immunoassay kit (Promega). The samples were acidified for measurement of total TGF-β1 (active plus latent). TGF-β1 standard curves were undertaken for each assay. All experiments were performed in triplicate. A t test was performed.
Production of Recombinant Fibrillin-1 Peptide and BIAcore Analysis
We used a full-length human fibrillin-1 expression plasmid6 to generate hFib1-49, a fragment encoding residues 1–1525 (N-terminal half of fibrillin-1). The conditioned medium of the stably transfected HEK293F cells was used to purify hFib1-49 via Ni2+-agarose chromatography, essentially as previously described.7 Purified hFib1-49 in 10 mM sodium acetate (pH 4.5) was immobilized on a BIAcore CM5 sensor chip (research grade) with the amine coupling kit according to the manufacturer's instructions (chip and kit from GE Health Care, Piscataway, NJ). The Resonance unit coupled to the chip was 1614 RU, and the analysis used a Biacore 3000 instrument (GE Health Care, Piscataway, NJ). The kinetic analysis was performed at 25°C in 10 mM HEPES buffer (pH 7.4) with 0.15 M NaCl, 2 mM CaCl2, and 0.005% (v/v) surfactant P20 at a flow rate of 20 μl/min. The purified mouse Adamtsl2 was diluted in the above buffer at different concentrations and injected both to an uncoupled control flow cell and the cell coupled with hFib1-49. The sample injection time was 2 min and was followed by a pause of 6 min for dissociation. A total of 10 μM NaOH solution was used for regeneration after each injection at a flow rate of 50 μl/min for 30 s. The stabilization time after regenerations was 3 min. All the data were corrected with reference to the background binding in the control flow cell. We calculated the kinetic constants by assuming a 1:1 (Langmuir) binding model with the BIAevaluation software (version 4.0.1, GE Health Care, Piscataway, NJ).
Homology Modeling
Homology modeling of the cbEGF-TB5-cbEGF25 region was carried out with the coordinates of the fibrillin-1 cbEGF22-TB4-cbEGF23 structure (PDB 1UZJ) and Modeler software.8 Figures were rendered with the PyMOL Molecular Graphics System (Schrödinger).
Results
Exome Analysis
Among the 19 GD cases with no ADAMTSL2 mutations, absence of consanguinity or recurrence in sibs prompted us to perform exome sequencing in two out of 19 unexplained GD cases. We also performed exome sequencing in three out of ten AD unrelated patients. We first focused our analyses on nonsynonymous variants, splice acceptor and donor site mutations, and coding indels because we anticipated that synonymous variants were far less likely to be pathogenic. We also defined variants as previously unidentified if they were absent from control populations and from all datasets including dbSNP129, the 1000 Genomes Project, and in-house exome data.
Considering the recessive mode of inheritance of GD, we selected 11 candidate genes on the basis of the presence of either two distinct mutations or one mutation present at the homozygote state. For AD, we selected 66 candidate genes considering the dominant mode of inheritance. Given the phenotypic overlap between AD and GD, we also searched for a shared mutated gene among the five exomes. We identified changes in three genes: MUC17 (MIM 608422), HYDIN (MIM 610812), and FBN1. The link between tall stature and Marfan syndrome caused by FBN1 mutations (MIM 154700) prompted us to consider FBN1 as the best candidate gene.9
Exome analysis detected three missense FBN1 mutations (c.5096A>G [p.Tyr1699Cys], c.5087A>G [p.Tyr1696Cys], and c.1414T>C [p.Tyr472His]) in both GD patients and three heterozygous FBN1 missense mutations (c.5182G>A [p.Ala1728Thr], c.5165C>G [p.Ser1722Cys], and c.5251T>G, [p.Ser1750Arg]) in AD patients. These results were confirmed by Sanger sequencing. However, Tyr472His was considered a polymorphism on the basis of information from the Marfan mutation database. FBN1 screening led to the identification of six distinct heterozygous mutations in 17 additional GD cases. These mutations were not observed in GD parents, confirming that they occurred de novo.
Subsequent analyses in AD isolated seven FBN1 mutations. In two out of ten AD cases, mutations were identified in the affected parent, whereas they occurred de novo in the remaining cases. All mutations were absent from alleles in 2000 ethnicity-matched controls and in the Marfan mutation database.10,11 All together, we identified a total of 16 distinct heterozygous FBN1 mutations in 29 GD and AD cases (15 missense and one insertion, Table 2). Importantly, all mutations were clustered in the same region (exons 41 and 42) encoding the TGFβ-binding protein-like 5 (TB5) domain of FBN1 (Figure 2A).
Table 2.
FBN1 Mutations Identified in Individuals with GD and AD
| Family | Origin | Diagnosis | Nucleotide Change | Amino Acid Change | Parent Tested: De Novo Event Confirmed |
|---|---|---|---|---|---|
| 1 | Belgium | GD | c.5087A>G | p.Tyr1696Cys | yes |
| 2 | France | GD | c.5096A>G | p.Tyr1699Cys | yes |
| 3 | Russia | GD | c.5284G>A | p.Gly1762Ser | no mutation in the mother; father not available |
| 4 | Switzerland | GD | c.5096A>G | p.Tyr1699Cys | Parents not available |
| 5 | Russia | GD | c.5284G>A | p.Gly1762Ser | yes |
| 6 | France | GD | c.5284G>A | p.Gly1762Ser | yes |
| 7 | UK | GD | c.5087A>G | p.Tyr1696Cys | yes |
| 8 | Turkey | GD | c.5096A>G | p.Tyr1699Cys | no mutation in the mother; father not available |
| 9 | Algeria | GD | c.5117G>A | p.Cys1706Tyr | yes |
| 10 | Lebanon | GD | c.5157C>G | p.Cys1719Trp | yes |
| 11 | USA | GD | c.5096A>G | p.Tyr1699Cys | no |
| 12 | Turkey | GD | c.5284G>A | p.Gly1762Ser | yes |
| 13 | Russia | GD | c.5284G>A | p.Gly1762Ser | yes |
| 14 | Iraq | GD | c.5182G>A | p.Ala1728Thr | yes |
| 15 | USA | GD | c.5183C>T | p.Ala1728Val | yes |
| 16 | Japan | GD | c.5095T>G | p.Tyr1699Asp | yes |
| 17 | Australia | GD | c.5198G>A | p.Cys1733Tyr | yes |
| 18 | USA | GD | c.5284G>A | p.Gly1762Ser | no |
| 19 | Korea/Japan | GD | c.5096A>G | p.Tyr1699Cys | no mutation in the mother; father not available |
| 20 | France | AD | c.5182G>A | p.Ala1728Thr | yes |
| 21 | France | AD | c.5165C>G | p.Ser1722Cys | yes |
| 22-a | France | AD | c.5251T>G | p.Ser1750Arg | inherited from 22-c |
| 22-b | France | AD | c.5251T>G | p.Ser1750Arg | inherited from 22-c |
| 22-c | France | AD | c.5251T>G | p.Ser1750Arg | yes |
| 23 | Belgium | AD | c.5177G>T | p.Gly1726Val | yes |
| 24 | Netherlands | AD | c.5096A>G | p.Tyr1699Cys | yes |
| 25 | France | AD | c.5202_5204dup | p.Gln1735dup | yes |
| 26 | Italy | AD | c.5273A>T | p.Asp1758Val | no |
| 27-a | China | AD | c.5099A>G | p.Tyr1700Cys | inherited from 27-b |
| 27-b | China | AD | c.5099A>G | p.Tyr1700Cys | yes |
| 28 | France | AD | c.5141T>G | p.Met1714Arg | yes |
| 29 | France | AD | c.5165C>G | p.Ser1722Cys | yes |
Figure 2.
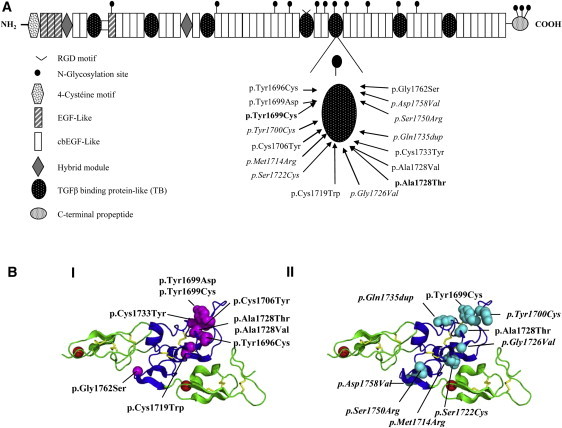
Location of FBN1 Mutations Identified in GD and AD Patients
(A) Functional domains of FBN1. The location of the amino change found in each family is shown (GD are families listed in roman font; AD families are in italics, and mutations shared by AD and GD are in bold).
(B) 3D modeling of the fibrillin-1 cbEGF24-TB5-cbEGF25 region showing residues affected by GD and AD mutations. GD substitutions (I) are shown in magenta and AD substitution sites (II) are shown in cyan. Note the clustering of disease-causing substitutions in the region of the TB domain previously associated with protein-protein interactions. cbEGF domains are shown in green, and the TB domain is in blue. Red spheres represent calcium ions bound to domains cbEGF24 and cbEGF25. Homology modeling created with Modeler software7 and the coordinates of the fibrillin-1 cbEGF22-TB4-cbEGF23 structure (PDB 1UZJ). The figure was generated with Pymol. GD substitutions are shown in roman font, whereas those found in AD or in both diseases are shown in italics and bold, respectively.
Consequences of Mutations on 3D TB5 Domain Structure
We used homology modeling to investigate the distribution of sites affected by heterozygous GD and AD mutations within the structure of the TB5 domain (Figure 2B) by using the known crystal structure of fibrillin-1 fragment cbEGF22-TB4-cbEGF23 as a template.12 GD substitutions appeared to alter structurally important residues such as cysteines involved in disulphide bond formation (Cys1706, Cys1719, Cys1733) or large aromatic components (Tyr1696, Tyr1699, Tyr1700). The GD substitutions were also clustered near the region of the TB domain known to be involved in intermolecular interactions on the basis of integrin-binding studies with fibrillin-1 TB4 domain fragments and LAP-LTBP interactions. AD substitutions appeared to be even more evenly distributed throughout the TB5 domain.
Microfibrillar Structure
To analyze the consequences of FBN1 mutations, we compared the microfibrillar structure in skin fibroblasts from GD and AD patients to that in control fibroblasts by indirect immunofluorescence. Staining revealed abundant long microfibrils in controls, but AD and GD fibroblasts demonstrated a reduced number of microfibrils and complete network disorganization (Figure 3A).
Figure 3.
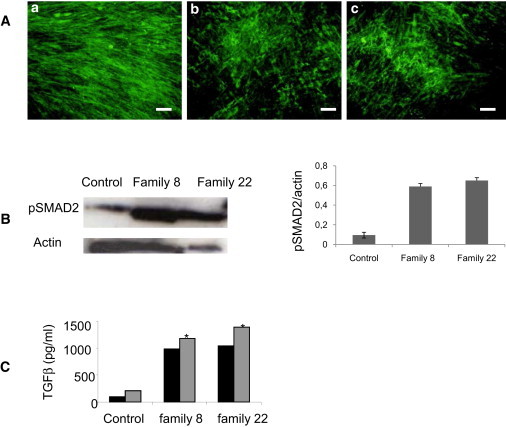
Microfibril and TGFβ-Signaling Analysis in GD and AD Skin Fibroblasts
(A) Microfibril analysis in skin fibroblasts from (a) control, (b) a GD patient, and (c) an AD patient. The microfibrillar network formation was detected by indirect immunofluorescence with fibrillin-1 antibody. (MAB019). The staining revealed abundant long microfibrils in the control fibroblasts (a). Conversely, the GD and AD patient fibroblasts showed a reduced number of microfibrils and a disorganization of the MF network (b and c). The scale bar represents 50 μm.
(B) Enhanced phosphorylation of SMAD2 (pSMAD2) in skin fibroblasts from one GD patient (family 8), one AD patient (family 22), and control. pSMAD2 was normalized to actin for comparison of pSMAD2 levels in affected and unaffected fibroblasts as shown in the right panel.
(C) Quantification of total (gray bars) and active (black bars) TGFβ in the conditioned medium of fibroblasts from individuals with GD (family 8) or AD (family 22) and controls. The conditioned medium from families 8 and 22 showed an amount of total TGFβ (∗p < 0,003) greater than the conditioned medium from the controls.
Analysis of TGFβ Signaling Pathway
To test the impact of TB5 domain substitutions on TGFβ signaling, we analyzed phospho-SMAD2 and -3 in the cell lysate of GD and AD fibroblasts and age- and passage-matched control skin fibroblasts by immunoblot (Figure 3B) and found an enhanced signal. Consistent with this observation, we quantified active and total TGF-β in the cultured medium of GD and AD skin fibroblasts by ELISA and found a 10-fold higher level of total TGF-β in the cultured medium of GD and AD fibroblasts than in the controls (p < 0.0003) (Figure 3C).
Link between FBN1 and ADAMTSL2
Because we have previously identified ADAMTSL2 mutations in a subset of GD patients, we hypothesized a direct link between FBN1, which is involved in AD and a portion of GD patients, and ADAMTSL2. To demonstrate this interaction, we performed surface plasmon resonance analysis by using FBN1 recombinant protein.
Although the fibrillin construct used did not contain TB5 domain, this analysis identified a specific direct interaction (KD = 60 nM) between the two proteins (Figure 4).
Figure 4.
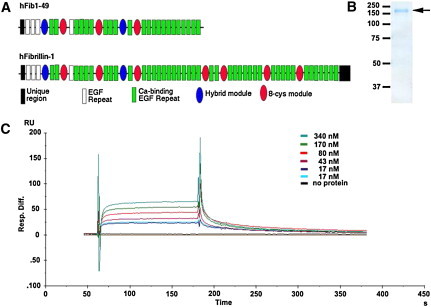
ADAMTSL2 Interacts Directly with Fibrillin-1
(A) The domain structure of the fibrillin peptide hFib1-49 is shown relative to the full-length fibrillin-1. The key to the fibrillin-1 modules is shown.
(B) Colloidal Coomassie-blue-stained-reducing polyacrylamide gel showing purification of hFib1-49 (arrow). The molecular weight markers (in kDa) are indicated on the left.
(C) SPR analysis of ADAMTSL2 (analyte) binding to hFib1-49 (ligand). The sensorgrams shown were obtained after injection of increasing concentrations of ADAMTSL2 as indicated. The y axis indicates the response difference obtained between the flow cell with bound hFib1-49 and the control flow cell without hFib1-49 when ADAMTSL2 was used as the analyte. The x axis shows time (s).
Discussion
Here, we report the identification of FBN1 mutations in 19 GD and ten AD patients; both GD and AD are clinically defined conditions combining short stature, short hands, and stiff joints.
Although GD has been described as an autosomal-recessive disorder, the identification of heterozygous FBN1 mutations demonstrates a dominant form of GD, strictly fulfilling the diagnostic criteria for GD (including progressive cardiac valvular thickening and early death in three out of 19 GD patients without ADAMTSL2 mutations). As previously reported,5 we did not find any significant difference in the main clinical and radiological features characteristic of GD, namely cardio-respiratory involvement, skin thickness, facial features (including a round full face, a small nose with anteverted nostrils, and a long philtrum) hepatomegaly, natural history of the disorder, and severe outcome between the two forms of GD. However, a broad nasal bridge, narrow palpebral fissures, and tip-toe walking were more consistently observed in the ADAMTSL2-mutated group.5 These minor phenotypic differences between the two forms of GD might guide the clinician diagnostically and help in the prioritization of the molecular screening.
Similarly, all AD cases fulfilled the diagnostic criteria of AD. None of them had cardiac involvement or early death. These data demonstrate that GD and AD appear clinically distinct but are allelic conditions.
We did not find any obvious differences in the nature of the mutations identified in GD compared to the mutations identified in AD. Among the 16 FBN1 mutations identified, seven were specifically identified in GD and seven were specifically identified in AD, but two mutations were found in either GD or AD cases. All mutations were located in exons 41 and 42, encoding the TB5 domain and were altering either large aromatic components or structurally important residues. Half of the mutations were creating or removing a cysteine residue within this domain, which is characterized, as are the other TB domains, by eight cysteines directly involved in FBN1 folding via intradomain disulfide linkage.13
Although FBN1 mutations have been identified in a wide range of disorders from Marfan syndrome to isolated ectopia lentis, our findings support that mutations of the TB5 domain are responsible for the phenotype of short stature, short hands, and stiff joints. A deletion in TB5 domain has been previously reported in Weill-Marchesani syndrome [MIM 608328], which is also characterized by short stature, short hands, and stiff joints but differs from AD and GD in the presence of microspherophakia.14 More recently, mutations in the TB4 domain have been reported in stiff skin [MIM 184900] patients, who differ from AD and GD by the absence of short stature and short hands.15
It is not known why mutations affecting the TB5 domain give rise to GD and AD rather than Marfan syndrome. Our findings of a specific interaction between FBN1 and ADAMTSL2 might support the hypothesis that a dysregulation of FBN1/ADAMTSL2/TGFβ interrelationship is the underlying mechanism of the short stature phenotypes The finding of similar enhancement of TGFβ signaling and microfibrillar structural changes in AD and GD with FBN1 or ADAMTSL2 mutations further supports the functional link between the TB5 domain and ADAMTSL2.
However, the finding of increased TGFβ signaling in fibroblasts from AD and GD and Marfan patients is still questionable and further illustrates the tissue dependence and the complexity of the TGFβ- and SMAD-signaling pathways with various levels of regulations. We hope that ongoing studies will document interaction of TB5 domain and ADAMTSL2 and contribute to a greater understanding of how enhanced TGFβ signaling caused by FBN1 mutations can trigger either tall stature, arachnodactyly, and hyperlaxity or severe short stature, short hands, and stiff joints.
Acknowledgments
We thank all the patients and their families for their contribution to this work. The work presented here was supported by the Medical Research Foundation (FRM to S.A.), a French National Research Agency (ANR) award (R09183KS to V.C.-D.), and a National Institutes of Health-National Institute of Arthritis and Musculoskeletal Skin Diseases grant (AR53890 to S.S.A). B.Z. is supported by the German Federal Ministry of Education and Research (BMBF, SKELNET project).
Web Resources
The URLs for data presented herein are as follows:
Marfan Mutation Database, http://www.umd.be/
Mutation Nomenclature, http://www.hgvs.org/mutnomen
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
Pymol, http://www.pymol.org
References
- 1.Spranger J.W., Gilbert E.F., Tuffli G.A., Rossiter F.P., Opitz J.M. Geleophysic dwarfism—a “focal” mucopolysaccharidosis? Lancet. 1971;2:97–98. doi: 10.1016/s0140-6736(71)92073-3. [DOI] [PubMed] [Google Scholar]
- 2.Maroteaux P., Stanescu R., Stanescu V., Rappaport R. Acromicric dysplasia. Am. J. Med. Genet. 1986;24:447–459. doi: 10.1002/ajmg.1320240307. [DOI] [PubMed] [Google Scholar]
- 3.Faivre L., Le Merrer M., Baumann C., Polak M., Chatelain P., Sulmont V., Cousin J., Bost M., Cordier M.P., Zackai E. Acromicric dysplasia: Long term outcome and evidence of autosomal dominant inheritance. J. Med. Genet. 2001;38:745–749. doi: 10.1136/jmg.38.11.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Le Goff C., Morice-Picard F., Dagoneau N., Wang L.W., Perrot C., Crow Y.J., Bauer F., Flori E., Prost-Squarcioni C., Krakow D. ADAMTSL2 mutations in geleophysic dysplasia demonstrate a role for ADAMTS-like proteins in TGF-beta bioavailability regulation. Nat. Genet. 2008;40:1119–1123. doi: 10.1038/ng.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allali S., Le Goff C., Pressac-Diebold I., Pfennig G., Mahaut C., Dagoneau N., Alanay Y., Brady A.F., Crow Y.J., Devriendt K. Molecular screening of ADAMTSL2 gene in 33 patients reveals the genetic heterogeneity of geleophysic dysplasia. J. Med. Genet. 2011;48:417–421. doi: 10.1136/jmg.2010.087544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kettle S., Card C.M., Hutchinson S., Sykes B., Handford P.A. Characterisation of fibrillin-1 cDNA clones in a human fibroblast cell line that assembles microfibrils. Int. J. Biochem. Cell Biol. 2000;32:201–214. doi: 10.1016/s1357-2725(99)00120-x. [DOI] [PubMed] [Google Scholar]
- 7.Hirohata S., Wang L.W., Miyagi M., Yan L., Seldin M.F., Keene D.R., Crabb J.W., Apte S.S. Punctin, a novel ADAMTS-like molecule, ADAMTSL-1, in extracellular matrix. J. Biol. Chem. 2002;277:12182–12189. doi: 10.1074/jbc.M109665200. [DOI] [PubMed] [Google Scholar]
- 8.Eswar N., Webb B., Marti-Renom M.A., Madhusudhan M.S., Eramian D., Shen M.Y., Pieper U., Sali A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Protein Sci. 2007 doi: 10.1002/0471140864.ps0209s50. Chapter 2:Unit 2.9. [DOI] [PubMed] [Google Scholar]
- 9.Dietz H.C., Cutting G.R., Pyeritz R.E., Maslen C.L., Sakai L.Y., Corson G.M., Puffenberger E.G., Hamosh A., Nanthakumar E.J., Curristin S.M. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352:337–339. doi: 10.1038/352337a0. [DOI] [PubMed] [Google Scholar]
- 10.Frédéric M.Y., Lalande M., Boileau C., Hamroun D., Claustres M., Béroud C., Collod-Béroud G. UMD-predictor, a new prediction tool for nucleotide substitution pathogenicity — application to four genes: FBN1, FBN2, TGFBR1, and TGFBR2. Hum. Mutat. 2009;30:952–959. doi: 10.1002/humu.20970. [DOI] [PubMed] [Google Scholar]
- 11.Collod-Béroud G., Le Bourdelles S., Ades L., Ala-Kokko L., Booms P., Boxer M., Child A., Comeglio P., De Paepe A., Hyland J.C. Update of the UMD-FBN1 mutation database and creation of an FBN1 polymorphism database. Hum. Mutat. 2003;22:199–208. doi: 10.1002/humu.10249. [DOI] [PubMed] [Google Scholar]
- 12.Lee S.S., Knott V., Jovanović J., Harlos K., Grimes J.M., Choulier L., Mardon H.J., Stuart D.I., Handford P.A. Structure of the integrin binding fragment from fibrillin-1 gives new insights into microfibril organization. Structure. 2004;12:717–729. doi: 10.1016/j.str.2004.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yuan X., Downing A.K., Knott V., Handford P.A. Solution structure of the transforming growth factor beta-binding protein-like module, a domain associated with matrix fibrils. EMBO J. 1997;16:6659–6666. doi: 10.1093/emboj/16.22.6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Faivre L., Gorlin R.J., Wirtz M.K., Godfrey M., Dagoneau N., Samples J.R., Le Merrer M., Collod-Beroud G., Boileau C., Munnich A., Cormier-Daire V. In frame fibrillin-1 gene deletion in autosomal dominant Weill-Marchesani syndrome. J. Med. Genet. 2003;40:34–36. doi: 10.1136/jmg.40.1.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Loeys B.L., Gerber E.E., Riegert-Johnson D., Iqbal S., Whiteman P., McConnell V., Chillakuri C.R., Macaya D., Coucke P.J., De Paepe A. Mutations in fibrillin-1 cause congenital scleroderma: Stiff skin syndrome. Sci. Transl. Med. 2010;2:ra20. doi: 10.1126/scitranslmed.3000488. [DOI] [PMC free article] [PubMed] [Google Scholar]