

Abstract
The erythrocyte sedimentation rate (ESR), a commonly performed test of the acute phase response, is the rate at which erythrocytes sediment in vitro in 1 hr. The molecular basis of erythrocyte sedimentation is unknown. To identify genetic variants associated with ESR, we carried out a genome-wide association study of 7607 patients in the Electronic Medical Records and Genomics (eMERGE) network. The discovery cohort consisted of 1979 individuals from the Mayo Clinic, and the replication cohort consisted of 5628 individuals from the remaining four eMERGE sites. A nonsynonymous SNP, rs6691117 (Val→IIe), in the complement receptor 1 gene (CR1) was associated with ESR (discovery cohort p = 7 × 10−12, replication cohort p = 3 × 10−14, combined cohort p = 9 × 10−24). We imputed 61 SNPs in CR1, and a “possibly damaging” SNP (rs2274567, His→Arg) in linkage disequilibrium (r2 = 0.74) with rs6691117 was also associated with ESR (discovery p = 5 × 10−11, replication p = 7 × 10−17, and combined cohort p = 2 × 10−25). The two nonsynonymous SNPs in CR1 are near the C3b/C4b binding site, suggesting a possible mechanism by which the variants may influence ESR. In conclusion, genetic variation in CR1, which encodes a protein that clears complement-tagged inflammatory particles from the circulation, influences interindividual variation in ESR, highlighting an association between the innate immunity pathway and erythrocyte interactions.
Main Text
The erythrocyte sedimentation rate (ESR) is a commonly performed laboratory test of the acute phase response that was first introduced in the 1920s and was considered a great advance at that time.1 Although the clinical utility of ESR has diminished with the availability of direct measurement of acute phase response proteins such as C-reactive protein,2 it is still commonly used in clinical practice, and extreme elevation (>100 mm/hr) is a marker of serious underlying disease, such as infection, autoimmune disorder, or malignancy.3,4 An elevated ESR is associated with increased risk of coronary heart disease, although the mechanism underlying the association is unclear.5 The molecular basis of erythrocyte sedimentation is not known, although increased levels of acute phase proteins (especially fibrinogen and immunoglobulins)1 and alterations in the size, shape, and number of red blood cells (RBC)1 are thought to play a role. As part of the Electronic Medical Records and Genomics (eMERGE) network,6 we investigated whether common genetic variants are associated with RBC traits, including ESR.7
The electronic medical record (EMR)-based genome-wide association analyses in the eMERGE network have been approved by the institutional review boards of the participating sites. Data for ESR, which is measured by standardized assays in the clinical setting—most commonly the Westergen method—were obtained from the EMR. In the Westergen method, anticoagulated blood is allowed to sediment for 1 hr in a glass tube 200 mm in length. ESR values are generally < 10 mm in men and < 15 mm in women. Values increase with age and are higher in women and African Americans. Any ESR trait values obtained during an inpatient hospitalization were excluded.
Genotyping was performed with the Illumina Human 660W-Quadv1_A genotyping platform, consisting of 561,490 SNPs and 95,876 intensity-only probes, in 17,056 patients from the five eMERGE sites, including 14,105 patients of self-reported European ancestry. DNA samples from Mayo Clinic, Vanderbilt University Medical Center, and Northwestern University were genotyped at the Broad Institute's Center for Genotyping and Analysis. DNA samples from Marshfield Clinic and Group Health Cooperative were genotyped at the Center for Inherited Disease Research, Johns Hopkins University. Data were cleaned via the quality control (QC) pipeline developed by the eMERGE Genomics Working Group.8 The process includes evaluation of sample and marker call rates, gender mismatch and anomalies, duplicate and HapMap concordance, batch effects, Hardy-Weinberg equilibrium, sample relatedness, and population stratification. A total of 476,395 SNPs were used for analysis on the basis of the following QC criteria: SNP call rate > 98%, sample call rate > 98%, minor allele frequency > 0.05, Hardy-Weinberg equilibrium p value > 0.001, and 99.99% concordance rate in duplicates. The first two dimensions were derived from multidimensional decomposition analysis of the 1-IBS (identity-by-state) matrix based on all eMERGE samples. Samples greater than six standard deviations from the mean of self-reported White ancestry on dimensions 1 and 2 were excluded. After QC steps, 7607 patients (with 23,092 ESR values) were available for association analyses. The characteristics of this cohort are summarized in Table 1 (also see Figure S2, available online, for distribution of ESR by site). The lower mean ESR in the Mayo Clinic cohort could be partly attributed to a higher proportion of men in this cohort, the ESR being lower in men on average.
Table 1.
Sample Characteristics
|
Discovery Cohort (N = 1979) |
Replication Cohort (N = 5628) |
||||
|---|---|---|---|---|---|
| MAYO | GHC | MC | VUMC | NU | |
| Number of patients | 1979 | 1864 | 2631 | 743 | 390 |
| Number of ESR measurements | 6914 | 7528 | 5262 | 1554 | 1834 |
| Age (yrs) | 63.4 ± 10.0 | 77.8 ± 7.2 | 66.0 ± 12.1 | 53.1 ± 16.3 | 55.6 ± 13.2 |
| Women (%) | 808 (40.8%) | 1106 (59.3%) | 1562 (59.3%) | 477 (64.2%) | 232 (59.4%) |
| ESR (mm/h): total | 13.1 ± 15.0 | 22.1 ± 18.6 | 23.7 ± 19.0 | 20.8 ± 21.1 | 22.0 ± 25.8 |
| ESR (mm/h): men | 11.6 ± 15.2 | 21.0 ± 20.5 | 21.5 ± 19.4 | 20.5 ± 23.6 | 23.8 ± 29.0 |
| ESR (mm/h): women | 15.3 ± 14.2 | 22.9 ± 17.1 | 25.2 ± 18.5 | 21.0 ± 19.6 | 20.7 ± 23.3 |
| Hemoglobin (g/dL): total | 13.8 ± 1.3 | 13.3 ± 1.3 | 13.5 ± 1.3 | 13.4 ± 1.5 | 12.8 ± 1.7 |
| Hemoglobin (g/dL): men | 14.2 ± 1.3 | 13.7 ± 1.4 | 14.0 ± 1.4 | 14.0 ± 1.7 | 13.1 ± 2.0 |
| Hemoglobin (g/dL): women | 13.2 ± 1.1 | 13.0 ± 1.1 | 13.1 ± 1.1 | 13.1 ± 1.2 | 12.6 ± 1.4 |
MAYO, Mayo Clinic; GHC, Group Health Cooperative; MC, Marshfield Clinic; VUMC, Vanderbilt University Medical Center; NU, Northwestern University. Age, ESR, and hemoglobin data are shown as means ± standard deviation.
When multiple measurements of ESR were available for an individual patient, we chose the median ESR, which was subjected to natural logarithm transformation to reduce skewness, prior to genetic analyses. We used an efficient mixed-model association expedited (EMMAX) algorithm9 to correct for sample relatedness and cryptic population substructure. In this model, the IBS matrix was calculated for each pair of individuals with the use of the genome-wide genotype data, then the generalized least-squares F-test was used to estimate the regression coefficient (β) after adjustment for covariates. Because of sex differences in ESR, we also assessed genotype-sex interactions by using PLINK.10 No such interactions were present; therefore, analyses were not stratified by sex.
A three-step experimental design was used to conduct a genome-wide association study (GWAS) for ESR. We assigned the Mayo Clinic cohort (n = 1979) as the discovery cohort and patients from the other four sites (n = 5628) as the replication cohort. We performed a GWAS for (natural logarithm transformed) ESR in the discovery cohort, then in the replication cohort, and finally, we performed a joint analysis that included all patients. Adjustment variables included age, sex, and site, as well as hemoglobin level, because ESR is inversely related to hemoglobin level.11 Our power was 0.49, 1, and 1, for detecting a quantitative trait locus that explained 1.5% variance in the discovery cohort (n = 1979), the replication cohort (n = 5628), and the entire cohort (n = 7607), respectively, at a significance level of 5 × 10−8.
The Manhattan and quantile-quantile plots summarizing the results of genetic association analyses for ESR are shown in Figure 1. Five common SNPs in complement receptor 1 gene (CR1 [MIM 120620]) were associated with ESR in the discovery cohort at a genome-wide significance level, and the SNPs were replicated in patients from the four remaining eMERGE sites (Table 2). Of the five SNPs associated with ESR, one was a nonsynonymous SNP (rs6691117, Ile→Val; p = 7 × 10−12 and p = 3 × 10−14 in the discovery and replication cohorts, respectively). The minor allele G was associated with lower ESR. When we combined the cohorts in a joint analysis, the significance level increased (p = 9 × 10−24; Table 2). We imputed 61 additional SNPs (MAF ≥ 0.05 and p value for HWE ≥ 0.001) in CR1 (± 5 kb) on the basis of the phased chromosome 1 from the HapMap II (release 21) CEU population (Utah residents with ancestry from northern and western Europe), using MACH1.12 The imputation qualities (i.e., the average posterior probability for the most likely genotype) and R2 (the squared correlation between imputed and true genotypes) of the 61 imputed SNPs in CR1 were 0.98 and 0.95, respectively. Imputation-based association analysis indicated that the G allele in the “possibly damaging” SNP rs2274567 (His→Arg) was associated with lower ESR in the discovery cohort (p = 5 × 10−11), the replication cohort (p = 7 × 10−17), and the combined cohort (p = 2 × 10−25). The pattern of linkage disequilibrium (LD) and negative logarithm of p values for CR1 SNPs associated with ESR are shown in Figure 2, demonstrating that the five significant genotyped SNPs and rs2274567 are located in a single LD block of ∼125 kb. Haplotype-based association analysis based on 11 genotyped SNPs revealed that six specific haplotypes (frequency ≥ 0.01) were associated with ESR (Table S2).
Figure 1.
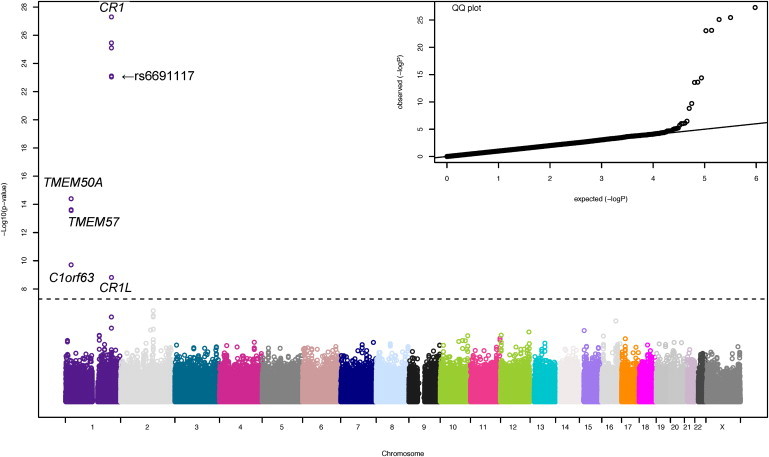
Manhattan and Quantile-Quantile Plots for Genome-wide Association Analysis of ESR in the Entire Cohort
The vertical axis indicates (–log10 transformed) observed P values, and the horizontal line indicates the genome-wide significance level of p = 5 × 10−8. In the quantile-quantile (QQ) plot, the horizontal axis shows (–log10 transformed) expected p values, and the vertical axis indicates (–log10 transformed) observed p values.
Table 2.
SNPs in CR1 Associated with ESR
| Cohortsa |
rs650877(G/A, 205,815,416)d |
rs11118131(T/C, 205,827,819) |
rs677066(G/A, 205,840,614) |
rs6691117(G/A, 205,849,554) |
rs12034383(G/A, 205,870,218) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MAFb | βc | p Value | MAF | β | p Value | MAF | β | p Value | MAF | β | p Value | MAF | β | p Value | |
| Discovery | |||||||||||||||
| MAYO (n = 1979) | 0.19 | −0.23 | 2 × 10−11 | 0.19 | −0.23 | 5 × 10−11 | 0.22 | −0.22 | 1 × 10−11 | 0.22 | −0.23 | 7 × 10−12 | 0.42 | −0.21 | 4 × 10−13 |
| Replication | |||||||||||||||
| GHC (n = 1864) | 0.18 | −0.16 | 1 × 10−06 | 0.18 | −0.17 | 4 × 10−07 | 0.21 | −0.16 | 3 × 10−07 | 0.20 | −0.17 | 2 × 10−07 | 0.39 | −0.15 | 2 × 10−08 |
| MC (n = 2631) | 0.18 | −0.14 | 1 × 10−08 | 0.18 | −0.14 | 3 × 10−08 | 0.21 | −0.10 | 2 × 10−05 | 0.21 | −0.10 | 2 × 10−05 | 0.41 | −0.10 | 4 × 10−08 |
| VUMC (n = 743) | 0.19 | −0.21 | 9 × 10−04 | 0.19 | −0.21 | 2 × 10−03 | 0.22 | −0.18 | 3 × 10−03 | 0.22 | −0.17 | 6 × 10−03 | 0.39 | −0.16 | 3 × 10−03 |
| NU (n = 390) | 0.22 | −0.22 | 0.013 | 0.21 | −0.24 | 0.008 | 0.24 | −0.21 | 0.016 | 0.24 | −0.20 | 0.019 | 0.45 | −0.18 | 0.012 |
| Total (n = 5628) | 0.18 | −0.16 | 4 × 10−17 | 0.18 | −0.16 | 3 × 10−17 | 0.21 | −0.14 | 2 × 10−14 | 0.21 | −0.14 | 3 × 10−14 | 0.41 | −0.13 | 2 × 10−18 |
| Combined | |||||||||||||||
| (n = 7607) | 0.18 | −0.18 | 3 × 10−26 | 0.18 | −0.18 | 8 × 10−26 | 0.22 | −0.16 | 8 × 10−24 | 0.21 | −0.16 | 9 × 10−24 | 0.41 | −0.15 | 5 × 10−28 |
MAYO, Mayo Clinic; GHC, Group Health Cooperative; MC, Marshfield Clinic; VUMC, Vanderbilt University Medical Center; NU, Northwestern University.
MAF, minor allele frequency.
The β is the effect size of the major allele.
The first allele is the minor allele, and the number indicates the physical location of the SNP (NCBI 36).
Figure 2.
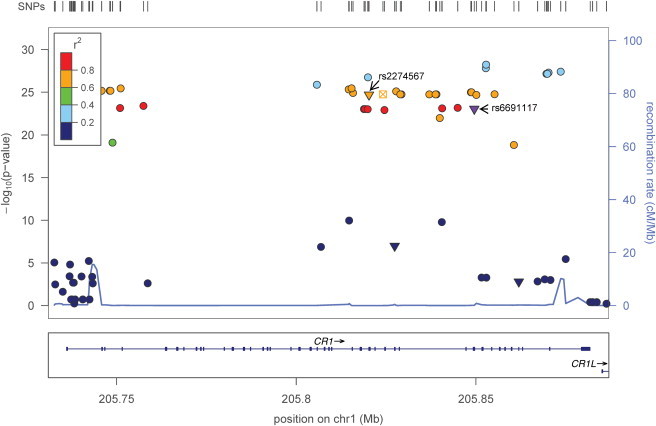
Regional Plot of CR1 Association with ESR
The top panel shows the association of CR1 SNPs (both genotyped and imputed) with ESR. Different colors represent LD (r2) between rs6691117 and other SNPs. The LD (r2) is 0.74 between the nonsynonymous SNPs rs6691117 and rs2274567 (labeled with arrows). Different point characters indicate different functional classes: ▾, nonsynonymous; ●, intronic or intergenic; ⊠, conserved in mammals. The bottom panel indicates the gene structure of CR1.
Because ESR can be affected by a wide array of medical conditions, we developed an algorithm based on billing codes and natural language processing (NLP) of unstructured clinical notes to exclude values affected by comorbidities, medications, or recent blood loss (Figure S1).7 We compiled the International Classification of Disease 9 Clinical Management (ICD-9 CM), procedural ICD-9, and Current Procedural Terminology (CPT-4) codes for hematologic and solid-organ malignancies, bone marrow and solid-organ transplantation, cirrhosis, hereditary anemias, malabsorption disorders, connective-tissue disorders, inflammatory bowel disease, chronic infectious diseases, and surgical procedures associated with significant blood loss. The medications included chemotherapeutic and immunosuppressive drugs. To validate the above algorithm, we randomly chose 50 ESR values that were included on the basis of the algorithm and 50 that were excluded. The algorithm correctly included all 50 and correctly excluded 49 out of 50 values. We then exported the algorithm to the other eMERGE sites to extract ESR values from the EMR. The algorithm led to the exclusion of 2358 patients. In the remaining 5249 patients, the five common SNPs in Table 2 remained significant although the significance levels decreased (Table S1). The CR1 SNPs were associated with ESR in the excluded patients (n = 2358) at a lower significance level (p = 1 × 10−5).
We noted five additional SNPs that were associated with ESR in the replication or combined cohorts (Table 3), including four SNPs in LD on chromosome 1p36—a nonsynonymous SNP (rs1043879, Glu→Gly) in chromosome 1 open reading frame (C1orf63), an intronic SNP (rs3091242) in transmembrane protein 50A (TMEM50A), and two intronic SNPs (rs873308 and rs10903129) in transmembrane protein 57 (TMEM57)—and one intronic SNP (rs7527798) in complement receptor 1-like (CR1L) on chromosome 1q32. The functions of TMEM50A and TMEM57 are unknown, but the proteins have transmembrane domains, supporting the view that these integral membrane proteins could influence erythrocyte interactions. Of note, CR1L encodes a transmembrane protein, is in proximity to CR1, and is expressed in hematopoietic and fetal lymphoid tissue.13
Table 3.
Additional SNPs Associated with ESR in the Replication and Combined Cohorts
| SNP | Chr | Physical Position | Gene | Class |
Discovery Cohort |
Replication Cohort |
Entire Cohort |
|||
|---|---|---|---|---|---|---|---|---|---|---|
| βa | p Value | β | p Value | β | p Value | |||||
| rs1043879 | 1p36 | 25442668 | C1orf63 | nonsyn | −0.09 | 2 × 10−03 | −0.10 | 3 × 10−08 | −0.09 | 2 × 10−09 |
| rs3091242 | 1p36 | 25547372 | TMEM50A | intronic | −0.12 | 6 × 10−06 | −0.10 | 7 × 10−10 | −0.10 | 2 × 10−13 |
| rs873308 | 1p36 | 25631242 | TMEM57 | intronic | −0.11 | 7 × 10−05 | −0.10 | 4 × 10−10 | −0.10 | 1 × 10−12 |
| rs10903129 | 1p36 | 25641524 | TMEM57 | intronic | −0.11 | 5 × 10−05 | −0.10 | 2 × 10−10 | −0.10 | 5 × 10−13 |
| rs7527798 | 1q32.2 | 205938913 | CR1L | intronic | 0.11 | 3 × 10−04 | 0.09 | 1 × 10−06 | 0.10 | 2 × 10−09 |
Chr, chromosome; nonsyn, nonsynonymous.
The β is the effect size of the major allele.
CR1, also known as erythrocyte complement component (3b/4b) receptor 1, CD35, or immune adhesion receptor, is a member of the receptors of complement activation (RCA) family that spans 21.45 cM on chromosome 1q32, contains more than 60 genes of which 15 are complement-related genes, and is thought to have evolved as a result of a series of gene-duplication events.14 CR1 encodes a monomeric single-pass type I membrane glycoprotein found on a variety of cells, including erythrocytes. The primary role of CR1 is to prevent immune complex deposition in the vessel wall by binding complement-tagged inflammatory particles and facilitating their clearance (Figure 3).15,16
Figure 3.
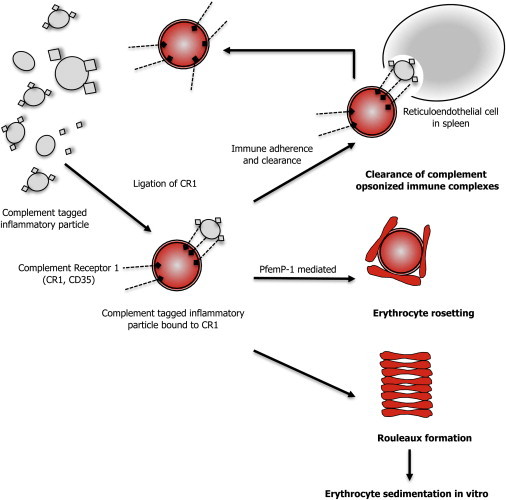
Role of CR1 in Clearing Opsonized Immune Particles, Erythrocyte Rosetting, and Rouleaux Formation
Pfemp-1: P. falciparum erythrocyte membrane protein 1.
CR1 is organized in tandem structural units called short consensus repeats (SCRs)17 that are grouped into four long homologous repeats (LHR-A to LHR-D) (Figure S3). Each SCR consists of 60–70 amino acids that are highly conserved (Figure S4).18,19 The location of the nonsynonymous SNPs rs2274567 and rs6691117 within the three-dimensional structure of CR1 (PDB 2Q7Z) is shown in Figure S2.20 The nonsynonymous SNP rs6691117 is located within the conserved β sheet (β2) of the 25th SCR domain (LHR-C) and changes an isoleucine residue to valine (Figure S2 and Figure S3). The (imputed) nonsynonymous SNP rs2274567 is located in the conserved β sheet (β2) of the 19th SCR (LHR-B) (see Figure S3 and Figure S4) and results in change from histidine to arginine. We hypothesize that the nonsynonymous SNPs rs6691117 and/or rs2274567 may alter the secondary structure of one of the sheets (β2) thereby affecting the binding affinity of CR1 to C3b and/or C4b. When C3b or C4b binds to CR1, it induces clustering of CR1 and its subsequent complex formation with a scaffolding protein, Fas-associated phosphatase 1 (FAP-1), resulting in an increase in erythrocyte membrane deformability.21,22 Thus, altered binding of circulating complement-opsonized particles to RBC CR1 may in turn influence ESR (Figure 3).
PolyPhen-223 and SNPeffect24 databases were used to elucidate the potential functional consequences of the nonsynonymous SNPs rs6691117 and rs2274567. PolyPhen-2, a tool that predicts the possible impact of an amino acid substitution on the structure and function of a human protein by using physical and comparative considerations, predicted the nonsynonymous SNP rs2274567 to be “probably damaging” and rs6691117 to be “benign.” SNPeffect indicated that rs6691117 may affect Hsp70 binding, which could affect the folding of CR1 and thereby affect structural stability and induce changes in functions mediated by subsequent protein-protein interactions.
To assess whether soluble CR1 levels in the serum differed by genotypes at SNPs rs6691117 and rs2274567, we measured CR1 levels by ELISA (USCN Life Science, Wuhan, China) in 50 patients with the genotype GG at rs669117 and 50 age- and sex-matched patients with genotypes AA and AG at that SNP. All standards and patient samples were assayed in duplicate. The limit of detection with the use of this assay was 0.014 ng/ml. In interassay precision testing, the mean of the low control was 57.2 ng/ml (coefficient of variation = 15.3%) and the mean of the high control was 90.8 ng/ml (coefficient of variation = 10.7%). Intraassay precision for the low and high controls was 13.1% and 7.3%, respectively. We did not find soluble serum CR1 levels to differ by genotypes at rs6691117 (GG = 69.4 ± 21.7 ng/ml, AG = 69.4 ± 24.7 ng/ml, and AA = 69.5 ± 18.4 ng/ml; n = 50 each; p = 0.99) or at rs2274567 (GG = 68.1 ± 21.4 ng/ml, AG = 70.1 ± 25.1 ng/ml, and AA = 69.8 ± 18.2 ng/ml; n = 40, 54, and 56, respectively; p = 0.77).
CR1 is known to mediate a variety of molecular functions and biological processes.18,19 Gene Ontology (GO) annotations of proteins interacting with CR1 revealed these to be involved in protein binding, signal transduction, receptor activity, and enzyme regulator activity, as well as the biological processes of cell communication and cell differentiation (Figure S5). Membrane-complement regulatory proteins modulate tissue injury in autoimmune and inflammatory diseases.25,26 CR1 mediates inhibitory signals in human lymphocytes and has emerged as a candidate gene26 for susceptibility to immune and/or inflammatory disorders.27,28 Gene expression studies indicate that CR1 is significantly upregulated in malaria, acute lymphoblastic leukemia, and Duchenne muscular dystrophy. In mice, knocking out the ortholog of CR1 affects embryogenesis and immune, reproductive, and renal and/or urinary systems.29,30 A two-SNP (rs6656401 and rs3818361) haplotype (AA) in CR1 was associated with Alzheimer disease (OR = 1.22, p = 3E-10) in a GWAS.31 However, these two intronic SNPs were not strongly associated with ESR in the present study (p = 0.003 and p = 0.0005 for rs6656401 and rs3818361, respectively), and it is likely that different variants in CR1 may be associated with different phenotypes.
Whether CR1 variants affect erythrocyte interactions in vivo needs additional investigation. CR1, by binding P. falciparum erythrocyte membrane protein-1 (PfEMP-1),32 leads to erythrocyte rosette formation in cerebral malaria, a phenomenon that may be dependent on CR1 copy number (Figure 3).27 Genes that mediate cyto-adherence by P. falciparum-infected erythrocytes, such as CR1, might exhibit signatures of natural selection.33,34 In support of this hypothesis, up to 80% of the population of a malarious region of Papua New Guinea have a high frequency of the derived (G) allele of the nonsynonymous SNP rs2274567,35 suggestive of recent positive selection. The frequency of the derived G allele of the imputed nonsynonymous SNP rs2274567 was not high (0.19) in our cohort; however, extended haplotype homozygosity (EHH) analysis36 indicated that the overall EHH of the derived G allele decayed more slowly than that of the ancestral allele (Figure S6). Additional studies are needed to confirm whether natural selection has operated on CR1 in various geographic locales.
In conclusion, several common SNPs in CR1 were associated with interindividual variation in ESR, suggesting that CR1 is involved in the molecular mechanisms that determine erythrocyte sedimentation. CR1 plays a key role in immune recognition and clearance of immune complexes and complement-opsonized particles from the circulation. Two nonsynonymous SNPs (rs2274567 and rs6691117) associated with ESR may cause conformational changes in CR1 that could affect its ability to bind C3b and/or C4b. Our findings indicate that a complement receptor protein that has a role in innate immunity also influences erythrocyte interactions.
Acknowledgments
The authors acknowledge Elizabeth Pugh for helpful discussions. The eMERGE Network was initiated and funded by the National Human Genome Research Institute (NHGRI), with additional funding from National Institute of General Medical Sciences (NIGMS) through the following grants: U01-HG-04599 (Mayo Clinic), U01-HG-004610 (Group Health Cooperative), U01-HG-004608 (Marshfield Clinic), U01HG004609 (Northwestern University), and U01-HG-04603 (Vanderbilt University, also serving as the Administrative Coordinating Center).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Array Express, http://www.ebi.ac.uk/arrayexpress
Ensembl, http://www.ensembl.org
HapMap, http://hapmap.ncbi.nlm.nih.gov/
Online Mendelian Inheritance in Man, http://www.omim.org
SNPeffect, http://snpeffect.vib.be
References
- 1.Fabry T.L. Mechanism of erythrocyte aggregation and sedimentation. Blood. 1987;70:1572–1576. [PubMed] [Google Scholar]
- 2.Gabay C., Kushner I. Acute-phase proteins and other systemic responses to inflammation. N. Engl. J. Med. 1999;340:448–454. doi: 10.1056/NEJM199902113400607. [DOI] [PubMed] [Google Scholar]
- 3.Brigden M.L. Clinical utility of the erythrocyte sedimentation rate. Am. Fam. Physician. 1999;60:1443–1450. [PubMed] [Google Scholar]
- 4.Sox H.C., Jr., Liang M.H. The erythrocyte sedimentation rate. Guidelines for rational use. Ann. Intern. Med. 1986;104:515–523. doi: 10.7326/0003-4819-104-4-515. [DOI] [PubMed] [Google Scholar]
- 5.Danesh J., Wheeler J.G., Hirschfield G.M., Eda S., Eiriksdottir G., Rumley A., Lowe G.D., Pepys M.B., Gudnason V. C-reactive protein and other circulating markers of inflammation in the prediction of coronary heart disease. N. Engl. J. Med. 2004;350:1387–1397. doi: 10.1056/NEJMoa032804. [DOI] [PubMed] [Google Scholar]
- 6.McCarty C.A., Chisholm R.L., Chute C.G., Kullo I.J., Jarvik G.P., Larson E.B., Li R., Masys D.R., Ritchie M.D., Roden D.M., eMERGE Team The eMERGE Network: a consortium of biorepositories linked to electronic medical records data for conducting genomic studies. BMC Med. Genomics. 2011;4:13. doi: 10.1186/1755-8794-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kullo I.J., Ding K., Jouni H., Smith C.Y., Chute C.G. A genome-wide association study of red blood cell traits using the electronic medical record. PLoS ONE. 2010;5:e13011. doi: 10.1371/journal.pone.0013011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turner S., Armstrong L.L., Bradford Y., Carlson C.S., Crawford D.C., Crenshaw A.T., de Andrade M., Doheny K.F., Haines J.L., Hayes G. Current Protocols in Human Genetics. Wiley-Blackwell; 2011. Quality control procedures for genome wide association studies. 68, 1.19.1–1.19.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kang H.M., Sul J.H., Service S.K., Zaitlen N.A., Kong S.Y., Freimer N.B., Sabatti C., Eskin E. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 2010;42:348–354. doi: 10.1038/ng.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bain B.J. Some influences on the ESR and the fibrinogen level in healthy subjects. Clin. Lab. Haematol. 1983;5:45–54. doi: 10.1111/j.1365-2257.1983.tb00495.x. [DOI] [PubMed] [Google Scholar]
- 12.Li Y., Willer C., Sanna S., Abecasis G. Genotype imputation. Annu. Rev. Genomics Hum. Genet. 2009;10:387–406. doi: 10.1146/annurev.genom.9.081307.164242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Logar C.M., Chen W., Schmitt H., Yu C.Y., Birmingham D.J. A human CR1-like transcript containing sequence for a binding protein for iC4 is expressed in hematopoietic and fetal lymphoid tissue. Mol. Immunol. 2004;40:831–840. doi: 10.1016/j.molimm.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 14.Krushkal J., Bat O., Gigli I. Evolutionary relationships among proteins encoded by the regulator of complement activation gene cluster. Mol. Biol. Evol. 2000;17:1718–1730. doi: 10.1093/oxfordjournals.molbev.a026270. [DOI] [PubMed] [Google Scholar]
- 15.Li J., Wang J.P., Ghiran I., Cerny A., Szalai A.J., Briles D.E., Finberg R.W. Complement receptor 1 expression on mouse erythrocytes mediates clearance of Streptococcus pneumoniae by immune adherence. Infect. Immun. 2010;78:3129–3135. doi: 10.1128/IAI.01263-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weisman H.F., Bartow T., Leppo M.K., Marsh H.C., Jr., Carson G.R., Concino M.F., Boyle M.P., Roux K.H., Weisfeldt M.L., Fearon D.T. Soluble human complement receptor type 1: in vivo inhibitor of complement suppressing post-ischemic myocardial inflammation and necrosis. Science. 1990;249:146–151. doi: 10.1126/science.2371562. [DOI] [PubMed] [Google Scholar]
- 17.Norman D.G., Barlow P.N., Baron M., Day A.J., Sim R.B., Campbell I.D. Three-dimensional structure of a complement control protein module in solution. J. Mol. Biol. 1991;219:717–725. doi: 10.1016/0022-2836(91)90666-t. [DOI] [PubMed] [Google Scholar]
- 18.Krych-Goldberg M., Atkinson J.P. Structure-function relationships of complement receptor type 1. Immunol. Rev. 2001;180:112–122. doi: 10.1034/j.1600-065x.2001.1800110.x. [DOI] [PubMed] [Google Scholar]
- 19.Liu D., Niu Z.X. The structure, genetic polymorphisms, expression and biological functions of complement receptor type 1 (CR1/CD35) Immunopharmacol. Immunotoxicol. 2009;31:524–535. doi: 10.3109/08923970902845768. [DOI] [PubMed] [Google Scholar]
- 20.Furtado P.B., Huang C.Y., Ihyembe D., Hammond R.A., Marsh H.C., Perkins S.J. The partly folded back solution structure arrangement of the 30 SCR domains in human complement receptor type 1 (CR1) permits access to its C3b and C4b ligands. J. Mol. Biol. 2008;375:102–118. doi: 10.1016/j.jmb.2007.09.085. [DOI] [PubMed] [Google Scholar]
- 21.Glodek A.M., Mirchev R., Golan D.E., Khoory J.A., Burns J.M., Shevkoplyas S.S., Nicholson-Weller A., Ghiran I.C. Ligation of complement receptor 1 increases erythrocyte membrane deformability. Blood. 2010 doi: 10.1182/blood-2010-04-273904. Published online September 22 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghiran I., Glodek A.M., Weaver G., Klickstein L.B., Nicholson-Weller A. Ligation of erythrocyte CR1 induces its clustering in complex with scaffolding protein FAP-1. Blood. 2008;112:3465–3473. doi: 10.1182/blood-2008-04-151845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reumers J., Maurer-Stroh S., Schymkowitz J., Rousseau F. SNPeffect v2.0: a new step in investigating the molecular phenotypic effects of human non-synonymous SNPs. Bioinformatics. 2006;22:2183–2185. doi: 10.1093/bioinformatics/btl348. [DOI] [PubMed] [Google Scholar]
- 25.Wagner C., Ochmann C., Schoels M., Giese T., Stegmaier S., Richter R., Hug F., Hänsch G.M. The complement receptor 1, CR1 (CD35), mediates inhibitory signals in human T-lymphocytes. Mol. Immunol. 2006;43:643–651. doi: 10.1016/j.molimm.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 26.Khera R., Das N. Complement Receptor 1: disease associations and therapeutic implications. Mol. Immunol. 2009;46:761–772. doi: 10.1016/j.molimm.2008.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thomas B.N., Donvito B., Cockburn I., Fandeur T., Rowe J.A., Cohen J.H., Moulds J.M. A complement receptor-1 polymorphism with high frequency in malaria endemic regions of Asia but not Africa. Genes Immun. 2005;6:31–36. doi: 10.1038/sj.gene.6364150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nath S.K., Harley J.B., Lee Y.H. Polymorphisms of complement receptor 1 and interleukin-10 genes and systemic lupus erythematosus: a meta-analysis. Hum. Genet. 2005;118:225–234. doi: 10.1007/s00439-005-0044-6. [DOI] [PubMed] [Google Scholar]
- 29.Molina H., Holers V.M., Li B., Fung Y., Mariathasan S., Goellner J., Strauss-Schoenberger J., Karr R.W., Chaplin D.D. Markedly impaired humoral immune response in mice deficient in complement receptors 1 and 2. Proc. Natl. Acad. Sci. USA. 1996;93:3357–3361. doi: 10.1073/pnas.93.8.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miwa T., Zhou L., Kimura Y., Kim D., Bhandoola A., Song W.C. Complement-dependent T-cell lymphopenia caused by thymocyte deletion of the membrane complement regulator Crry. Blood. 2009;113:2684–2694. doi: 10.1182/blood-2008-05-157966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lambert J.C., Heath S., Even G., Campion D., Sleegers K., Hiltunen M., Combarros O., Zelenika D., Bullido M.J., Tavernier B., European Alzheimer's Disease Initiative Investigators Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat. Genet. 2009;41:1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 32.Tham W.H., Wilson D.W., Lopaticki S., Schmidt C.Q., Tetteh-Quarcoo P.B., Barlow P.N., Richard D., Corbin J.E., Beeson J.G., Cowman A.F. Complement receptor 1 is the host erythrocyte receptor for Plasmodium falciparum PfRh4 invasion ligand. Proc. Natl. Acad. Sci. USA. 2010;107:17327–17332. doi: 10.1073/pnas.1008151107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kwiatkowski D.P. How malaria has affected the human genome and what human genetics can teach us about malaria. Am. J. Hum. Genet. 2005;77:171–192. doi: 10.1086/432519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Teeranaipong P., Ohashi J., Patarapotikul J., Kimura R., Nuchnoi P., Hananantachai H., Naka I., Putaporntip C., Jongwutiwes S., Tokunaga K. A functional single-nucleotide polymorphism in the CR1 promoter region contributes to protection against cerebral malaria. J. Infect. Dis. 2008;198:1880–1891. doi: 10.1086/593338. [DOI] [PubMed] [Google Scholar]
- 35.Cockburn I.A., Mackinnon M.J., O'Donnell A., Allen S.J., Moulds J.M., Baisor M., Bockarie M., Reeder J.C., Rowe J.A. A human complement receptor 1 polymorphism that reduces Plasmodium falciparum rosetting confers protection against severe malaria. Proc. Natl. Acad. Sci. USA. 2004;101:272–277. doi: 10.1073/pnas.0305306101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sabeti P.C., Reich D.E., Higgins J.M., Levine H.Z., Richter D.J., Schaffner S.F., Gabriel S.B., Platko J.V., Patterson N.J., McDonald G.J. Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002;419:832–837. doi: 10.1038/nature01140. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.