

Abstract
Inherited retinal degenerations, including retinitis pigmentosa (RP) and Leber congenital amaurosis (LCA), comprise a group of disorders showing high genetic and allelic heterogeneity. The determination of a full catalog of genes that can, when mutated, cause human retinal disease is a powerful means to understand the molecular physiology and pathology of the human retina. As more genes are found, remaining ones are likely to be rarer and/or unexpected candidates. Here, we identify a family in which all known RP/LCA-related genes are unlikely to be associated with their disorder. A combination of homozygosity mapping and exome sequencing identifies a homozygous nonsense mutation, c.496C>T (p.Arg166X), in a gene, KCNJ13, encoding a potassium channel subunit Kir7.1. A screen of a further 333 unrelated individuals with recessive retinal degeneration identified an additional proband, homozygous for a missense mutation, c.722T>C (p.Leu241Pro), in the same gene. The three affected members of the two families have been diagnosed with LCA. All have a distinct and unusual retinal appearance and a similar early onset of visual loss, suggesting both impaired retinal development and progressive retinal degeneration, involving both rod and cone pathways. Examination of heterozygotes revealed no ocular disease. This finding implicates Kir7.1 as having an important role in human retinal development and maintenance. This disorder adds to a small diverse group of diseases consequent upon loss or reduced function of inwardly rectifying potassium channels affecting various organs. The distinct retinal phenotype that results from biallelic mutations in KCNJ13 should facilitate the molecular diagnosis in further families.
Main Text
Retinitis pigmentosa (RP [MIM 268000]) is a heterogeneous group of genetic disorders that feature progressive loss of photoreceptor cells as a primary or secondary event.1 Consequent visual impairment usually manifests initially as night blindness and visual field constriction. However, great variability in age of onset, progression, fundus appearance, and final visual outcome is observed. Leber congenital amaurosis (LCA [MIM 204000]) represents the most severe end of the phenotypic spectrum, causing severe visual impairment from the first year of life.1,2 Genetic heterogeneity of RP and LCA is striking; inheritance is predominantly monogenic (autosomal and X linked), but polygenic and mitochondrial inheritance patterns, as well as cases with de novo mutations, have also been reported (RetNet and OMIM). The most prevalent and genetically heterogeneous RP subtype is the autosomal recessive form, with more than 30 genes implicated, accounting for less than 60% of autosomal recessive RP cases.3 LCA is typically inherited in an autosomal recessive manner and has been linked to more than 15 genes, together explaining ∼70% of families.3 Half of the genes mutated in LCA exhibit clinical heterogeneity with recessive mutations giving rise to both LCA and early onset RP (RetNet and OMIM). Identifying novel genes associated with these conditions is a major challenge, particularly as they are likely to be less prevalent and less obvious candidates than those already known. Importantly, their identification will offer further insight into the mechanisms that alter photoreceptor cell death kinetics.
In order to characterize the clinical consequences of mutations in genes previously associated with human ocular disease, as well as to identify novel disease-associated genes, families with evidence of parental consanguinity, presenting to the inherited eye disease clinics at Moorfields Eye Hospital, are recruited as part of an ongoing study. More than 100 families have been analyzed using single-nucleotide polymorphism (SNP) arrays, on the assumption that affected individuals will be autozygous for a chromosomal region surrounding the mutated gene (homozygosity mapping).4 One such family of Middle-Eastern origin (family A), including two affected family members, aged 34 (subject A-2; V-3 in Figure 1A) and 32 (subject A-3; V-4 in Figure 1A), is the basis of this report. The study was approved by the local research ethics committee, and all investigations were conducted in accordance with the principles of the Declaration of Helsinki; informed consent was obtained from all participating individuals. Both affected siblings in family A were noticed to have nystagmus and were diagnosed with LCA shortly after birth. Poor night vision and difficulty reading print from an early age was reported for both patients A-2 and A-3; gradual progression of visual problems affecting central and peripheral vision was also noted. Both had bilateral cataract surgery in their 20s. No other family history of retinal disease was reported. Visual acuities were 2.0 logMAR (logarithm of the minimal angle of resolution) in each eye of patient A-2 and 1.78 logMAR for the right and 1.48 for the left eye of patient A-3. Fundoscopy revealed significant pigment at the level of the retinal pigment epithelium (RPE) and in a configuration unlike that of typical RP (A-2 and A-3, Figure 2A).
Figure 1.

Identification of KCNJ13 Mutations in Individuals from Two Families with LCA
(A) Pedigree of family A, exome sequencing data from patient A-3, and Sanger sequencing results in affected brother A-2 and unaffected sister A-1. Pedigree reveals a recent common ancestor. Exome sequencing was performed in DNA from patient A-3. An R script assessing and plotting homozygous or heterozygous state of detected SNPs was composed. In chromosome 2, long regions of homozygosity were detected; SNPs within them are represented as red lines. Within the largest region of homozygosity, a nonsense mutation was identified in KCNJ13 (c.496C>T [p.Arg166X]). Gene structure of KCNJ13 is presented (reverse strand), comprising of three exons. Coverage depth distribution of the mapped reads along the three exons is shown (Savant Genome Browser; forward reads are in dark blue, reverse in light blue). Sequencing reads showing the homozygous nonsense mutation are presented (IGV viewer; 80 reads total: 11 forward and 69 reverse, 100% adenine). Electropherograms of DNA sequence surrounding the p.Arg166X mutation are shown for affected brother A-2 and unaffected sister A-1.
(B) Pedigree of family B, electropherograms of DNA sequence surrounding the c.722T>C (p.Leu241Pro) mutation in subjects B-3 and B-2, multiple alignment of ten KCNJ13 orthologs around the mutated amino acid, and schematic representation of Kir7.1. The alignment was performed with ClustalW using the following Ensemble transcripts: Homo sapiens ENST00000233826, Pan troglodytes ENSPTRT0000002418, Macaca mulatta ENSMMUT00000030719, Bos taurus ENSBTAT00000007700, Canis familiaris ENSCAFT00000018359, Mus musculus ENSMUST00000113212, Rattus norvegicus ENSRNOT00000021507, Gallus gallus ENSGALT00000002259, Xenopus tropicalis ENSXETT00000030393, and Danio rerio ENSDART00000063777. The schematic representation of Kir7.1 highlights the structural domains, predicted membrane topology, and the location of mutated residue identified.
Figure 2.
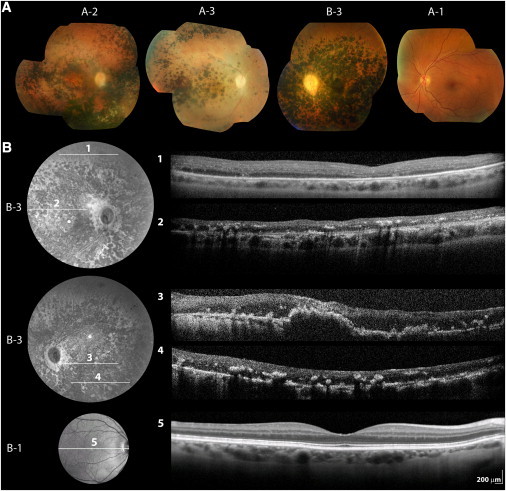
Retinal Imaging of Patients and Unaffected Family Members Carrying KCNJ13 Mutations
(A) Color photographs of right fundi of patient A-2 (aged 34) and patient A-3 (aged 32) in family A as well as left fundus of patient B-3 in family B (aged 33). Mormal left fundus of 39-year old unaffected c.496C>T (p.Arg166X) carrier (A-1, family A) is also shown.
(B) Infrared image and linear spectral domain optical coherence tomography scans (SD-OCT; Spectralis HRA+OCT, Heidelberg Engineering, Heidelberg, Germany) of right (1, 2) and left (3, 4) retina of patient B-3 in family B. Right retina of his 64-year-old unaffected mother (B-1) is also shown for comparison (5).
On some occasions, image quality was compromised by unstable fixation due to nystagmus or posterior capsule opacification.
Initially, DNA from patient A-2 was analyzed using an arrayed primer extension (APEX) microarray chip containing previously identified disease-causing variants in six LCA genes (LCA Chip, Asper Biotech, Tartu, Estonia); no disease-associated alleles were detected. As the phenotype exhibited some features previously reported in disease due to mutations in RDH12 ([MIM 608830], heavy pigmentation and macular atrophy) and CRB1 ([MIM 604210], nummular pigmented lesions), the coding region and intron-exon boundaries of these genes were sequenced and found to be normal. Subsequently, homozygosity mapping was performed. Genotypes of both patients A-2 and A-3 were generated using a microarray containing 1.8 million markers of genetic variation (more than 906,000 SNPs and more than 946,000 probes for copy number variant detection; Genome-Wide Human SNP Array 6.0, Affymetrix, Santa Clara, CA, USA) according to the manufacturer's recommendations. The birdsuite set of algorithms was used to detect and report SNP genotypes (Birdseed v2, threshold 0.0025), common copy number polymorphisms, and novel or rare copy number variants in the processed samples.5 A python script interacting with a MySQL database was written (PS) to detect shared regions of homozygosity and rank them by genetic distance; the Marshfield linkage map was used.6 Four chromosomal segments over 5 cM were identified (Table S1 available online), the largest being a 45 cM region located on 2q (flanked by SNPs rs10192834 and rs10199178).
As mutations were not detected in candidate genes observed within shared regions of homozygosity (SAG [MIM 181031] and PDE6D [MIM 602676] from the largest, chromosome 2q, region and WDR17 [MIM 609005] from a smaller region on 4q) and no pathogenic copy number variants were identified, exome sequencing was undertaken. Solution-phase hybrid capture with biotinylated RNA baits was used to target 38 Mb of genomic sequence from patient A-3 (SureSelect Human All Exon Kit, Agilent, Santa Clara, CA, USA). The enriched library was amplified and then sequenced on a single lane of a HiSeq2000 (Illumina, San Diego, CA, USA), generating 12.2 Gb of mappable sequence as 100 bp paired-end reads. Novoalign version 2.05 (Novocraft, Selangor, Malaysia) was used to align these reads to the hg19 reference sequence. The average sequencing depth on the 38 Mb target region was 61, with 85% of the bases covered with a depth of at least 10×. The ANNOVAR tool was used to annotate single-nucleotide variants and small insertions/deletions.7 A total of 16,649 calls with respect to the reference sequence were detected (Table 1). We used the exome data to identify regions of homozygosity, and these results were consistent with the findings from SNP genotyping. Owing to the prior belief that the causal variant is rare, we filtered calls that were found in the 1000 genomes data set. We subsequently filtered the remaining variants on the basis of the linkage data and the regions of homozygosity shared by both affected individuals (Table 1). Given the level of consanguinity in this family, we focused on homozygous calls. We identified a single loss-of-function homozygous rare variant, c.496C>T (p.Arg166X) in KCNJ13 (RefSeq accession number NM_002242.4). The presence of this mutation in homozygous state was confirmed by direct Sanger sequencing in DNA from both affected siblings (subjects A-2 and A-3). Parental DNA was not available for testing, but we were able to obtain samples from unaffected siblings. Subject A-1 (V-1 in Figure 1A), a 39-year-old female, was found to carry the mutation in heterozygous state. She was asymptomatic, and fundus examination, 55 degree fundus autofluorescence imaging, and spectral domain optical coherence tomography (SD-OCT; Spectralis HRA+OCT, Heidelberg Engineering, Heidelberg, Germany) were normal (A-1, Figure 2A).
Table 1.
Number of Variants Identified in Exome Sequencing Data of Patient A-3 and Filtering on the Basis of Genotyping Data
| Total | Nonsynonymous Heterozygous Variants Not in 1000 Genomes | Nonsynonymous Homozygous Variants Not in 1000 Genomes | Nonsense/Splice Site/Frameshifting Indel Heterozygous Variants Not in 1000 Genomes | Nonsense/Splice Site/Frameshifting Indel Homozygous Variants Not in 1000 Genomes | |
|---|---|---|---|---|---|
| Full exome | 16,649 | 329 | 42 | 80 | 37 |
| Within segments where both parental haplotypes are shared with affected sibling | 2,914 | 69 | 12 | 26 | 10 |
| Within regions of homozygosity | 577 | 2 | 6 | 0 | 4 |
| Within regions of homozygosity shared with affected sibling | 161 | 2 | 2a | 0 | 1 |
A threshold of 5 cM was used for segments where both parental haplotypes are shared in the two affected siblings. Regions of homozygosity were considered significant if larger than 3 cM. The 20100804 sequence and alignment release of the 1000 genomes project was used (628 individuals).
These two variants (p.Glu180Val in ARMC9 [rs1626451] and p.Arg302His in GEN1) cannot be fully excluded, but on the basis of physiological relevance, the loss-of-function KCNJ13 change is much more likely disease causing.
KCNJ13 is a three-exon gene encoding Kir7.1, a 360 amino acid low-conductance inwardly rectifying potassium channel (Kir) that functions as a homotetramer.8–10 Like other Kir subunits, Kir7.1 contains two transmembrane regions (M1 and M2), one pore-forming loop (H5), and cytosolic −NH2 and −COOH termini.11 The protein is localized at the plasma membrane of a variety of ion-transporting epithelia; high gene expression has been found in intestines, thyroid, choroidal plexus, prostate, kidney, and retina (Unigene,9,10,12). Within the retina, Kir7.1 is primarily localized to the apical membranes of RPE.12–14 The mutant allele, if the transcript is not subjected to nonsense-mediated decay, would produce a peptide lacking almost the entire −COOH intracellular segment of 204 amino acids. Such a protein product is unlikely to form functional homotetramers, as this portion is known to be critical for Kir channel subunit assembly.11 Additionally, the −COOH terminus is required for correct targeting of Kir7.1 to the apical membrane of epithelial cells15, and its absence in this mutant would likely lead to mislocalization.
Previously, a missense mutation in KCNJ13 (c.484C>T [p.Arg162Trp]) has been associated with a distinct ocular condition, autosomal dominant snowflake vitreoretinal degeneration (SVD [MIM 193230]), in a large pedigree comprised of 14 affected family members.14 The disorder's name is derived from the presence of small inner retinal crystalline-like deposits that are often observed in the fundi of affected individuals.16 Other ocular features include corneal guttae, early onset cataract, fibrillar vitreous degeneration, optic nerve head dysmorphism, normal macular appearance, and granular changes near the equatorial fundus.17 Increased predisposition to retinal detachment has also been reported.18 Visual acuity is normal, and retinal function testing reveals only mild abnormalities.18 Although the term SVD has been used in another eight families, currently, these do not appear to be the same condition by clinical or genetic evaluation.16,17,19–23 Clearly, the phenotype in family A is very different to that of SVD and includes severe visual impairment from an early age and a grossly abnormal fundus examination.
To assess whether mutations in KCNJ13 are a common cause of autosomal recessive retinal dystrophy, we performed bidirectional Sanger sequencing of the entire coding region and intron-exon boundaries of the gene in 333 additional unrelated patients (primer details are listed in Table S2). Among the patients, 132 were affected with recessive LCA or childhood onset retinal dystrophy; this panel was enriched for mutations in novel genes, as the patients' DNA had been screened and excluded for previously reported LCA-causing variants through a microarray chip (LCA Chip, Asper Biotech; 35% mutation detection rate; D.S.M, unpublished data). The remaining 201 individuals were diagnosed with autosomal recessive adult onset rod-cone or cone-rod dystrophy and had unknown molecular diagnosis.
A homozygous missense variant (c.722T>C [p.Leu241Pro]) was identified in a patient of white European descent (subject B-3, III-2 in Figure 1A; proband of family B) with a remarkably similar phenotype to that observed in family A. Segregation analysis confirmed heterozygosity for the p.Leu241Pro change in maternal (subject B-1, II-4 in Figure 1A) and paternal (subject B-2, II-5 in Figure 1A) DNA. This sequence change has not been previously reported (dbSNP and 1000 genomes) and was found to be absent from 382 ethnically matched control chromosomes (Human Random Control DNA Panels, Salisbury, UK). Although the patient is not knowingly consanguineous, his parents originate from the same isolated geographic area. It is therefore likely that the homozygous state at this locus is consequent upon the presence of a recent common ancestor. The mutated amino acid is highly conserved across a broad range of species (Figure 1B); among other human Kir family members, it is either conserved or replaced by another nonpolar hydrophobic residue (Ile, Met or Val; Figure S1). In order to further evaluate the physiological significance of the p.Leu241Pro change, we performed homology modeling (Figure 3). Leu241 is located in the cytosolic −COOH terminus of Kir7.1 and is the first of six amino acids to form a β sheet (βI).24 The mutant Pro241 residue does not take part in this β sheet formation, alters the orientation of the β sheet, and distorts a proximal similar structure, thus inducing a considerable conformational change (Figure 3).
Figure 3.

Predicted Protein Structure of a Human Kir7.1 Subunit
(A) Overlay of wild-type (WT; green) and mutant (yellow) Kir7.1 monomer models. Similar to other Kir family members, the Kir7.1 monomer is comprised of an α-helical membrane domain and an intracellular domain primarily composed of β sheets (asterisk highlights the position of the c.722T>C [p.Leu241Pro]).
(B) Detail of the region around the 241 amino acid. Distortion of the protein secondary structure is evident in the mutant when compared to the wild-type monomer.
(C and D) The side chains of the Leu241 (wild-type; blue) and Pro241 (mutant; red) residues are highlighted; the cyclic R group structure of proline bends the amino acid chain. (D) presents detail of (C).
The structural model was generated by using the SWISS-MODEL protein homology modeling server; the crystal structure of KirBac1.1 (Protein Data Bank code 1q7b) was used as a template. Similar findings were observed when the structure of cKir2.2 (Protein Data Bank code 3jycA) was used as a template. PyMOL (Delano Scientific, Portland, OR) was used to view the three-dimensional molecular structures.
The patient carrying this p.Leu241Pro homozygous variant (subject B-3), a 33-year old male, was noticed to have strabismus, nystagmus, and poor vision before the age of 1. At 2 years of age, the diagnosis of LCA was made. Only mild progression was reported, with night vision having most noticeably deteriorated. He is a moderate myope and had strabismus surgery at age 2, and an electroretinogram recorded at 10 years of age showed no consistent rod- or cone-driven responses. Bilateral cataract surgery was undertaken in his early 20s. There was no family history of retinal disease. Visual acuities were 1.45 logMAR in each eye. There was bilateral nystagmus, and confrontational visual field testing showed severe field loss but relative preservation of the inferior field. Fundus examination showed areas of nummular pigment at the level of the RPE, especially over the posterior pole (B-3, Figure 2A). In vivo cross-sectional imaging using SD-OCT revealed loss of outer retinal structures, thinning of the hyperreflective band corresponding to RPE/Bruch's membrane, and a coarse lamination pattern (B-3, Figure 2B). The distorted retinal microanatomy in an area of the fundus in which cell death was not evident may suggest that the early development of the human neurosensory retina is disturbed. The proband's 64-year-old father (subject B-2) and 62-year-old mother (subject B-1) were also examined, and fundoscopy, 55 degree fundus autofluorescence imaging, and SD-OCT were normal (B-1, Figure 2B).
Other sequence alterations identified in our patient cohort included three missense variants in the heterozygous state not present in dbSNP or 1000 Genomes Project (Table 2). A c.350A>G (p.Gln117Arg) change was found in a patient of white European ancestry with early onset retinal dystrophy and heavily pigmented fundi. Gln117 is located in an evolutionarily conserved region of the protein that corresponds to the pore-forming loop; this glutamine is also conserved among the majority of other Kir family members.9 As this patient's phenotype closely resembled that of affected individuals in families A and B, further molecular analysis was performed to investigate the possibility of a variant on the other allele. Initially, the untranslated exon 1 of KCNJ13 was screened by direct Sanger sequencing (Table S2) and found to be normal. Second, to investigate the possibility of a small heterozygous insertion or deletion spanning the translated region of KCNJ13, long-range PCR using primers encompassing exons 2 and 3 was performed (Table S2). Single PCR amplimers were obtained at the expected size of 3460 bp from both patient and control samples. Direct Sanger sequencing of the patient amplimer confirmed heterozygous variants in both exons 2 (p.Glu117Leu) and 3 (c.524C>T [p.Thr175Ile]; rs1801251), thus excluding the possibility of a large heterozygous deletion. Lastly we aimed to investigate whether monoallelic expression of the p.Gln117Arg variant was occurring by performing a series of reverse-transcriptase PCRs using a patient-derived leucoyte RNA sample. Unfortunately, KCNJ13 was not found to be expressed in either the patient or any control leukocyte cDNA sample tested.
Table 2.
Summary of DNA Variants in KCNJ13 Coding Region Reported Here and Elsewhere
|
Coding DNA Variants |
SIFT |
PolyPhen2 |
Blosum 62 Score (−4 to 4) |
Frequency (Chromosomes) |
Reference | ||||
|---|---|---|---|---|---|---|---|---|---|
| Nucleotide | Protein | Prediction | Tolerance Index (0 to 1) | Prediction | HumVar Score (0 to 1) | In patients with recessive retinal disease (n = 666+4) | In European controls (n = 382) | ||
| c.208G>T | p.Val70Phe | tolerated | 0.5 | benign | 0.043 | −1 | 0/670 | 0/382 | dbSNP 132 (rs79005659) |
| c.214G>T | p.Ala72Ser | intolerant | 0.02 | PRD | 0.978 | 1 | 0/670 | 0/382 | dbSNP 132 (rs77818131) |
| c.344A>G | p.Glu115Gly | intolerant | 0 | PRD | 0.991 | −2 | 0/670 | 0/382 | dbSNP 132 (rs112079468) |
| c.350A>G | p.Gln117Arg | intolerant | 0 | PRD | 0.945 | 1 | 1/670 | 0/382 | this study (LCA panel) |
| c.484C>T | p.Arg162Trp | intolerant | 0 | PRD | 0.972 | −3 | 0/670 | 0/382 | Hejtmancik et al., AJHG 2008 |
| c.485G>A | p.Arg162Gln | tolerated | 0.2 | PRD | 0.937 | 1 | 2/670 | 0/382 | this study (ARRP panel) |
| c.496C>T | p.Arg166X | not applicable | −4 | 4/670 | 0/382 | this study (family A) | |||
| c.524C>T | p.Thr175Ile | tolerated | 0.38 | benign | 0.001 | −1 | 212/670 | 109/382 | dbSNP 132 (rs1801251) |
| c.652T>C | p.Tyr218His | tolerated | 0.74 | benign | 0 | −2 | 0/670 | 0/382 | 1000 genomes |
| c.722T>C | p.Leu241Pro | intolerant | 0.02 | PRD | 0.997 | −3 | 2/670 | 0/382 | this study (LCA panel; family B) |
| c.827A>C | p.Glu276Ala | intolerant | 0.03 | PRD | 0.987 | −2 | 1/670 | 0/382 | this study (ARRP panel) |
| c.869C>A | p.Pro290Gln | intolerant | 0.02 | PRD | 0.945 | −1 | 0/670 | 0/382 | dbSNP 132 (rs17853727) |
| c.925G>T | p.Gly309Cys | intolerant | 0 | PRD | 0.99 | −3 | 0/670 | 0/382 | dbSNP 132 (rs17857137) |
SIFT results are reported to be tolerant if tolerance index ≥ 0.05 or intolerant if tolerance index < 0.05. Polyphen-2 appraises mutations qualitatively as benign, possibly damaging (POS), or probably damaging (PRD) based on the model's false positive rate. Blosum62 substitution matrix score positive numbers indicate a substitution more likely to be tolerated evolutionarily, and negative numbers suggest the opposite. The cDNA is numbered according to Ensembl transcript ID ENST00000233826, in which +1 is the A of the translation start codon.
A c.485G>A (p.Arg162Gln) was identified in heterozygous state in two unrelated male patients of Turkish ancestry (33 and 42 years old). Interestingly, this change occurs in the same amino acid as the dominant mutation causing SVD (p.Arg162Trp).14 Both patients carrying heterozygous p.Arg162Gln have been diagnosed with adult onset RP, are night blind, and have field of vision reduced to less than 10 degrees. Notably, one of the two (42 year old) is the product of a first cousin marriage; he is the only affected family member, and fundoscopy on his parents was unremarkable. The identification of a heterozygous KCNJ13 variant in this proband suggests that the region is not autozygous and is therefore unlikely to be the disease locus.4 Moreover, the phenotype is very different to that seen in families A and B, suggesting a different molecular pathology. A third missense variant (c.827A>C [p.Glu276Ala]), altering a highly conserved amino acid in the cytosolic −COOH terminus, was identified in a patient of South Asian origin with adult onset RP. We could not find further convincing evidence that biallelic mutations of KCNJ13 were responsible for the retinal degeneration in any of these three families.
Based on the remarkable similar phenotypic presentation of affected individuals in families A (homozygous p.Arg166X) and B (homozygous p.Leu241Pro), we hypothesize that the LCA phenotype exhibited is consequent upon lack of Kir7.1 channel function. This phenotype is very different from that of dominantly inherited SVD, previously reported in patients carrying a heterozygous p.Arg162Trp variant in KCNJ13. Therefore, we speculate that the p.Arg162Trp change in the heterozygous state has an entirely different in vivo effect on Kir7.1 function, perhaps exerting a gain-of-function effect. In vitro patch-clamp studies have demonstrated that the SVD-associated mutation, p.Arg162Trp, produces a nonselective cation current, unlike its wild-type counterpart, when overexpressed in CHO-K1 cells.14 However, it remains to be determined how this mutant protein behaves in the presence of wild-type Kir7.1. It is noteworthy that unaffected family members (subjects A-1, B-1, and B-2) carrying the LCA-associated variants in heterozygous state display no retinal abnormality or signs of SVD; this suggests that haploinsufficiency in KCNJ13 does not cause ocular disease. Future in-depth patch clamp experiments are necessary to further investigate the underlying pathophysiology of Kir7.1-related retinopathies, in particular the electrophysiological effects of p.Arg162Trp and p.Leu241Pro on Kir7.1 channel function with and without the presence of wild-type Kir7.1.
Retinal disease due to KCNJ13 mutations adds to a small group of disorders caused by reduced or abrogated expression of genes encoding inwardly rectifying potassium channel subunits. These diseases involve glucose homeostasis (familial hyperinsulinaemia hypoglycaemia type II [MIM 601820], KCNJ11 [MIM 600937]),25 renal sodium and potassium reabsorption (Bartter syndrome type II [MIM 241200], KCNJ1 [MIM 600359]),26 glial cell membrane resting potential (SeSAME/EAST syndrome [MIM 612780], KCNJ10 [MIM 602208])27, and excitability of cardiac and skeletal muscle (Andersen-Tawil syndrome [MIM 170390], KCNJ2 [MIM 600681]).28
As part of a large program of work on consanguineous families presenting with retinal disease, one family showed homozygosity for a nonsense mutation in KCNJ13. An extensive screen of 333 families identified a proband with a similar retinal phenotype to harbor a homozygous missense mutation. This strongly suggests that absence or deficiency of Kir7.1 causes LCA, and further studies of its function in the developing and adult retina of vertebrates, mammals, and humans will be of interest. Additionally, the particular, distinct retinal appearance and course of retinal degeneration in these two families will hopefully simplify the identification of further families presenting to other physicians.
Acknowledgments
We acknowledge the cooperation and help provided by the family members in this study. We thank Thomas Daskalakis for his key contribution in developing the python program, Geoffrey Maher and Eva Lenassi for their help with homology modeling and patient phenotyping, respectively, and Jill Urqhart and Sarah Daly at NIHR Manchester BRC for their technical assistance with Affymetrix array genotyping. We are grateful to colleagues who referred patients to us at MEH and those who contributed to the assembly of large panels of probands, particularly Louise Ocaka, Naushin Waseem, Bev Scott, Genevieve Wright, Sophie Devery, Michel Michaelides, Cathy Egan, Alan Bird, Graham Holder, Tony Robson, and Shomi Bhattacharya. We acknowledge the following sources of funding: British Retinitis Pigmentosa Society, Fight for Sight, Moorfields Eye Hospital Special Trustees, National Institute for Health Research UK (Moorfields Eye Hospital and Institute of Ophthalmology, London, UK), and the Foundation Fighting Blindness (USA). None of the authors declare a conflict of interest.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 genomes (628 individuals from the 20100804 sequence and alignment release of the 1000 genomes project), ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data/
Blossum62, http://www.ncbi.nlm.nih.gov/Class/FieldGuide/BLOSUM62.txt
IGV (Interactive Genomics Viewer), http://www.broadinstitute.org/software/igv/
Online Mendelian Inheritance in Man (OMIM), www.omim.org
Polymorphism Phenotyping (PolyPhen) version 2, http://genetics.bwh.harvard.edu/pph2/
Protein Data Bank, http://www.pdb.org/pdb/home/home.do
PyMOL, http://www.pymol.org/
Retinal Information Network (RetNet), http://www.sph.uth.tmc.edu/retnet/
SAMtools, http://samtools.sourceforge.net/
Savant Genome Browser, http://genomesavant.com/
Sorting intolerant from tolerant (SIFT), http://sift.bii.a-star.edu.sg/
Swiss-Model, http://swissmodel.expasy.org/
Unigene, http://www.ncbi.nlm.nih.gov/UniGene/
UniProt, http://www.uniprot.org/
References
- 1.Wright A.F., Chakarova C.F., Abd El-Aziz M.M., Bhattacharya S.S. Photoreceptor degeneration: genetic and mechanistic dissection of a complex trait. Nat. Rev. Genet. 2010;11:273–284. doi: 10.1038/nrg2717. [DOI] [PubMed] [Google Scholar]
- 2.den Hollander A.I., Roepman R., Koenekoop R.K., Cremers F.P.M. Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog. Retin. Eye Res. 2008;27:391–419. doi: 10.1016/j.preteyeres.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 3.den Hollander A.I., Black A., Bennett J., Cremers F.P. Lighting a candle in the dark: advances in genetics and gene therapy of recessive retinal dystrophies. J. Clin. Invest. 2010;120:3042–3053. doi: 10.1172/JCI42258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lander E.S., Botstein D. Homozygosity mapping: a way to map human recessive traits with the DNA of inbred children. Science. 1987;236:1567–1570. doi: 10.1126/science.2884728. [DOI] [PubMed] [Google Scholar]
- 5.Korn J.M., Kuruvilla F.G., McCarroll S.A., Wysoker A., Nemesh J., Cawley S., Hubbell E., Veitch J., Collins P.J., Darvishi K. Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat. Genet. 2008;40:1253–1260. doi: 10.1038/ng.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Broman K.W., Murray J.C., Sheffield V.C., White R.L., Weber J.L. Comprehensive human genetic maps: individual and sex-specific variation in recombination. Am. J. Hum. Genet. 1998;63:861–869. doi: 10.1086/302011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Döring F., Derst C., Wischmeyer E., Karschin C., Schneggenburger R., Daut J., Karschin A. The epithelial inward rectifier channel Kir7.1 displays unusual K+ permeation properties. J. Neurosci. 1998;18:8625–8636. doi: 10.1523/JNEUROSCI.18-21-08625.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krapivinsky G., Medina I., Eng L., Krapivinsky L., Yang Y., Clapham D.E. A novel inward rectifier K+ channel with unique pore properties. Neuron. 1998;20:995–1005. doi: 10.1016/s0896-6273(00)80480-8. [DOI] [PubMed] [Google Scholar]
- 10.Partiseti M., Collura V., Agnel M., Culouscou J.M., Graham D. Cloning and characterization of a novel human inwardly rectifying potassium channel predominantly expressed in small intestine. FEBS Lett. 1998;434:171–176. doi: 10.1016/s0014-5793(98)00972-7. [DOI] [PubMed] [Google Scholar]
- 11.Hibino H., Inanobe A., Furutani K., Murakami S., Findlay I., Kurachi Y. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol. Rev. 2010;90:291–366. doi: 10.1152/physrev.00021.2009. [DOI] [PubMed] [Google Scholar]
- 12.Nakamura N., Suzuki Y., Sakuta H., Ookata K., Kawahara K., Hirose S. Inwardly rectifying K+ channel Kir7.1 is highly expressed in thyroid follicular cells, intestinal epithelial cells and choroid plexus epithelial cells: implication for a functional coupling with Na+,K+-ATPase. Biochem. J. 1999;342:329–336. [PMC free article] [PubMed] [Google Scholar]
- 13.Kusaka S., Inanobe A., Fujita A., Makino Y., Tanemoto M., Matsushita K., Tano Y., Kurachi Y. Functional Kir7.1 channels localized at the root of apical processes in rat retinal pigment epithelium. J. Physiol. 2001;531:27–36. doi: 10.1111/j.1469-7793.2001.0027j.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hejtmancik J.F., Jiao X., Li A., Sergeev Y.V., Ding X., Sharma A.K., Chan C.C., Medina I., Edwards A.O. Mutations in KCNJ13 cause autosomal-dominant snowflake vitreoretinal degeneration. Am. J. Hum. Genet. 2008;82:174–180. doi: 10.1016/j.ajhg.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tateno T., Nakamura N., Hirata Y., Hirose S. Role of C-terminus of Kir7.1 potassium channel in cell-surface expression. Cell Biol. Int. 2006;30:270–277. doi: 10.1016/j.cellbi.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 16.Hirose T., Lee K.Y., Schepens C.L. Snowflake degeneration in hereditary vitreoretinal degeneration. Am. J. Ophthalmol. 1974;77:143–153. doi: 10.1016/0002-9394(74)90665-5. [DOI] [PubMed] [Google Scholar]
- 17.Edwards A.O. Clinical features of the congenital vitreoretinopathies. Eye (Lond.) 2008;22:1233–1242. doi: 10.1038/eye.2008.38. [DOI] [PubMed] [Google Scholar]
- 18.Lee M.M., Ritter R., III, Hirose T., Vu C.D., Edwards A.O. Snowflake vitreoretinal degeneration: follow-up of the original family. Ophthalmology. 2003;110:2418–2426. doi: 10.1016/S0161-6420(03)00828-5. [DOI] [PubMed] [Google Scholar]
- 19.Lang G.E., Laudi B., Pfeiffer R.A. [Autosome dominant vitreoretinal dystrophy with skeletal dysplasia in one generation] Klin. Monatsbl. Augenheilkd. 1991;198:207–214. doi: 10.1055/s-2008-1045954. [DOI] [PubMed] [Google Scholar]
- 20.Gheiler M., Pollack A., Uchenik D., Godel V., Oliver M. Hereditary snowflake vitreoretinal degeneration. Birth Defects Orig. Artic. Ser. 1982;18:577–580. [PubMed] [Google Scholar]
- 21.Robertson D.M., Link T.P., Rostvold J.A. Snowflake degeneration of the retina. Ophthalmology. 1982;89:1513–1517. doi: 10.1016/s0161-6420(82)34609-6. [DOI] [PubMed] [Google Scholar]
- 22.Pollack A., Uchenik D., Chemke J., Oliver M. Prophylactic laser photocoagulation in hereditary snowflake vitreoretinal degeneration. A family report. Arch. Ophthalmol. 1983;101:1536–1539. doi: 10.1001/archopht.1983.01040020538005. [DOI] [PubMed] [Google Scholar]
- 23.Chen C.J., Everett T.K., Marascalco D. Snowflake degeneration: an independent entity or a variant of retinitis pigmentosa? South. Med. J. 1986;79:1216–1223. doi: 10.1097/00007611-198610000-00006. [DOI] [PubMed] [Google Scholar]
- 24.Tao X., Avalos J.L., Chen J., MacKinnon R. Crystal structure of the eukaryotic strong inward-rectifier K+ channel Kir2.2 at 3.1 A resolution. Science. 2009;326:1668–1674. doi: 10.1126/science.1180310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thomas P., Ye Y., Lightner E. Mutation of the pancreatic islet inward rectifier Kir6.2 also leads to familial persistent hyperinsulinemic hypoglycemia of infancy. Hum. Mol. Genet. 1996;5:1809–1812. doi: 10.1093/hmg/5.11.1809. [DOI] [PubMed] [Google Scholar]
- 26.Simon D.B., Karet F.E., Rodriguez-Soriano J., Hamdan J.H., DiPietro A., Trachtman H., Sanjad S.A., Lifton R.P. Genetic heterogeneity of Bartter's syndrome revealed by mutations in the K+ channel, ROMK. Nat. Genet. 1996;14:152–156. doi: 10.1038/ng1096-152. [DOI] [PubMed] [Google Scholar]
- 27.Scholl U.I., Choi M., Liu T., Ramaekers V.T., Häusler M.G., Grimmer J., Tobe S.W., Farhi A., Nelson-Williams C., Lifton R.P. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc. Natl. Acad. Sci. USA. 2009;106:5842–5847. doi: 10.1073/pnas.0901749106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Plaster N.M., Tawil R., Tristani-Firouzi M., Canún S., Bendahhou S., Tsunoda A., Donaldson M.R., Iannaccone S.T., Brunt E., Barohn R. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen's syndrome. Cell. 2001;105:511–519. doi: 10.1016/s0092-8674(01)00342-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.