

Abstract
To identify rare causal variants in late-onset Parkinson disease (PD), we investigated an Austrian family with 16 affected individuals by exome sequencing. We found a missense mutation, c.1858G>A (p.Asp620Asn), in the VPS35 gene in all seven affected family members who are alive. By screening additional PD cases, we saw the same variant cosegregating with the disease in an autosomal-dominant mode with high but incomplete penetrance in two further families with five and ten affected members, respectively. The mean age of onset in the affected individuals was 53 years. Genotyping showed that the shared haplotype extends across 65 kilobases around VPS35. Screening the entire VPS35 coding sequence in an additional 860 cases and 1014 controls revealed six further nonsynonymous missense variants. Three were only present in cases, two were only present in controls, and one was present in cases and controls. The familial mutation p.Asp620Asn and a further variant, c.1570C>T (p.Arg524Trp), detected in a sporadic PD case were predicted to be damaging by sequence-based and molecular-dynamics analyses. VPS35 is a component of the retromer complex and mediates retrograde transport between endosomes and the trans-Golgi network, and it has recently been found to be involved in Alzheimer disease.
Main Text
Parkinson's disease (PD [MIM 168600]) is the second-most common neurodegenerative disorder; it affects 1%–2% of the population above the age of 60.1 It is characterized by degeneration of dopaminergic neurons in the nigrostriatal pathway and other monoaminergic cell groups in the brainstem. This degeneration leads to bradykinesia, resting tremor, muscular rigidity, and postural instability as well as nonmotor symptoms. Up to 20% of cases with PD are reported to be familial,2,3 but extended pedigrees with clear Mendelian inheritance are rare. Genetic studies have so far revealed mutations in five genes causing autosomal-recessive (PARK2 [MIM 602544], PINK1 [MIM 608309], PARK7 [MIM 602533]) or autosomal-dominant (SNCA [MIM 163890], LRRK2 [MIM 609007]) forms of PD.4–9 Whereas the autosomal-recessive forms with early onset and SNCA missense mutations or duplications10 are rare, a single LRRK2 mutation (RefSeq number NM_198578.3: c.6055G>A [p.Gly2019Ser]) accounts for approximately 1% of sporadic cases of European origin.11–13 A recent study revealed a strong association of PD with glucocerebrosidase (GBA) mutations in carriers for Gaucher [MIM 230800] disease, thus implicating a lysosomal enzyme in the pathogenesis of PD.14,15 Genome-wide association studies revealed several low-risk susceptibility loci, among them LAMP3 [MIM 605883] and HIP1R [MIM 605613], which have been reported to be implicated in the lysosomal pathway.16–18
We identified an Austrian family in which 16 members were affected by PD (family A, Figure 1). PD seemed to be inherited in an autosomal-dominant mode with high penetrance. Seven affected members were available for clinical and DNA investigations. Six of them exhibited at least three of the four cardinal signs of PD (akinesia, resting tremor, rigidity, and postural instability) and showed improvement after dopaminergic treatment. A single affected individual had displayed action tremors since childhood but developed L-Dopa-responsive resting tremors and akinesia only at the age of 62 years. The mean age of onset was 53 years (range 40–68 years) (Table 1). The clinical diagnosis of idiopathic PD was made by movement-disorder specialists who used UK brain bank criteria for PD.19 All participants gave written informed consent. The study was approved by the institutional review board of the Medizinische Universität Wien and the Hessische Landesärztekammer Wiesbaden.
Figure 1.
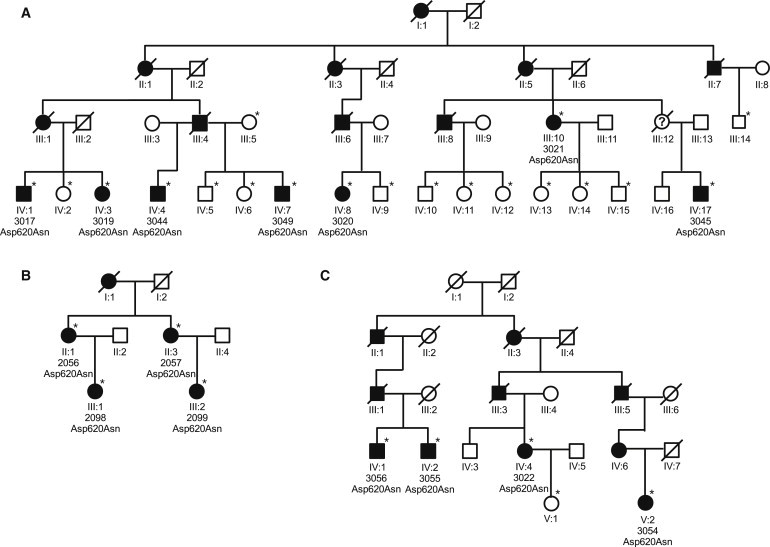
Pedigrees of Families A, B, and C
Unaffected family members are indicated by open symbols, affected members by closed symbols. Asterisks denote individuals genotyped for p.Asp620Asn. To maintain confidentiality, we have not shown genotypes of unaffected individuals. A question mark within a symbol denotes an unknown phenotype. Diagonal bars through symbols denote deceased individuals.
Table 1.
Clinical Findings for PD Patients Carrying Variants in VPS35
| Family | Patient | Variation | AaO | DD | IS | B | R | RT | PI | L-Dopa/DA | Other Features |
|---|---|---|---|---|---|---|---|---|---|---|---|
| A | 3017 | p.Asp620Asn | 48 | 7 | B | + | + | − | + | + | |
| A | 3019 | p.Asp620Asn | 40 | 5 | B | + | + | + | + | + | |
| A | 3020 | p.Asp620Asn | 46 | 7 | PI | + | + | − | + | + | |
| A | 3021 | p.Asp620Asn | 68 | 16 | PI | + | + | + | + | + | |
| A | 3049 | p.Asp620Asn | 49 | 4 | RT | + | + | + | − | + | |
| A | 3044 | p.Asp620Asn | 64 | 3 | PI | + | + | + | + | + | |
| A | 3045 | p.Asp620Asn | 63 | 1 | RT | + | − | + | − | + | action tremor since childhood |
| B | 2056 | p.Asp620Asn | 61 | 15 | RT | + | + | + | + | + | fluctuations, dyskinesias |
| B | 2057 | p.Asp620Asn | 56 | 8 | RT | + | + | + | + | + | fluctuations, dyskinesias |
| B | 2098 | p.Asp620Asn | 46 | 0.5 | RT | − | − | + | − | untreated | depression, action tremor, pathologic DAT SPECT |
| B | 2099 | p.Asp620Asn | 51 | 5 | B | + | + | + | − | + | fluctuations, pathologic DAT SPECT |
| C | 3022 | p.Asp620Asn | 61 | 5 | RT | + | + | + | − | + | dyskinesias |
| C | 3055 | p.Asp620Asn | 46 | 12 | RT | + | + | + | − | + | |
| C | 3054 | p.Asp620Asn | 53 | 9 | B | + | + | − | − | + | dyskinesias |
| C | 3056 | p.Asp620Asn | 43 | 10 | B | + | + | + | + | + | dyskinesias |
| 211 | p.Arg524Trp | 37 | 9 | MG | + | + | + | − | + | mild action tremor since youth; 75% motor improvement on levodopa-test; DBS for fluctuations and dyskinesias; pathologic DAT SPECT | |
| 524 | p.Leu774Met | 51 | 7 | RT | + | + | + | − | + | marked postural tremor | |
| 243 | p.Leu774Met | 73 | 9 | RT | + | + | + | + | + | dyskinesias, pathologic DAT SPECT | |
| 806 | p.Ile241Met | 72 | 2 | Postural tremor | + | − | + | + | + | hyposmia (6/12 sniffing sticks), DAT SPECT pathologic, pathologic crying | |
| 90/05 | p.Met57Ile | 62 | 13 | RT | + | + | + | + | + | dementia (MMSE 23), dysphagia and dysarthria, hyposmia by history, depression |
Abbreviations are as follows: AaO, age at onset; DD, disease duration in years; IS, initial symptoms; B, bradykiesia; R, rigidity; RT, resting tremor; PI, postural instability; L-Dopa/DA, response to L-Dopa and/or dopamine agonist; MG, micrographia; DBS, deep brain stimulation.
To identify the disease-causing variant, we selected two second cousins (#3017 and #3020) for exome sequencing. We assumed that any rare variants common in both individuals would be disease-causing candidates. Selecting distantly related members of the pedigree should minimize the proportion of alleles shared by descent. Exome sequencing was performed on a Genome Analyzer IIx system (Illumina) after in-solution enrichment of exonic sequences (SureSelect Human All Exon 38 Mb kit, Agilent). We sequenced two lanes of a flowcell for both samples, each as 54 bp paired-end runs. Read alignment was performed with BWA (version 0.5.8) to the human genome assembly hg19 (Table S1, available online). Single-nucleotide variants and small insertions and deletions (indels) were detected with SAMtools (v 0.1.7). We filtered called variants to exclude those present in 72 control exomes from patients with other unrelated diseases. We further excluded all variants that were present in dbSNP 131 and had an average heterozygosity of more than 0.02. Variant annotation was performed with custom scripts. This approach left ten heterozygous nonsynonymous variants shared by both affected individuals (Table 2; see also Table S2).
Table 2.
Exome Sequencing: Rare, Heterozygous, Nonsynonymous Variations Shared by Two Individuals of Pedigree A
| Gene | Position (hg19) | dbSNP | Transcript |
Variations |
Control Genotypes |
Segregation | |||
|---|---|---|---|---|---|---|---|---|---|
| Nucleotide | Amino Acid | 1/1 | 1/2 | 2/2 | |||||
| PLK3 | chr1:45270359 | NM_004073.2 | c.1543T>A | p.Ser515Thr | 669 | 0 | 0 | 4 of 7 | |
| C8A | chr1:57383357 | rs41285938 | NM_000562.2 | c.1723C>T | p.Pro575Ser | 5 of 7 | |||
| ADCY10 | chr1:167787479 | rs41270737 | NM_018417.4 | c.4313A>G | p.Asn1438Ser | 2 of 7 | |||
| LAMB2 | chr3:49166460 | NM_002292.3 | c.1724G>A | p.Arg575Gln | 647 | 28 | 0 | 5 of 7 | |
| NOM1 | chr7:156762317 | NM_138400.1 | c.2503G>A | p.Ala835Thr | 670 | 0 | 0 | 3 of 7 | |
| KIF22 | chr16:29816237 | NM_007317.1 | c.1780G>A | p.Asp594Asn | 665 | 6 | 0 | 6 of 7 | |
| SEZ6L2 | chr16:29899021 | NM_012410.2 | c.947G>A | p.Arg316His | 660 | 4 | 0 | 7 of 7 | |
| VPS35 | chr16:46696364 | NM_018206.4 | c. 1858G>A | p.Asp620Asn | 1069a | 0 | 0 | 7 of 7 | |
| NLRP1 | chr17:5421150 | NM_001033053.2 | c.3985G>A | p.Val1329Ile | 666 | 4 | 0 | 3 of 7 | |
| NEURL4 | chr17:7221197 | NM_001005408.1 | c.4109G>A | p.Arg1370Gln | 3 of 7 | ||||
Rare variations revealed by exome sequencing were checked in 670 controls (KORA S4) by MALDI-TOF analysis. The variant allele was denoted as “2,” the reference allele as “1.”
This number includes additional 554 Austrian control individuals investigated by a TaqMan genotyping assay. Segregation shows the number of affected pedigree A individuals who carry the variant allele.
Only a single heterozygous variant in the VPS35 gene (RefSeq number NM_018206.4: c.1858G>A [p.Asp620Asn]) fulfilled two further criteria of being possibly causative: (1) it was found in all seven affected members investigated and (2) was absent in approximately 680 KORA S4 general-population samples (Tables 2 and 3).20 We next screened 486 unrelated PD patients from Austria for the p.Asp620Asn variant by MALDI-TOF mass spectroscopy (Sequenom MassArray system). We detected two additional index patients carrying this mutation (families B and C; Figure 1 and Table 1). The variant was detected in all eight affected individuals investigated in both families. It was not present in a second set of 554 Austrian controls or in an additional 1014 KORA-AGE controls (Table 3). The variant was further detected in three clinically unaffected family members in families A, B, and C. Because the unaffected individuals are all younger than 60 years of age, either they are all presymptomatic or the mutation is nonpenetrant in these subjects.
Table 3.
Summary of the Samples Used in This Study
| Cohort | Sample Size | Mean Age (SD) | Females/Males |
|---|---|---|---|
| Austrian PD casesa | 486 | 58.7 (11.3) | 172/314 |
| German PD casesb | 376 | 71.1 (9.4) | 119/257 |
| KORA S4 controlsc | 680 | 54.7 (11.9) | 280/400 |
| KORA-AGE controlsd | 1014 | 76.0 (6.6) | 508/505 |
| Austrian controlse | 554 | 46 (15.2) | 254/300 |
Patients presenting with atypical or secondary (e.g., vascular) parkinsonian disorders as well as patients with known mutations were excluded.
The Austrian cases were recruited at the Department of Neurology, Medizinische Universität Wien, Vienna, as well as in affiliated departments on a consecutive basis. A positive family history for PD was reported from 131 patients. A positive family history was defined by at least one other affected first- or second-degree related family member.
The German PD population originated from the Paracelsus-Elena Klinik, Kassel, a hospital specializing in movement disorders.
This control population was recruited from the KORA S4 survey, comprising individuals who were aged 25–74 years and were examined during 1999–2001.
The KORA-AGE samples were collected in 2009 as a gender- and age-stratified subsample of the KORA S1–S4 studies comprising participants born before 1944. KORA S1–S4 surveys comprise four independent cross-sectional population-based studies in the region of Augsburg, Southern Germany, and were conducted in 5 year intervals. Patients for whom PD was suspected on the basis of questionnaire data were excluded.
These control samples were recruited through the Department of Neurology, Medical University of Vienna, as subjects without known history of a neurological disorder and included, for example, blood donors or unrelated companions or spouses of patients.
Cross-species alignment of VPS35 from plants, fungi, invertebrates, and vertebrates showed complete conservation of amino acid Asp620 (Figure S1). The likely consequence of the p.Asp620Asn variant was predicted to be damaging by PolyPhen2,21 SNAP,22 and SIFT.23 We therefore concluded that the variant p.Asp620Asn is indeed very likely to be causative for PD in families A, B, and C.
To determine whether the variant p.Asp620Asn occurred on the same haplotype, we genotyped 20 individuals from families A–C with oligonucleotide SNP arrays (HumanOmni2.5-Quad, Illumina). Haplotyping and linkage analysis were performed with the Merlin software.24 The haplotypes carrying the variant p.Asp620Asn in families A–C are depicted in Table S3. Family A and B shared a common haplotype across 21 Mb between markers rs1072594 and rs4444336. Family C, however, showed only a common region of 65 kb across VPS35. Different alleles were located at markers rs56168099 and rs74459547, 25 kb upstream and 11 kb downstream of VPS35, respectively (Table S3). Because the two intragenic markers did not differ, we could not determine whether the three families shared an old common haplotype or whether the mutation has recently arisen on two different haplotypes.
To assess the prevalence of other VPS35 mutations among PD cases and the general population, we screened all 17 coding exons for variations by dye-binding/high-resolution DNA melting curve analysis (LightScanner HR I 384, Idaho Technology) in 860 cases (484 Austrian and 376 German cases) and 1014 controls. For controls, we used a population-based cohort (KORA AGE) with a mean age of 76 years but excluded eight individuals known to be on medications for PD (Table 3). Exons 2 to 12 are located within a region that is duplicated 12 Mb upstream. Primers were designed to specifically amplify these exons (Table S4). The screening revealed six further rare coding SNVs in addition to p.Asp620Asn (Table 4). Including p.Asp620Asn, we identified four different nonsynonymous missense variants only present in cases, two only present in controls, and one present in cases and controls. Two of the variants unique to PD cases were predicted to be damaging by all three methods (c.1858G>A [p.Asp620Asn]; c.1570C>T [p.Arg524Trp]), and one was predicted by PolyPhen2 to be possibly damaging (c.723T>G, p.Ile241Met). The other variants were predicted to be benign by all methods. Family information was only available for the patient carrying the p.Arg524Trp variant. The only available family member was her mother, aged 74 years. She was found to also carry the variant and showed mild extrapyramidal signs, including intermittent resting tremor of the left fingers and mild postural tremor of both upper limbs, but no bradykinesia. However, a DAT SPECT examination showed normal striatal binding, excluding the possibility of an early stage of PD in this subject. Of note, the screening did not reveal any common nonsynonymous coding SNVs. Furthermore, common nonsynonymous coding SNVs were not found in the 72 control exomes from patients with other unrelated diseases, nor were any recorded in the dbSNP database (version 131).
Table 4.
Rare VPS35 Variants in Cases and Controls
| ID Cases | KORA AGE Controls | Heterozygous Nucleotide Change | Amino Acid Change | Predicted Impact on Protein | Exon/ Intron | Genomic Position (hg19, chr16) |
KORA S4 Controls |
||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1/1 | 1/2 | 2/2 | |||||||||
| Nonsynonymous | (i) | (ii) | (iii) | ||||||||
| - | 1 | c.151G>A | p.Gly51Ser | + | + | + | 3 | 46,716,,039 | |||
| 90/05 | - | c.171G>A | p.Met57Ile | + | + | + | 3 | 46,716,019 | 670 | 0 | 0 |
| - | 1 | c.245C>G | p.Thr82Arg | + | + | + | 4 | 46,715,367 | |||
| 806 | - | c.723T>G | p.Ile241Met | ± | + | + | 7 | 46,711,308 | 667 | 0 | 0 |
| [211] | - | c.1570C>T | p.Arg524Trp | − | − | − | 13 | 46,702,919 | 671 | 0 | 0 |
| [Families A-C] | - | c.1858G>A | p.Asp620Asn | − | − | − | 15 | 46,696,364 | 669 | 0 | 0 |
| 243, 524 | 2 | c.2320C>A | p.Leu774Met | + | + | + | 17 | 46,694,455 | |||
| Synonymous | |||||||||||
| 53097 | - | c.492A>G | p.Glu164Glu | 5 | 46,714,597 | 671 | 0 | 0 | |||
| - | 1 | c.954A>T | p.Gly315Gly | 9 | 46,708,542 | ||||||
| 53496 | - | c.1881C>T | p.Ala627Ala | 15 | 46,696,341 | 668 | 5 | 0 | |||
| 45, 117, 53626 | 1 | c.2145A>G | p.Leu715Leu | 16 | 46,695,696 | 666 | 2 | 0 | |||
| 53667 | - | c.2241C>T | p.Ile747Ile | 17 | 46,694,534 | 667 | 2 | 0 | |||
| 53063 | - | c.2346A>G | p.Glu782Glu | 17 | 46,694,429 | 671 | 0 | 0 | |||
| - | 1 | c.2361G>A | p.Glu787Glu | 17 | 46,694,414 | ||||||
| Noncoding | |||||||||||
| 2212 | 2 | c.1-35C>T | 5′UTR | 46,723,080 | 667 | 2 | 0 | ||||
| - | 2 | c.1-29C>T | 5′UTR | 46,723,074 | |||||||
| 95, 2206 | 3 | c.3+24A>G | 1 | 46,723,019 | 662 | 6 | 0 | ||||
| 159, 528 | 1 | c.102+33G>A | 2 | 46,717,387 | 668 | 2 | 0 | ||||
| [157, 2023] | - | c.103-77T>C | 3 | 46,716,164 | 668 | 0 | 0 | ||||
| - | 1 | c.199+9T>G | 3 | 46,715,982 | |||||||
| 213 | - | c.506+6T>C | 5 | 46,714,577 | 644 | 0 | 0 | ||||
| 53093 | - | c.720+18C>T | 6 | 46,712,773 | |||||||
| - | 1 | c.914+38T>C | 8 | 46,710,457 | |||||||
| 52824 | - | c.1161-87A>C | 10 | 46,706,471 | |||||||
| 52791 | - | c.1161-70G>A | 10 | 46,706,454 | 668 | 0 | 0 | ||||
| - | 1 | c.1368+16C>T | 11 | 46,706,161 | |||||||
| [2028] | - | c.1369-11G>A | 12 | 46,705,783 | 669 | 0 | 0 | ||||
| - | 1 | c.1525-17delT | 12 | 46,702,985 | |||||||
| - | 1 | c.1647+14T>C | 13 | 46,702,828 | |||||||
| 320 | - | c.2212-45T>C | 16 | 46,694,608 | 670 | 0 | 0 | ||||
| [352] | - | c.2391+7A>G | 3′UTR | 46,694,377 | |||||||
| - | 1 | c.2391+8A>G | 3′UTR | 46,694,376 | |||||||
Variants for 863 cases and 1014 KORA AGE controls were determined by dye-binding/high-resolution DNA melting curve analysis and confirmed by Sanger sequencing. The table lists the case ID and the number of detected variant alleles of the cases and KORA AGE samples, respectively. Genotypes of identified variants were further investigated by MALDI-TOF analysis in approximately 680 KORA S4 controls. For the KORA S4 samples, the variant allele was denoted as “2,” the reference allele as “1.” cDNA numbering is based on reference gene NM_018206.4 for VPS35, where +1 corresponds to the A of ATG start translation codon. Familial cases are given in square brackets. Three methods were used for predicting the impact of SNPs on the protein. (1) PolyPhen2, (2) SNAP, and (3) SIFT; “+” indicates a benign impact, “±” indicates a possibly damaging impact, and “−” indicates a damaging impact. We detected a further nonsynonymous variant (c.1093C>T [p.Arg365Cys], genomic position 46,708,293) in a patient carrying two PARKIN variants (c.exon3_4del and p.Arg275Trp). This variant was not present in 670 KORA S4 and 1014 KORA AGE controls. It is predicted to be possibly damaging by all three methods. This patient's brother is also affected by PD. He carries the 2 PARKIN variants but not the VPS35 variant.
VPS35 is a component of the retromer complex and is involved in retrograde transport from the endosomes back to the trans-Golgi network.25 This multi-protein complex consists of the cargo-recognition VPS26-VPS29-VPS35 heterotrimer and a membrane-targeting heterodimer or homodimer of SNX1 and/or SNX2 (vps5).25,26 All proteins involved are evolutionarily conserved and have been previously described in Saccharomyces cerevisiae. The best characterized cargo proteins of the retromer complex are the cation-independent mannose 6-phosphate receptor (CI-MPR)27 and Vps10p in mammals and Saccharomyces cerevisiae, respectively; these proteins transport hydroxylases to the lysosomes or lysosomal vacuoles. Recently, additional cargo proteins and functions of VPS35 have been described.28,29 Most interesting in our context is the involvement of the retromer into the retrograde transport of SORL1, a VPS10P-domain receptor protein that has been implicated in Alzheimer disease.30,31 The crystal structure of the C-terminal part of VPS35 has been resolved.32 The three variants p.Asp620Asn, p.Arg524Trp, and p.Leu774Met are located in this part of the protein, and we have investigated their impact on protein stability by using molecular dynamics (MD) simulations. We manually introduced the mutations to the crystal structure and modeled the side chains by using scwrl 4.0.33 All MD simulations were performed via GROMACS 4.5,34 with the all-atom force field AMBER0335 and the water model TIP3P36 as parameters. All three proteins are found on the edge of helices interacting with VPS29. Wild-type residue Asp620 forms frequent hydrogen bonds (HBs) to Lys622, but these bonds are less frequent in the p.Asp620Asn variant (Figure 2A). Similarly, Arg524 is involved in a triple HB network together with residues Asp483 and Asp486, but this network is broken by the introduction of p.Arg524Trp (Figure 2B). Both changes result in the loss of salt bridges and cause the protein to be locally more flexible, as shown by root-mean-square fluctuation (RMSF) profiles (Figure S2). In contrast to the effect predicted for p.Arg524Trp and p.Asp620Asn, the p.Leu774Met variant was not predicted to have a strong impact on protein stability.
Figure 2.

Hydrogen-Bonding Capacities for Wild-Type Asp620 and Arg524 and the Variants p.Asp620Asn and p.Arg524Trp
Hydrogen bonds (HB) are shown as red dashed lines. Asp60 and Arg524 are in green; p.Asp620Asn and p.Arg524Trp are in orange.
(A) Asp620 forms a HB to Lys622 and shows an additional salt-bridge interaction. p.Asp620Asn forms fewer HBs, and no electrostatic interaction is possible.
(B) Arg524 forms a HB network with Asp483 and Asp486. This network is broken by the p.Arg524Trp substitution.
In summary, we identified rare VPS35 missense variants that are potentially pathogenic. One of these variants, p.Asp620Asn, cosegregates with late-onset PD in three unrelated families. The observation that the three families share only a small common haplotype across VPS35, the high conservation of VPS35, the predicted structural changes, and the protein's known involvement in lysosomal trafficking together provide strong support for the p.Asp620Asn variant's being causative for late-onset PD, although we identified only a single familial mutation. The penetrance of p.Asp620Asn is high but not complete and might be lower for the other variants. The proportion of PD caused by VPS35 variants is expected to be low. Although exome sequencing provides perfect access to rare-variant detection, both large families and large collections of cases and controls remain a crucial resource for the identification of disease genes.
Acknowledgments
We thank all patients and their families for participating in this study. We also thank C. Fischer and B. Schmick for technical assistance and S. Schmidegg, S. Hoedl, and M. Guger for clinical examination of family A. This work was supported by a grant from the German Ministry for Education and Research (01GR0804-4). The KORA study was financed by the Helmholtz Zentrum München, the German Federal Ministry of Education and Research, the State of Bavaria, the German National Genome Research Network (NGFNplus: 01GS0823), and the Munich Center of Health Sciences (MCHealth) as part of LMUinnovativ. M.N.O., S.C.R., and B.R. were supported by the Alexander von Humboldt Foundation.
Contributor Information
Alexander Zimprich, Email: alexander.zimprich@meduniwien.ac.at.
Tim M. Strom, Email: timstrom@helmholtz-muenchen.de.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
ExonPrimer, http://ihg.helmholtz-muenchen.de/exonprimer.html
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Lang A.E., Lozano A.M. Parkinson's disease. First of two parts. N. Engl. J. Med. 1998;339:1044–1053. doi: 10.1056/NEJM199810083391506. [DOI] [PubMed] [Google Scholar]
- 2.Bonifati V., Fabrizio E., Vanacore N., De Mari M., Meco G. Familial Parkinson's disease: A clinical genetic analysis. Can. J. Neurol. Sci. 1995;22:272–279. doi: 10.1017/s0317167100039469. [DOI] [PubMed] [Google Scholar]
- 3.Payami H., Larsen K., Bernard S., Nutt J. Increased risk of Parkinson's disease in parents and siblings of patients. Ann. Neurol. 1994;36:659–661. doi: 10.1002/ana.410360417. [DOI] [PubMed] [Google Scholar]
- 4.Zimprich A., Biskup S., Leitner P., Lichtner P., Farrer M., Lincoln S., Kachergus J., Hulihan M., Uitti R.J., Calne D.B. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 5.Bonifati V., Rizzu P., van Baren M.J., Schaap O., Breedveld G.J., Krieger E., Dekker M.C., Squitieri F., Ibanez P., Joosse M. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–259. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- 6.Kitada T., Asakawa S., Hattori N., Matsumine H., Yamamura Y., Minoshima S., Yokochi M., Mizuno Y., Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 7.Paisán-Ruíz C., Jain S., Evans E.W., Gilks W.P., Simón J., van der Brug M., López de Munain A., Aparicio S., Gil A.M., Khan N. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 8.Polymeropoulos M.H., Lavedan C., Leroy E., Ide S.E., Dehejia A., Dutra A., Pike B., Root H., Rubenstein J., Boyer R. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 9.Valente E.M., Abou-Sleiman P.M., Caputo V., Muqit M.M., Harvey K., Gispert S., Ali Z., Del Turco D., Bentivoglio A.R., Healy D.G. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science. 2004;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- 10.Johnson J., Hague S.M., Hanson M., Gibson A., Wilson K.E., Evans E.W., Singleton A.A., McInerney-Leo A., Nussbaum R.L., Hernandez D.G. SNCA multiplication is not a common cause of Parkinson disease or dementia with Lewy bodies. Neurology. 2004;63:554–556. doi: 10.1212/01.wnl.0000133401.09043.44. [DOI] [PubMed] [Google Scholar]
- 11.Gilks W.P., Abou-Sleiman P.M., Gandhi S., Jain S., Singleton A., Lees A.J., Shaw K., Bhatia K.P., Bonifati V., Quinn N.P. A common LRRK2 mutation in idiopathic Parkinson's disease. Lancet. 2005;365:415–416. doi: 10.1016/S0140-6736(05)17830-1. [DOI] [PubMed] [Google Scholar]
- 12.Nichols W.C., Pankratz N., Hernandez D., Paisán-Ruíz C., Jain S., Halter C.A., Michaels V.E., Reed T., Rudolph A., Shults C.W., Parkinson Study Group-PROGENI investigators Genetic screening for a single common LRRK2 mutation in familial Parkinson's disease. Lancet. 2005;365:410–412. doi: 10.1016/S0140-6736(05)17828-3. [DOI] [PubMed] [Google Scholar]
- 13.Di Fonzo A., Rohé C.F., Ferreira J., Chien H.F., Vacca L., Stocchi F., Guedes L., Fabrizio E., Manfredi M., Vanacore N., Italian Parkinson Genetics Network A frequent LRRK2 gene mutation associated with autosomal dominant Parkinson's disease. Lancet. 2005;365:412–415. doi: 10.1016/S0140-6736(05)17829-5. [DOI] [PubMed] [Google Scholar]
- 14.Sidransky E., Nalls M.A., Aasly J.O., Aharon-Peretz J., Annesi G., Barbosa E.R., Bar-Shira A., Berg D., Bras J., Brice A. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N. Engl. J. Med. 2009;361:1651–1661. doi: 10.1056/NEJMoa0901281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aharon-Peretz J., Rosenbaum H., Gershoni-Baruch R. Mutations in the glucocerebrosidase gene and Parkinson's disease in Ashkenazi Jews. N. Engl. J. Med. 2004;351:1972–1977. doi: 10.1056/NEJMoa033277. [DOI] [PubMed] [Google Scholar]
- 16.Simón-Sánchez J., Schulte C., Bras J.M., Sharma M., Gibbs J.R., Berg D., Paisan-Ruiz C., Lichtner P., Scholz S.W., Hernandez D.G. Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat. Genet. 2009;41:1308–1312. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Satake W., Nakabayashi Y., Mizuta I., Hirota Y., Ito C., Kubo M., Kawaguchi T., Tsunoda T., Watanabe M., Takeda A. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson's disease. Nat. Genet. 2009;41:1303–1307. doi: 10.1038/ng.485. [DOI] [PubMed] [Google Scholar]
- 18.Nalls M.A., Plagnol V., Hernandez D.G., Sharma M., Sheerin U.M., Saad M., Simón-Sánchez J., Schulte C., Lesage S., Sveinbjörnsdóttir S., International Parkinson Disease Genomics Consortium Imputation of sequence variants for identification of genetic risks for Parkinson's disease: A meta-analysis of genome-wide association studies. Lancet. 2011;377:641–649. doi: 10.1016/S0140-6736(10)62345-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hughes A.J., Daniel S.E., Kilford L., Lees A.J. Accuracy of clinical diagnosis of idiopathic Parkinson's disease: A clinico-pathological study of 100 cases. J. Neurol. Neurosurg. Psychiatry. 1992;55:181–184. doi: 10.1136/jnnp.55.3.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wichmann H.E., Gieger C., Illig T. KORA-gen–resource for population genetics, controls and a broad spectrum of disease phenotypes. Gesundheitswesen. 2005;67(Suppl. 1):26–30. doi: 10.1055/s-2005-858226. [DOI] [PubMed] [Google Scholar]
- 21.Ramensky V., Bork P., Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bromberg Y., Rost B. SNAP: predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res. 2007;35:3823–3835. doi: 10.1093/nar/gkm238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin—Rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 25.Bonifacino J.S., Rojas R. Retrograde transport from endosomes to the trans-Golgi network. Nat. Rev. Mol. Cell Biol. 2006;7:568–579. doi: 10.1038/nrm1985. [DOI] [PubMed] [Google Scholar]
- 26.Rojas R., Kametaka S., Haft C.R., Bonifacino J.S. Interchangeable but essential functions of SNX1 and SNX2 in the association of retromer with endosomes and the trafficking of mannose 6-phosphate receptors. Mol. Cell. Biol. 2007;27:1112–1124. doi: 10.1128/MCB.00156-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Damen E., Krieger E., Nielsen J.E., Eygensteyn J., van Leeuwen J.E. The human Vps29 retromer component is a metallo-phosphoesterase for a cation-independent mannose 6-phosphate receptor substrate peptide. Biochem. J. 2006;398:399–409. doi: 10.1042/BJ20060033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Braschi E., Goyon V., Zunino R., Mohanty A., Xu L., McBride H.M. Vps35 mediates vesicle transport between the mitochondria and peroxisomes. Curr. Biol. 2010;20:1310–1315. doi: 10.1016/j.cub.2010.05.066. [DOI] [PubMed] [Google Scholar]
- 29.Korolchuk V.I., Schütz M.M., Gómez-Llorente C., Rocha J., Lansu N.R., Collins S.M., Wairkar Y.P., Robinson I.M., O'Kane C.J. Drosophila Vps35 function is necessary for normal endocytic trafficking and actin cytoskeleton organisation. J. Cell Sci. 2007;120:4367–4376. doi: 10.1242/jcs.012336. [DOI] [PubMed] [Google Scholar]
- 30.Willnow T.E., Petersen C.M., Nykjaer A. VPS10P-domain receptors—Regulators of neuronal viability and function. Nat. Rev. Neurosci. 2008;9:899–909. doi: 10.1038/nrn2516. [DOI] [PubMed] [Google Scholar]
- 31.Rogaeva E., Meng Y., Lee J.H., Gu Y., Kawarai T., Zou F., Katayama T., Baldwin C.T., Cheng R., Hasegawa H. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat. Genet. 2007;39:168–177. doi: 10.1038/ng1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hierro A., Rojas A.L., Rojas R., Murthy N., Effantin G., Kajava A.V., Steven A.C., Bonifacino J.S., Hurley J.H. Functional architecture of the retromer cargo-recognition complex. Nature. 2007;449:1063–1067. doi: 10.1038/nature06216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krivov G.G., Shapovalov M.V., Dunbrack R.L., Jr. Improved prediction of protein side-chain conformations with SCWRL4. Proteins. 2009;77:778–795. doi: 10.1002/prot.22488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hess B., Kutzner C., van der Spoel D., Lindahl E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 35.Duan Y., Wu C., Chowdhury S., Lee M.C., Xiong G., Zhang W., Yang R., Cieplak P., Luo R., Lee T. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003;24:1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- 36.Mahoney M.W. A five-site model for liquid water and the reproduction of the density anomaly by rigid, nonpolarizable potential functions. J. Chem. Phys. 2000;112:8910–8922. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.