

Abstract
Genetic mutations responsible for oblique facial clefts (ObFC), a unique class of facial malformations, are largely unknown. We show that loss-of-function mutations in SPECC1L are pathogenic for this human developmental disorder and that SPECC1L is a critical organizer of vertebrate facial morphogenesis. During murine embryogenesis, Specc1l is expressed in cell populations of the developing facial primordial, which proliferate and fuse to form the face. In zebrafish, knockdown of a SPECC1L homolog produces a faceless phenotype with loss of jaw and facial structures, and knockdown in Drosophila phenocopies mutants in the integrin signaling pathway that exhibit cell-migration and -adhesion defects. Furthermore, in mammalian cells, SPECC1L colocalizes with both tubulin and actin, and its deficiency results in defective actin-cytoskeleton reorganization, as well as abnormal cell adhesion and migration. Collectively, these data demonstrate that SPECC1L functions in actin-cytoskeleton reorganization and is required for proper facial morphogenesis.
Introduction
Orofacial clefts are a common congenital facial defect that affect on average one in 800 live births.1 Cleft lip with or without cleft palate (CL/P) comprises the majority of orofacial clefts, and a number of contributory genes, including several from recent genome wide association studies,2 have been identified.1 In contrast, having been definitively described by Tessier in 1976,3 oblique facial clefts (ObFCs) represent a rare form of orofacial clefts (0.25% of the total), for which the genetic basis is largely unknown. In fact, it has even been postulated that ObFCs occur only from mechanical tearing and not from primary defects in facial morphogenesis.4
Facial morphogenesis requires coordinated morphogenetic movements in the three dimensions of the developing facial prominences and the precise control of basic cellular processes, including cell proliferation, differentiation, migration, and adhesion. Membranous bones of the head and facial skeleton, as well as cartilage, dental pulp, adipocytes, dermis, and some connective tissue, derive from cranial neural crest (CNC), one of four types of neural crest (reviewed by Hall5). Recent vertebrate studies show that patterning of the mesoderm-derived facial musculature is regulated by the CNC during facial development.6 During the fourth week of gestation, CNC migrates from the dorsal neural tube into the branchial arches and facial region to comprise the ectomesenchyme of the facial prominences.5 Although the cellular basis for ObFC remains unclear, it is attractive to hypothesize that a failure of CNC migration, proliferation or maxillary (MxP) and lateral nasal prominence (LNP) fusion could be responsible.
CNC migration commences with the epithelial-mesenchymal transition and delamination of ectomesenchymal neural crest (NC) cells from the neural folds, and it involves their movement along paths dictated by anatomical constraints, the local extracellular matrix (ECM), and differentiation cues.5 Mechanistically, this migration has both intrinsic and extrinsic components. Intrinsic components include the ability of CNC cells to reorganize their cytoskeleton in response to their environment and thus alter cell shape and motility. The migratory behavior of NC cells is initiated and maintained by several signaling pathways, which include noncanonical Wnt, integrin, ephrinB, neuregulin, and endothelin molecules (reviewed in Nelms and Labosky7). The extrinsic component, which includes the ECM composition and the physical environment through which the CNC migrates, provides the directional cues to the integrin-bearing CNC cell as it moves from the neural tube toward the branchial arches.5–7 The growth of the branchial arches then gives rise to the facial prominences, which eventually fuse to form the face.
The analysis of balanced chromosomal rearrangements that are linked to human facial dysmorphoses such as ObFC offers an unbiased approach to identifying genes that are required for proper formation of the human face. Here, we report identification of SPECC1L mutations in two patients with ObFC. SPECC1L is a previously uncharacterized gene predicted to encode a coiled-coil domain (CCD) containing protein. Our cellular analyses show that SPECC1L is a “cross-linking” protein that functionally interacts with both microtubules and with the actin cytoskeleton. Furthermore, SPECC1L knockdown analysis in zebrafish, Drosophila, and mammalian cells shows a role for SPECC1L in cell migration and adhesion, downstream of the integrin, Ca2+ and non-canonical Wnt signaling pathways, each of which engenders reorganization of the actin cytoskeleton. Collectively, these data provide a genetic basis for ObFC and implicate defective SPECC1L-mediated cell migration and adhesion as a potential underlying mechanism.
Material and Methods
ObFC Patients and Controls
DGAP177 was previously described8 with a balanced translocation between chromosomes 1 and 22, [46,XX,t(1;22)(q21;q12)], which has been revised herein to [46,XX,t(1;22)(q21.3;q11.23)]. This white female from the U.S. exhibited bilateral oromedial-canthal (Tessier IV) clefts, and a head CT scan revealed normal brain parenchyma but severe bilateral ocular hypoplasia. DGAP177 also had a unilateral calcaneovarus foot deformity. Twenty-three other patients with ObFC were also ascertained: seven from Brazil, 12 from the Philippines, three from the U.S., and one from Denmark. The two sequence variants identified in two different male Brazilian patients are NM_015330.2 (SPECC1L_v001) c.569C>T (p.Thr190Met) and c.1244A>C (p.Gln415Pro). DNA samples from 258 population-matched Brazilian and 50 North American normal individuals were used as controls. Parental DNA samples were obtained for the Brazilian patients with the two missense changes (p.Thr190Met and p.Gln415Pro). The identity of the parental samples to the patient harboring the de novo c.1244A>C mutation was confirmed with a panel of microsatellite markers from the Combined DNA Index System (CODIS) database.
Cytogenetics
Peripheral blood specimens were collected with institutional-review-board-approved informed consent for the Developmental Genome Anatomy Project (DGAP) through the Partner's Healthcare System, as well as through the Universidade de Saõ Paulo (Brazil), the University of Iowa, and Rigshospitalet (Copenhagen, Denmark). Cell transformation aimed at generating DGAP177 lymphoblastoid cells was performed via standard protocols.9 Metaphase preparations from the cell lines were used in all fluorescence in situ hybridization (FISH) experiments. Probes were derived from bacterial artificial chromosomes (BACs), fosmids, or long-range PCR products. Probes mapping to the region of the cytogenetically determined breakpoints were selected from the University of California Santa Cruz Human Genome Browser. Probe DNA was labeled by nick translation with SpectrumGreen or SpectrumOrange (Abbott, Abbott Park, IL), denatured, and hybridized to chromosomal preparations fixed to microscope slides.9 At least ten metaphases were scored per hybridization. Comparative genome hybridization (CGH) microarray analysis was performed with the Spectral Genomics (Houston, TX) 1 Mb BAC array.
Reverse-Transcriptase Polymerase Chain Reaction
Total RNA was extracted from DGAP177 lymphoblasts with the RNeasy Mini Kit (QIAGEN, Germantown, MD), and cDNA was synthesized with the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA). This cDNA was subsequently used for conventional polymerase chain reaction (PCR) as well as quantitative reverse-transcriptase PCR (qRT-PCR) for SPECC1L transcripts with the iQ SYBR Green Supermix (Bio-Rad, Hercules, CA) on an iQ LightCycler (Bio-Rad, Hercules, CA). Primers used resided in exons 8 and 14 (see Table S1 available online).
In Situ Hybridization
Whole-mount and section in situ experiments were performed with two different probes for Specc1l. Primers that incorporated either SP6 or T7 promoter sequence upstream of the gene-specific sequences were used for amplification of the cDNA products that were subsequently used as templates for riboprobe generation via in vitro transcription coupled with UTP-digoxigenin labeling. All primer sequences are provided in Table S1.
Zebrafish Husbandry and Morpholino Knockdown
Zebrafish were maintained according to protocols of the institutional animal care and use committee. Morpholinos (MOs) (GeneTools, Philomath, OR) designed against the ATG and exon 1 splice sites of Specc1l-chr5, Specc1l-chr8, and Specc1l-chr21 (Table S1) were injected into one-cell-stage embryos.
Drosophila Knockdown
The Drosophila conditional RNAi fly for the SPECC1L ortholog, CG13366, was obtained from the Vienna Drosophila Resource Center (stock #108092). The stock is from the RNAi library generated by insertion of the antisense sequence via phiC integrase.10 The conditional fly was crossed with the daughterless-GAL4 strain (Bloomington stock #5460) for a ubiquitous knockdown and with A9-GAL4 (Bloomington stock #8761) for a wing-imaginal-disc-specific knockdown.
Cloning and Mutagenesis
Full-length and C-terminal calponin homology domain (CHD) truncated cDNA (designated ΔCHD) of Specc1l (NM_153406) were cloned into a Gateway pENTR/SD/D-TOPO vector (Invitrogen, Carlsbad, CA) and transferred to a Gateway-compatible destination vector with a C-terminal GFP tag (pcDNA-DEST47, Invitrogen, Carlsbad, CA). p.Thr190Met and p.Gln415Pro mutations were created with the QuikChange Mutagenesis kit (Stratagene, La Jolla, CA) according to the manufacturer's protocol. The cDNA fragment encoding amino acids 887–997 of SPECC1L was cloned into the pGEX6p-2 vector to create a GST-tagged peptide that was then used to generate a rabbit polyclonal antibody (Covance, Princeton, NJ). The specificity of the SPECC1L antibody was confirmed in two different experiments: (1) overexpressed Specc1l-GFP was specifically detected by the antibody, and (2) immunoblotting of SPECC1L-knockdown cell lysate showed a decrease in a protein band of the expected size of approximately 120 kDa (see Supplemental Data).
Cell Culture
DGAP177 lymphoblastoid cells were grown in RPMI 1640 media (GIBCO-Invitrogen, Carlsbad, CA) supplemented with 10% FBS. Human 293T kidney epithelial and U2OS osteosarcoma cells (kindly provided by the late Dr. Priscilla A. Schaffer, University of Arizona) were cultured in standard DMEM supplemented with 10% FBS. Cell transfections were carried out in 24-well plates with Superfect (QIAGEN, Germantown, MD) reagent according to the manufacturer's protocol. Immunostaining was carried out on coverslips coated with poly-L-lysine. In brief, cells were fixed in 4% PFA for 10 min, followed by blocking in PBS with 1% goat serum and 0.1% Tween. SPECC1L and γ-tubulin antibodies were used at a 1:4000 dilution, and remaining antibodies were used at a 1:1000 dilution. F-actin staining was carried out with rhodamine-labeled phalloidin (Cytoskeleton Inc., Denver, CO), diluted 1:500 in PBS, for 30 min. After staining, coverslips were mounted in VectaShield containing DAPI (Vector Labs, Burlingame, CA). Nocodazole treatment aimed at reducing the microtubule-associated background was carried out as described.11
Lentiviral Knockdown
Two lentiviral preparations, each containing a different short hairpin, V2LHS_41578 (B8) or TRCN0000112962 (C1) against human SPECC1L (Open Biosystems, Huntsville, AL), were used for transduction of 293T and U2OS cells. A lentiviral preparation containing a short hairpin against GFP was used as a control. In brief, 1.5 × 106 cells seeded onto a 10 cm dish were transduced with ∼5 × 105 TU of lentiviral supernatant in the presence of 8 μg/ml polybrene for 6 hr at 37°C. Transduced cells were then selected by culture in 3 μg/ml puromycin for approximately two weeks. Upon confluence, cells were plated at very low densities (∼1:5000 dilution) so that clonal cell lines would be created. Single colonies were isolated with sterile cloning disks (Thermo Fisher Scientific, Waltham, MA) and expanded.
Wound-Repair, or Scratch, Assay
U2OS wild-type and SPECC1L-kd cells (5 × 105) were plated in a 6-well plate and grown to confluence. Cells were then scratched with a pipette tip, and the media were replaced. Images were taken at pre-marked locations after defined periods of time. For Wnt treatments, we used conditioned media obtained from cells overexpressing Wnt5a or Wnt3a (ATCC, Washington, D.C.) to treat cells for 30 min after scratch treatment.12 Cells were then fixed and stained as described above.
Results
Identification of SPECC1L Mutations in ObFC Patients
We ascertained a white female (DGAP177) with the previously reported apparently balanced chromosomal translocation 46,XX,t(1;22)(q21;q12). She was born with bilateral, symmetrical ObFC (oromedial-canthal, Tessier IV) and also exhibited microphthalmia, cleft palate, and talipes calcaneovarus deformity.8 FISH analyses with probes generated from BAC and fosmid clones showed that the 1q21 breakpoint region (chr1:149,713,417–149,752,534, identified with the March 2006 NCBI36/hg18 assembly), identified from a split signal with fosmid G248P84603E5, does not span any known genes. In contrast, the breakpoint on 22q, reassigned as 22q11.23 after FISH analyses, was identified from a split signal with fosmid G248P81272C11, which spans the 3′-region of SPECC1L (chr22:22,996,866–23,143,221) and part of the downstream gene, ADORA2A (chr22:23,121,123–23,158,636) (Figures 1A and 1B). The breakpoint region was refined to exclude ADORA2A by Southern blot analysis with probes generated from the proximal end of the split fosmid G248P81272C11. Aberrant bands in DGAP177 DNA after AvaII, PstI, and SacI digestion narrowed the breakpoint to a 5 kb region in intron 14 of the calponin homology and coiled-coil-domain-containing gene, SPECC1L (chr22:23,122,059–23,128,018) (Figure 1B).
Figure 1.
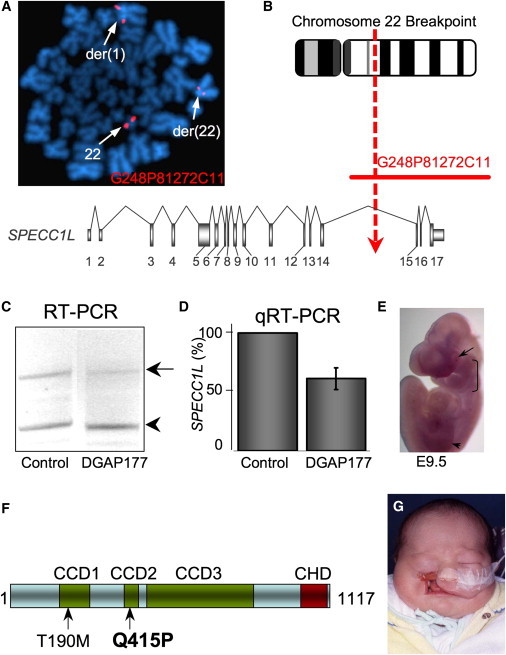
SPECC1L Disruption and Mutation in ObFC Patients
(A and B) FISH analysis of metaphase chromosomes from DGAP177 lymphoblasts and schematic of fosmid G248P81272C11 spanning the 22q breakpoint, which disrupts SPECC1L in a 5 kb region of intron 14.
(C and D) RT-PCR and qRT-PCR show haploinsufficiency of SPECC1L transcripts (arrow; relative to ACTB transcripts, arrowhead) in RNA from DGAP177 and control lymphoblasts.
(E) Whole-mount in situ hybridization shows Specc1l expression in maxillary and lateral nasal prominences (bracket), eyes (arrow), and limbs (arrowhead) at E9.5.
(F) SPECC1L representation showing the position of the p.Thr190Met polymorphism and p.Gln415Pro missense mutation. CCD, coiled coil domain; CHD, calponin homology domain.
(G) Tessier IV type cleft in the patient with the p.Gln415Pro mutation.
Comparative genomic hybridization (CGH) microarray analysis at an average 1 Mb genome-wide resolution did not reveal any copy-number variants (CNVs) of clinical significance. Because of the proximity of SPECC1L to genomic regions for DiGeorge (DGS [MIM 188400]), Velocardiofacial (VCFS [MIM 192430]), and distal 22q11.2 deletion (MIM 611867) syndromes, we performed a detailed literature search and did not find any reported cases of deletions that extended into SPECC1L.13 Using the Database of Genomic Variants and DECIPHER, we found two reports of small duplications14,15 and one instance of a small deletion15 in normal samples. This deletion encompassed only exon 2 of SPECC1L (Variation_32455). Interestingly, exon 2 of SPECC1L encodes only a part of the 5′ UTR and is shown to be spliced variably in the RefSeq database.
We confirmed SPECC1L haploinsufficiency at the RNA level in DGAP177 lymphoblastoid cells by using conventional and quantitative real-time RT-PCR (Figures 1C and 1D). In situ hybridization in embryonic day 9.5–10.5 (E9.5–E10.5) mouse embryos showed Specc1l expression in the developing maxillary prominence (MxP) and the lateral nasal process (LNP), which fail to fuse in ObFC, as well as in the limbs and eye (Figure 1E; see also Figure S1).
To establish further involvement of SPECC1L in the pathogenesis of ObFC, we sequenced its coding region in 23 ObFC patients, including seven from Brazil, 12 from the Philippines, one from Denmark, and three from the U.S. The sequencing analysis identified heterozygous missense changes, c.569C>T (p.Thr190Met) and c.1244A>C (p.Gln415Pro), in two Brazilian male patients with Tessier IV clefting. These variants, p.Thr190Met and p.Gln415Pro, occur, respectively, in the first and second of three predicted CCDs of SPECC1L (Figure 1F). Parental samples were subsequently obtained and sequenced for both cases. The p.Thr190Met change was found in the clinically unaffected maternal sample and was thus assigned as a likely nonfunctional polymorphism. However, the p.Gln415Pro change was not detected in either unaffected parent and therefore arose de novo. The p.Gln415Pro variant was also not present in 258 population-matched Brazilian controls (516 chromosomes), in 50 North American controls, or in any of the SNP databases. Moreover, p.Gln415Pro is predicted by PolyPhen, SIFT, and pMUT mutation evaluation algorithms16 to have a deleterious effect on protein function. The patient with the p.Gln415Pro mutation has severe unilateral Tessier IV clefting on the right and mild Tessier VII clefting on the left, normal eyes, and no other birth defects (Figure 1G).
Knockdown of a Zebrafish SPECC1L Homolog Results in Loss of Facial Structures
To obtain further evidence of a role for SPECC1L in vertebrate facial morphogenesis, we performed a morpholino (MO)-based knockdown of the three previously uncharacterized SPECC1L homologs in zebrafish; these homologs are located on fish chromosomes 5, 8, and 21 (Figure S2). Knockdown of the zebrafish chromosome 5 homolog specifically resulted in a “faceless” phenotype characterized by a MO-dose-dependent loss of jaw and facial structures, whereas knockdown of either of the other two homologs alone did not produce an observable phenotype even at the highest MO concentration tested (Figures 2A–2F and not shown).
Figure 2.
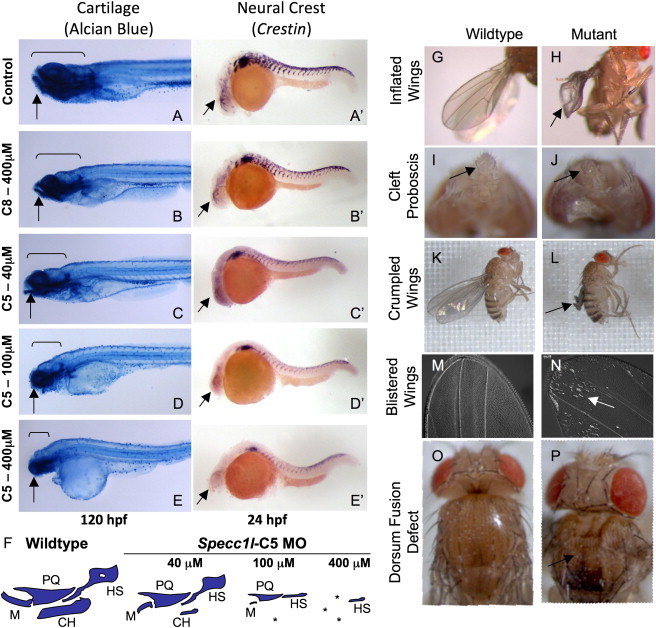
Specc1l Knockdown Results in Zebrafish “Faceless” and Drosophila “Split Discs” Phenotypes
(A–E and A′–E′) Lateral views of 120 hpf fish stained with alcian blue for cartilage (A–E) and of 24 hpf fish stained for crestin (A′–E′), a neural crest (NC) marker, after morpholino (MO) treatment. Control (A and A′) and chromosome-8-homolog (C8) MO at the highest dose (B and B′) appear normal. In contrast, increasing doses of the zebrafish chromosome-5-homolog (C5) MO results in diminishing sizes of facial structures (C–E; arrows and brackets) and poor NC migration to the anterior branchial arches (C′–E′; arrows).
(F) Schematic representation of the effect on jaw cartilages after MO treatment. M, Meckel's; PQ, palatoquadrate; CH, ceratohyal; HS, hyosymplectic; ∗, missing cartilage.
(G–P) The Drosophila SPECC1L-ortholog (CG13366) conditional RNAi line (108092) was crossed with da-GAL4 and A9-GAL4 flies for ubiquitous (G–J) or wing-imaginal-disc-specific (K–P) knockdown. Resulting flies were flightless with inflated (G and H), crumpled (K and L), or blistered (M and N) wings. Eclosing flies with ubiquitous knockdown failed to survive, probably as a result of feeding problems from the defective proboscis (I and J). Some flies with severely affected wings also showed a mild cleft dorsum (O and P).
Skeletal components of the vertebrate jaw and face derive from CNC that migrates to the first and second branchial arches.17,18 In zebrafish, the first mandibular arch gives rise to Meckel's (M) and palatoquadrate (PQ) cartilages that form the lower and upper jaws, respectively. The second hyoid arch, in contrast, gives rise to cartilages that support the jaw, including ceratohyal (CH) and hyosymplectic (HS) cartilages.18 In our faceless morphant, at low concentrations of the chromosome 5 MO, both Meckel's and ceratohyal cartilages were reduced in size. However, at high concentrations, all facial cartilage was absent and only a small remnant of the hyosymplectic cartilage remained (Figure 2F). We also examined NC migration in the zebrafish by using crestin as a marker. As expected from the facial cartilage defects, NC migration into the branchial arches was significantly impaired. At more posterior embryonic levels, crestin expression was also reduced, but the underlying pattern of NC migration was retained (Figures 2A′–2E′). These data suggest a function for SPECC1L in facial development across at least two vertebrate classes, mammals and fish.
Knockdown of a SPECC1L Ortholog in Drosophila Phenocopies Integrin Signaling Mutants
To identify molecular pathways perturbed by SPECC1L deficiency, we knocked down the previously uncharacterized Drosophila ortholog of SPECC1L, CG13366, by using an inducible RNA hairpin10 driven by the ubiquitous daughterless-Gal4 driver. A small number of severely affected flies failed to eclose and died as pharate adults. Eclosing flies were infertile and flightless and had a range of wing defects, including inflated, crumpled, or blistered wings (Figures 2G and 2H). These flies died 2–3 days after eclosion as a result of distal proboscis defects (Figures 2I and 2J) and feeding difficulties. Using a A9-Gal4 driver, we also knocked down CG13366 specifically in wing imaginal discs. All resulting flies were flightless; approximately 70% had partially or completely uninflated, crumpled wings (Figures 2K and 2L), and the remaining 30% exhibited wings that were “held out” and had blisters (Figures 2M and 2N). Among flies with uninflated, crumpled wings, 70% also had a mild cleft dorsum (Figures 2O and 2P).
The Drosophila wing blade forms when two layers of cells derived from the dorsal and ventral regions of the wing imaginal disc migrate, appose, and subsequently adhere via integrin-mediated cell adhesion. Indeed, the completely penetrant wing phenotypes in CG13366-knockdown flies strikingly phenocopy Drosophila mutants that are known to affect the integrin signaling pathway; such mutants include inflated (integrin αPS2), multiple edematous wings (integrin αPS1), wing blister (laminin α1,2), and blistery (tensin) (reviewed in references19–21). Together with the zebrafish and mouse data, these results suggest a role for SPECCIL in integrin-mediated migration and adhesive interactions of cell populations in the developing human facial prominences.
Specc1l-GFP Expression Stabilizes Microtubules
We extended our functional analysis by expressing a GFP-tagged mSpecc1l in U2OS osteosarcoma cells. Expression of Specc1l-GFP stabilized a subset of microtubules, as detected by enhanced β-tubulin immunostaining, and this effect was prevented by pretreatment with the microtubule-depolymerizing agent nocodazole (Figure 3A; see also Figure S3A). Immunostaining also showed that the Specc1l-stabilized microtubules contained acetylated α-tubulin (Figure 3B), a known component of stabilized microtubules.22 Because Specc1l contains a C-terminal CHD that in other contexts can facilitate actin binding,23 we also examined actin staining. Specc1l-GFP expression severely altered the cellular actin cytoskeleton and the overall cell shape (Figure 3C). Next, we tested whether the Specc1l Thr190Met-GFP and Gln415Pro-GFP variants also stabilized acetylated α-tubulin-containing microtubules. Consistent with the view that c.569C > T represents a neutral polymorphism, Thr190Met-GFP expression produced no visible effect. In contrast, expression of a truncated form of Specc1l lacking the CHD (ΔCHD-GFP) nearly abolished formation of stabilized microtubules, suggesting that Specc1l stabilizes microtubules via an interaction with the actin cytoskeleton (Figures 3D–3F). Strikingly, Gln415Pro-GFP expression severely impaired formation of stabilized acetylated-α-tubulin-containing microtubules, although not to the same degree as ΔCHD-GFP, supporting the view that this variant constitutes a pathologic mutation (Figure 3G).
Figure 3.
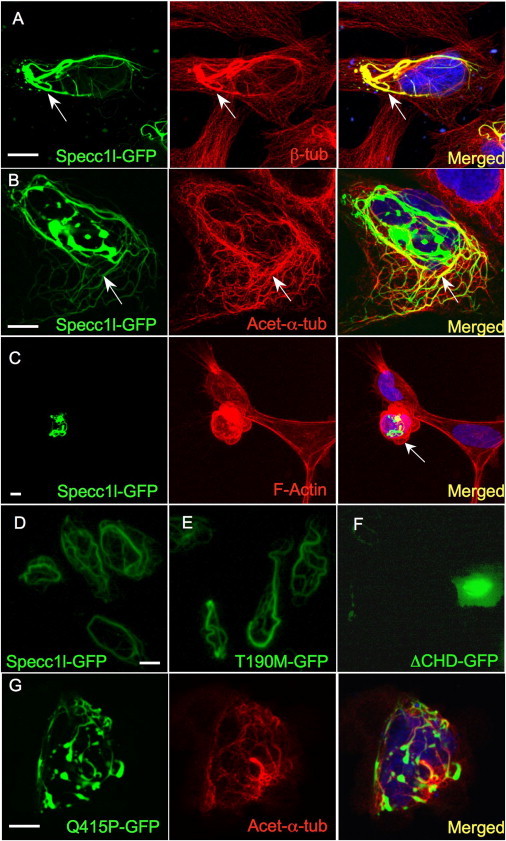
Specc1l-GFP Stabilizes Acetylated α-Tubulin-Containing Microtubules
(A) GFP-tagged Specc1l (green) in U2OS cells stabilizes a subset of microtubules (arrows) stained with β-tubulin (red).
(B) The stabilized subset colocalizes with acetylated α-tubulin-containing microtubules (yellow, arrows).
(C) Specc1l-GFP distorts the actin cytoskeleton (red) and cell shape (arrow).
(D–F) In contrast to wild-type Specc1l-GFP (A and D) and Thr190Met-GFP (E), which show no apparent difference in microtubule stabilization, a C-terminal truncation (ΔCHD) completely abolishes stabilization (F).
(G) Notably, the Gln415Pro-GFP mutant significantly reduces stabilization of acetylated α-tubulin-containing microtubules. Scale bars represent 10 μm.
Cellular SPECC1L Colocalizes with Microtubules and the Actin Cytoskeleton
We generated an antibody to SPECC1L (Figure S2B) to determine its cellular expression. As predicted by Specc1l-GFP expression, endogenous SPECC1L in U2OS cells showed a microtubule-type cytoplasmic expression pattern (Figures 4A and 4B; arrows). We also coimmunostained U2OS cells for SPECC1L and for acetylated α-tubulin. Indeed, SPECC1L localization overlapped with that of acetylated α-tubulin in the mitotic spindles during cell division and in gap junctions, which are involved in cell adhesion24 (Figures 4C and 4D). SPECC1L also showed punctate expression (Figures 4A and 4B; arrowheads) that localized in a ring around γ-tubulin-labeled centrioles in the presumptive microtubule-organizing center (MTOC) (Figures 4E and 4F). Such a pattern suggests association with the negative ends of spindle microtubules and a role in spindle orientation and cell polarity, both critical factors in directional migration.25
Figure 4.
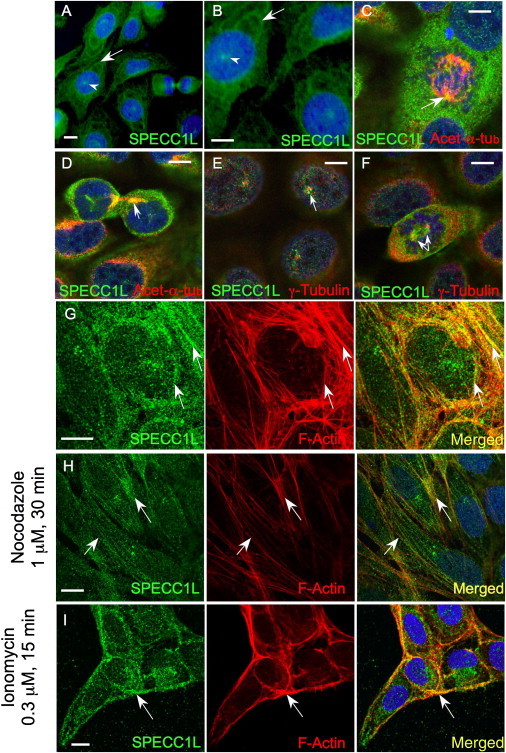
SPECC1L Colocalizes with Tubulin and Actin
(A and B) Endogenous SPECC1L shows a microtubule-type cytoplasmic expression pattern (arrows); a site of intranuclear expression is also detected (arrowheads; also see [E] below).
(C and D) SPECC1L colocalizes with acetylated α-tubulin (yellow and orange) in the mitotic spindle during cytokinesis (C) and in gap junctions (D).
(E and F) Punctate expression of SPECC1L is seen in the presumptive microtubule-organizing center (MTOC) surrounding centrioles stained with γ-tubulin.
(G and H) Colocalization with F-actin (G, arrows) is better visualized after nocodazole treatment (H, arrows).
(I) After ionomycin treatment, SPECC1L relocates with F-actin to the cell membrane. Scale bars represent 10 μm.
Given that SPECC1L contains a CHD that facilitates actin binding23 and that Specc1l overexpression distorts the actin cytoskeleton (Figure 3C), we asked whether SPECC1L also localizes to the actin cytoskeleton. First, we pretreated U2OS cells with nocodazole to minimize expression from microtubule-associated SPECC1L. This manipulation revealed a filamentous pattern of SPECC1L, which partly colocalized with phalloidin-stained F-actin fibers (Figures 4G and 4H). Second, to assess whether the actin cytoskeletal association was functional, we treated the cells with ionomycin, which increases intracellular calcium and rapidly reorganizes the actin cytoskeleton, with F-actin fibers translocating to the cell membrane. Ionomycin treatment produced a concomitant translocation of SPECC1L to the cell membrane (Figure 4I), suggesting a role in actin-cytoskeleton reorganization. These cellular localization studies implicate SPECC1L as a novel “cross-linking” protein that interacts both with microtubules and with the actin cytoskeleton.26 Moreover, taken together with the zebrafish and Drosophila data, the findings suggest that SPECC1L function in actin-cytoskeleton reorganization is necessary for cell adhesion and migration.
Knockdown of SPECC1L in Mammalian Cells Results in Cell-Adhesion and -Migration Defects
To test this hypothesis further, we analyzed DGAP177 lymphoblastoid cells and RNAi-based SPECC1L-knockdown clonal lines established from 293T and U2OS cells (Figure S4). First, we noted that DGAP177 lymphoblastoid cells failed to form the large clumps that characterize Epstein-Barr virus (EBV)-transformed cells27 (Figures 5A and 5B). EBV transformation causes increased adhesion as a result of upregulation of integrin LFA-1 (leukocyte function antigen-1), which consists of integrins αL (ITGAL) and β2 (ITGB2).27 Indeed, consistent with a defect in integrin-based cell adhesion, ITGAL expression was reduced by more than 2-fold (p < 0.03) in DGAP177 cells compared with control lymphoblastoid cells (Figure 5C). Second, we observed that although 293T SPECC1L-knockdown (SPECC1L-kd) cells attached well to culture plates when initially seeded (Figures 5F and 5G), they failed—in contrast to control cells—to remain adherent upon reaching confluency (Figures 5H and 5I). To test whether this adhesion failure was due to a defect in extracellular matrix (ECM) production or to an inability of the cells to bind ECM, we plated the cells on Matrigel, which contains all major ECM substrates. 293T knockdown cells initially attached (Figures 5J and 5K) but, in contrast to control cells, failed to remain adherent upon confluency (Figures 5L and 5M). Lastly, analysis of actin staining both in DGAP177 patient lymphoblastoid cells and in 293T knockdown cells showed reduced numbers of F-actin microspikes and filopodia (Figures 5D, 5E, 5N, and 5O); reductions in each have been linked to reduced cell adhesion and motility.28,29
Figure 5.
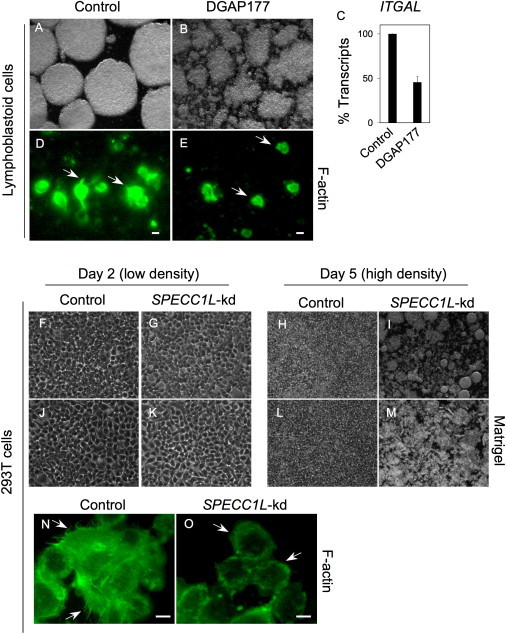
Cell-Adhesion Defects in SPECC1L-Deficient Cells
(A–C) EBV-transformed control lymphoblasts show increased integrin LFA-1-dependent adhesion in culture and grow as large circular clumps (A). DGAP177 EBV-transformed lymphoblasts fail to form these large clumps (B) and show reduced levels of integrin αL (ITGAL) transcripts (C). Data represent average transcript level ± standard error of the mean from three independent experiments.
(D and E) The adhesion defect is accompanied by altered F-actin staining and reduced numbers of F-actin microspikes (arrows) when SPECCIL-deficient cells are compared to control cells.
(F–I) Similar to control cells (F), SPECC1L-knockdown (SPECC1L-kd) 293T clonal cells (G) adhere normally to the culture plate at low density on day 2. Upon reaching confluence at day 5, control cells continue to adhere to the culture plate (H), whereas knockdown cells fail to adhere and lift off the plate (I).
(J–M) Plating SPECC1L-kd cells on Matrigel does not prevent this loss of adhesion at confluency on day 5.
(N and O) Compared with control cells (N), SPECC1L-kd 293T cells show abnormal F-actin staining (O) with reduced numbers of microspikes (arrows). Scale bars represent 10 μm.
To test the direct involvement of SPECC1L in cell migration, we examined the ability of two independent SPECC1L-kd clonal U2OS cell lines to migrate and seal acellular gaps in a wound-repair protocol. SPECC1L-kd U2OS-knockdown cells (uB8-5, uB8-9) showed a reduced ability to close such gaps (Figure 6A); moreover, in the knockdown cells the central actin fibers were more prominent and were randomly organized (Figures 6B and 6C). To show that SPECC1L deficiency results in a functional defect in actin cytoskeleton reorganization, we treated wild-type and knockdown cells with either ionomycin or Wnt5a, both known to reorganize the actin cytoskeleton. In response to ionomycin, knockdown cells failed to reorganize the actin fibers rapidly to the cell membrane (Figures 6D and 6E). We treated cells with the noncanonical Wnt ligand Wnt5a in wound-repair assays to induce directional cell migration. In response to Wnt5a, control cells reorganized the actin cytoskeleton such that actin fibers aligned perpendicularly to the direction of migration12 (compare Figures 6F and 6H). In contrast, actin fibers in SPECC1L-kd cells failed to orient perpendicular to the direction of migration, indicating an inability to properly reorganize the actin cytoskeleton (Figure 6G). As a further control, Wnt3a, a canonical Wnt ligand that does not induce actin-cytoskeletal reorganization, produced no effect on control cells or on knockdown cells, which continued to show markedly concentrated central actin fibers (compare Figures 6H and 6I with Figures 6B and 6C).
Figure 6.
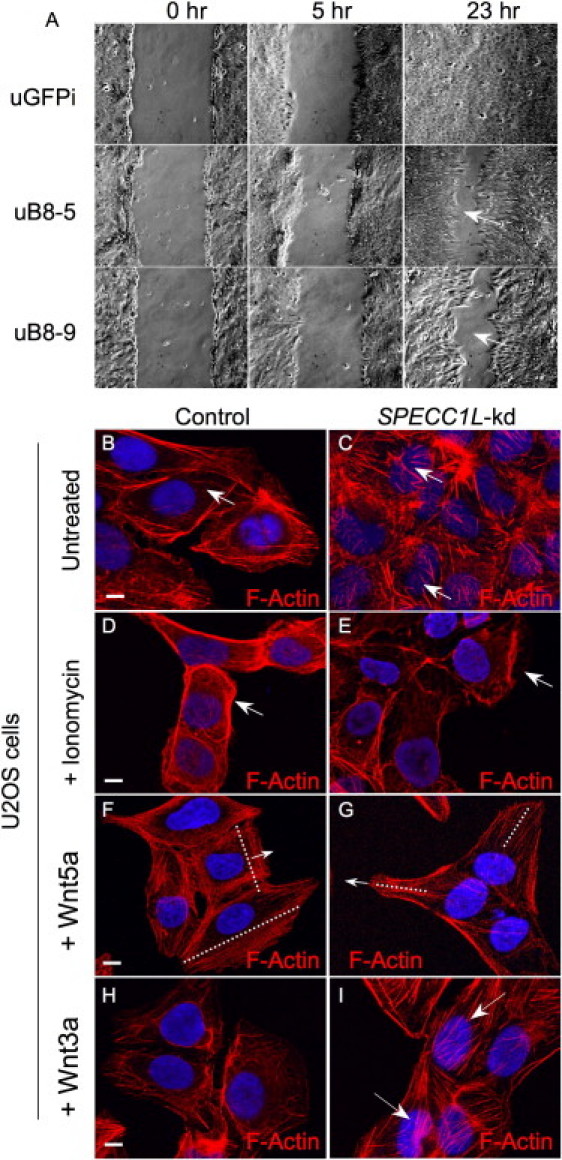
Migration and Actin-Cytoskeleton-Reorganization Defects in SPECC1L-Knockdown Cells
(A) Wound-repair (scratch) assays were used in comparisons of the ability of SPECC1L-kd U2OS clonal cells (uB8-5, uB8-9) versus control knockdown cells (uGFPi) to migrate. In severe cases, knockdown cells fail to close the wound (arrows) even after 23 hr.
(B and C) F-actin staining of SPECC1L-kd cells shows an increase in actin fibers in the center of the cells (overlying the nuclei) when these cells are compared to control cells (arrows).
(D and E) Ionomycin treatment causes rapid reorganization of the actin cytoskeleton to the cell membrane in control cells, whereas SPECC1L-kd cells respond poorly (arrows).
(F and G) Wound-repair assays with control and knockdown cells were treated with the noncanonical Wnt ligand, Wnt5a. In control cells, Wnt5a reorganizes the actin cytoskeleton toward the leading edge, perpendicular to the direction of migration (F). In contrast, knockdown cells show an abnormal alignment of actin fibers (G), indicating defective ability to reorganize the actin cytoskeleton. Dotted lines indicate alignment of actin filaments; arrows indicate the direction of migration.
(H and I) The canonical Wnt ligand, Wnt3a, which does not reorganize the actin cytoskeleton in control cells (H) (note similarity to [B]), was used as a control. Knockdown cells continue to show markedly concentrated central actin fibers (I, arrows) (note similarity to [C]). Scale bars represent 10 μm.
Discussion
Our data identify mutations in SPECC1L as contributory to the pathogenesis of ObFC. The evidence includes: (1) an intron 14 disruption of SPECC1L in DGAP177 and a functionally defective de novo SPECC1L missense variant, p.Gln415Pro, both associated with ObFC; (2) expression in developing murine facial prominences, and (3) a loss of facial structures in a zebrafish knockdown model. These data do not, however, rule out the possibility that variants in other genes might also contribute to ObFC.
SPECC1L encodes a novel cytoskeletal cross-linking protein. Interactions between microtubules and actin-cytoskeleton filaments are critical in many mechanical cellular processes, including cell movement, cell division, and wound healing. In response to stimuli, these processes require the rapid dynamic assembly of polarized cytoskeletal polymers of actin and tubulin to form higher-order structures such as radial arrays or bundles. Examples of these structures include the bipolar microtubular spindles that form during cell division and the actin-mediated protrusions located at the leading edge of migrating cells.30–33
Experiments in which Specc1l-GFP was overexpressed suggest a role for SPECC1L in microtubule stability and in actin cytoskeleton organization. In addition, the endogenous cellular expression of SPECC1L further indicates its presence in mitotic spindles and in the MTOC. In particular, the punctate pattern of SPECC1L expression around the centrioles suggests that SPECC1L might be associated with the negative ends of spindle microtubules and thereby be involved in spindle orientation and cell polarity, both critical for directional cell migration. These results place SPECC1L in a distinct functional class of cross-linking proteins that include adenomatous polyposis coli (APC),34,35 cytoplasmic dynein/dynactin,36 and MACF,37 each of which affects both spindle orientation and microtubule stability.
SPECC1L knockdown in mammalian cells demonstrated a profound impairment in the ability of cells to reorganize the actin cytoskeleton rapidly in response to stimuli such as Ca2+ and Wnt5a. The noncanonical Wnt5a/Ca2+ pathway induces actin cytoskeletal reorganization and plays a vital role in the polarized cell migratory movements that form the vertebrate body.38 In fact, Wnt5a−/− null mice lack structures that protrude out of the primary body axis, including the face and limbs.39 Combined with Specc1l expression in the developing MxP and LNP, this suggests a role for SPECC1L in mediating the effect of various stimuli that act during facial morphogenesis and require reorganization of the actin cytoskeleton.
Indeed, the knockdown-induced deficiency of the SPECC1L homolog in zebrafish and of the ortholog in Drosophila results in migration and adhesion defects that can be reconciled with the pathogenesis of human ObFC. The wing defects observed after SPECC1L ortholog knockdown in Drosophila strongly phenocopy known mutants of the integrin signaling pathway. These known integrin-pathway fly mutants also frequently manifest the cleft dorsum phenotype evident in some of our mutant flies.40 Whereas the wing blister phenotype results from a lack of cell adhesion, the cleft dorsum reflects a cell-migration defect whereby cells derived from the two wing imaginal discs fail to migrate properly and fuse. These migration and adhesion defects are thought to lead to improper flight-muscle attachment at the dorsum and thereby result in the flightless phenotype observed in mild alleles of integrin signaling mutants.20 Furthermore, pharate arrest combined with dorsum, proboscis, and feeding defects have been reported in Mnn-overexpressing flies with reduced Jun/JNK activity downstream of integrin signaling.41 Thus, almost all of the Drosophila CG13366 mutant phenotypes can be attributed to a failure of migration or adhesion of opposing imaginal-disc-derived cells, and we have accordingly given CG13366 the name split discs.
The zebrafish faceless phenotype is similar to that of the chinless mutant that lacks CNC-derived cartilage and mesoderm-derived muscles.42 Although the chinless gene remains unidentified, two genes known to play minor roles in zebrafish jaw development are integrinα543 and endothelin1.44 Mutants for both genes show missing or severely reduced ventral jaw cartilages (CH, HS), which are also lost in our faceless morphant. Future analyses of SPECC1L function will be required to elucidate whether our faceless phenotype results from a failure of CNC to properly migrate and populate the facial prominences or from an abnormal signaling environment within the prominences after they are populated by CNC. It is also important to note that although a facial phenotype was observed with the zebrafish chromosome 5 SPECC1L-homolog knockdown, the chromosome 8 and 21 homologs are actually more similar to SPECC1L and might be more functionally related. It will be of interest to determine the phenotypes resulting from combinatorial knockdowns of chromosome 5, 8, and 21 homologs of SPECC1L.
Together, the zebrafish and Drosophila mutants imply that SPECC1L might mediate endothelin and integrin signaling. The endothelin pathway leads to an intracellular increase in calcium, which affects cell shape and motility.45,46 Because SPECC1L-kd U2OS cells fail to reorganize the actin cytoskeleton properly after ionomycin-induced intracellular calcium increase, it is plausible that they could be impaired in responding to endothelin signaling as well. Integrins comprise a superfamily of cell-adhesion receptors that bind ECM or cell-surface ligands. Upon ligand binding, integrins can transduce a signal into the cell (via “outside-in signaling”); they can also receive intracellular signals that regulate their ligand-binding affinity (via “inside-out signaling”).20 A defect in integrin-mediated signaling is consistent with our observations that DGAP177 lymphoblasts fail to form integrin-dependent clusters and that upon reaching confluence, SPECC1L-kd 293T cells fail to bind to ECM.
Lastly, we have identified one disruption and one missense mutation in SPECC1L in two individuals with ObFC. The p.Gln415Pro amino acid change was not present in 258 population-matched or in 50 CEPH controls, nor in any of the extant SNP databases, and we have used an overexpression system to show that the p.Gln415Pro change causes a functional defect in microtubule stabilization. Our results thus provide a genetic basis for ObFC and demonstrate that ObFC can result from mutations in SPECC1L, which most likely regulates cell migration and adhesion in cells that comprise the developing facial prominences.
Acknowledgments
We thank J. Lund for assistance with cell lines, B. Gerami-Naini for help with Matrigel experiment, S. Daack-Hirsch for help with patient samples, and J. Shah for helpful suggestions. I.S. was supported by a Canadian Institutes of Health Research postdoctoral fellowship. R.P.E. is recipient of the Holsclaw Family Chair in Human Genetics and Inherited Disease. This work was supported by National Institutes of Health grants HD060050 (R.L.M.), GM061354 (C.C.M. and R.L.M.), DE08559 (J.C.M.), and HL090921 (P.L.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Bloomington Drosophila Stock Center, http://flystocks.bio.indiana.edu/
Combined DNA Index System (CODIS) database, http://www.cstl.nist.gov/strbase/fbicore.htm/
Constraint-based Multiple Alignment Tool (COBALT), http://www.ncbi.nlm.nih.gov/tools/cobalt/
Database of Genomic Variants, http://projects.tcag.ca/variation/
Developmental Genome Anatomy Project (DGAP), http://dgap.harvard.edu/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
RefSeq database, http://www.ncbi.nlm.nih.gov/RefSeq/
University of California Santa Cruz Human Genome Browser, http://www.genome.ucsc.edu/
Vienna Drosophila Resource Center, http://stockcenter.vdrc.at/
References
- 1.Jugessur A., Farlie P.G., Kilpatrick N. The genetics of isolated orofacial clefts: From genotypes to subphenotypes. Oral Dis. 2009;15:437–453. doi: 10.1111/j.1601-0825.2009.01577.x. [DOI] [PubMed] [Google Scholar]
- 2.Beaty T.H., Murray J.C., Marazita M.L., Munger R.G., Ruczinski I., Hetmanski J.B., Liang K.Y., Wu T., Murray T., Fallin M.D. A genome-wide association study of cleft lip with and without cleft palate identifies risk variants near MAFB and ABCA4. Nat. Genet. 2010;42:525–529. doi: 10.1038/ng.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tessier P. Anatomical classification facial, cranio-facial and latero-facial clefts. J. Maxillofac. Surg. 1976;4:69–92. doi: 10.1016/s0301-0503(76)80013-6. [DOI] [PubMed] [Google Scholar]
- 4.Eppley B.L., van Aalst J.A., Robey A., Havlik R.J., Sadove A.M. The spectrum of orofacial clefting. Plast. Reconstr. Surg. 2005;115:101e–114e. doi: 10.1097/01.prs.0000164494.45986.91. [DOI] [PubMed] [Google Scholar]
- 5.Hall B.K. Springer Science+Business Media; New York: 2009. The Neural Crest and Neural Crest Cells in Vertebrate Development and Evolution. [Google Scholar]
- 6.Rinon A., Lazar S., Marshall H., Büchmann-Møller S., Neufeld A., Elhanany-Tamir H., Taketo M.M., Sommer L., Krumlauf R., Tzahor E. Cranial neural crest cells regulate head muscle patterning and differentiation during vertebrate embryogenesis. Development. 2007;134:3065–3075. doi: 10.1242/dev.002501. [DOI] [PubMed] [Google Scholar]
- 7.Nelms B.L., Labosky P.A. Morgan and Claypool Publishers; San Francisco: 2010. Transcriptional Control of Neural Crest Development. [PubMed] [Google Scholar]
- 8.Dasouki M., Barr M., Jr., Erickson R.P., Cox B. Translocation (1;22) in a child with bilateral oblique facial clefts. J. Med. Genet. 1988;25:427–429. doi: 10.1136/jmg.25.6.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Higgins A.W., Alkuraya F.S., Bosco A.F., Brown K.K., Bruns G.A., Donovan D.J., Eisenman R., Fan Y., Farra C.G., Ferguson H.L. Characterization of apparently balanced chromosomal rearrangements from the developmental genome anatomy project. Am. J. Hum. Genet. 2008;82:712–722. doi: 10.1016/j.ajhg.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dietzl G., Chen D., Schnorrer F., Su K.C., Barinova Y., Fellner M., Gasser B., Kinsey K., Oppel S., Scheiblauer S. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature. 2007;448:151–156. doi: 10.1038/nature05954. [DOI] [PubMed] [Google Scholar]
- 11.Tsvetkov A.S., Samsonov A., Akhmanova A., Galjart N., Popov S.V. Microtubule-binding proteins CLASP1 and CLASP2 interact with actin filaments. Cell Motil. Cytoskeleton. 2007;64:519–530. doi: 10.1002/cm.20201. [DOI] [PubMed] [Google Scholar]
- 12.Witze E.S., Litman E.S., Argast G.M., Moon R.T., Ahn N.G. Wnt5a control of cell polarity and directional movement by polarized redistribution of adhesion receptors. Science. 2008;320:365–369. doi: 10.1126/science.1151250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jalali G.R., Vorstman J.A., Errami A., Vijzelaar R., Biegel J., Shaikh T., Emanuel B.S. Detailed analysis of 22q11.2 with a high density MLPA probe set. Hum. Mutat. 2008;29:433–440. doi: 10.1002/humu.20640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sharp A.J., Locke D.P., McGrath S.D., Cheng Z., Bailey J.A., Vallente R.U., Pertz L.M., Clark R.A., Schwartz S., Segraves R. Segmental duplications and copy-number variation in the human genome. Am. J. Hum. Genet. 2005;77:78–88. doi: 10.1086/431652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perry G.H., Ben-Dor A., Tsalenko A., Sampas N., Rodriguez-Revenga L., Tran C.W., Scheffer A., Steinfeld I., Tsang P., Yamada N.A. The fine-scale and complex architecture of human copy-number variation. Am. J. Hum. Genet. 2008;82:685–695. doi: 10.1016/j.ajhg.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thusberg J., Vihinen M. Pathogenic or not? And if so, then how? Studying the effects of missense mutations using bioinformatics methods. Hum. Mutat. 2009;30:703–714. doi: 10.1002/humu.20938. [DOI] [PubMed] [Google Scholar]
- 17.Le Douarin N.M. Cambridge University Press; Cambridge: 1982. The neural crest. 259 pp. [Google Scholar]
- 18.Schilling T.F., Kimmel C.B. Segment and cell type lineage restrictions during pharyngeal arch development in the zebrafish embryo. Development. 1994;120:483–494. doi: 10.1242/dev.120.3.483. [DOI] [PubMed] [Google Scholar]
- 19.Brown N.H., Gregory S.L., Martin-Bermudo M.D. Integrins as mediators of morphogenesis in Drosophila. Dev. Biol. 2000;223:1–16. doi: 10.1006/dbio.2000.9711. [DOI] [PubMed] [Google Scholar]
- 20.Bökel C., Brown N.H. Integrins in development: Moving on, responding to, and sticking to the extracellular matrix. Dev. Cell. 2002;3:311–321. doi: 10.1016/s1534-5807(02)00265-4. [DOI] [PubMed] [Google Scholar]
- 21.Delon I., Brown N.H. Integrins and the actin cytoskeleton. Curr. Opin. Cell Biol. 2007;19:43–50. doi: 10.1016/j.ceb.2006.12.013. [DOI] [PubMed] [Google Scholar]
- 22.Hammond J.W., Cai D., Verhey K.J. Tubulin modifications and their cellular functions. Curr. Opin. Cell Biol. 2008;20:71–76. doi: 10.1016/j.ceb.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gimona M., Djinovic-Carugo K., Kranewitter W.J., Winder S.J. Functional plasticity of CH domains. FEBS Lett. 2002;513:98–106. doi: 10.1016/s0014-5793(01)03240-9. [DOI] [PubMed] [Google Scholar]
- 24.Wei C.J., Xu X., Lo C.W. Connexins and cell signaling in development and disease. Annu. Rev. Cell Dev. Biol. 2004;20:811–838. doi: 10.1146/annurev.cellbio.19.111301.144309. [DOI] [PubMed] [Google Scholar]
- 25.Petrie R.J., Doyle A.D., Yamada K.M. Random versus directionally persistent cell migration. Nat. Rev. Mol. Cell Biol. 2009;10:538–549. doi: 10.1038/nrm2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rodriguez O.C., Schaefer A.W., Mandato C.A., Forscher P., Bement W.M., Waterman-Storer C.M. Conserved microtubule-actin interactions in cell movement and morphogenesis. Nat. Cell Biol. 2003;5:599–609. doi: 10.1038/ncb0703-599. [DOI] [PubMed] [Google Scholar]
- 27.Rothlein R., Springer T.A. The requirement for lymphocyte function-associated antigen 1 in homotypic leukocyte adhesion stimulated by phorbol ester. J. Exp. Med. 1986;163:1132–1149. doi: 10.1084/jem.163.5.1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kislauskis E.H., Zhu X., Singer R.H. beta-Actin messenger RNA localization and protein synthesis augment cell motility. J. Cell Biol. 1997;136:1263–1270. doi: 10.1083/jcb.136.6.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Webb D.J., Brown C.M., Horwitz A.F. Illuminating adhesion complexes in migrating cells: Moving toward a bright future. Curr. Opin. Cell Biol. 2003;15:614–620. doi: 10.1016/s0955-0674(03)00105-4. [DOI] [PubMed] [Google Scholar]
- 30.Waterman-Storer C.M., Duey D.Y., Weber K.L., Keech J., Cheney R.E., Salmon E.D., Bement W.M. Microtubules remodel actomyosin networks in Xenopus egg extracts via two mechanisms of F-actin transport. J. Cell Biol. 2000;150:361–376. doi: 10.1083/jcb.150.2.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Salmon W.C., Adams M.C., Waterman-Storer C.M. Dual-wavelength fluorescent speckle microscopy reveals coupling of microtubule and actin movements in migrating cells. J. Cell Biol. 2002;158:31–37. doi: 10.1083/jcb.200203022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kodama A., Lechler T., Fuchs E. Coordinating cytoskeletal tracks to polarize cellular movements. J. Cell Biol. 2004;167:203–207. doi: 10.1083/jcb.200408047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kunda P., Baum B. The actin cytoskeleton in spindle assembly and positioning. Trends Cell Biol. 2009;19:174–179. doi: 10.1016/j.tcb.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 34.McCartney B.M., McEwen D.G., Grevengoed E., Maddox P., Bejsovec A., Peifer M. Drosophila APC2 and Armadillo participate in tethering mitotic spindles to cortical actin. Nat. Cell Biol. 2001;3:933–938. doi: 10.1038/ncb1001-933. [DOI] [PubMed] [Google Scholar]
- 35.Barth A.I., Caro-Gonzalez H.Y., Nelson W.J. Role of adenomatous polyposis coli (APC) and microtubules in directional cell migration and neuronal polarization. Semin. Cell Dev. Biol. 2008;19:245–251. doi: 10.1016/j.semcdb.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dujardin D.L., Vallee R.B. Dynein at the cortex. Curr. Opin. Cell Biol. 2002;14:44–49. doi: 10.1016/s0955-0674(01)00292-7. [DOI] [PubMed] [Google Scholar]
- 37.Leung C.L., Green K.J., Liem R.K. Plakins: A family of versatile cytolinker proteins. Trends Cell Biol. 2002;12:37–45. doi: 10.1016/s0962-8924(01)02180-8. [DOI] [PubMed] [Google Scholar]
- 38.Slusarski D.C., Pelegri F. Calcium signaling in vertebrate embryonic patterning and morphogenesis. Dev. Biol. 2007;307:1–13. doi: 10.1016/j.ydbio.2007.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamaguchi T.P., Bradley A., McMahon A.P., Jones S. A Wnt5a pathway underlies outgrowth of multiple structures in the vertebrate embryo. Development. 1999;126:1211–1223. doi: 10.1242/dev.126.6.1211. [DOI] [PubMed] [Google Scholar]
- 40.Lee S.B., Cho K.S., Kim E., Chung J. blistery encodes Drosophila tensin protein and interacts with integrin and the JNK signaling pathway during wing development. Development. 2003;130:4001–4010. doi: 10.1242/dev.00595. [DOI] [PubMed] [Google Scholar]
- 41.Cerrato A., Parisi M., Santa Anna S., Missirlis F., Guru S., Agarwal S., Sturgill D., Talbot T., Spiegel A., Collins F. Genetic interactions between Drosophila melanogaster menin and Jun/Fos. Dev. Biol. 2006;298:59–70. doi: 10.1016/j.ydbio.2006.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schilling T.F., Walker C., Kimmel C.B. The chinless mutation and neural crest cell interactions in zebrafish jaw development. Development. 1996;122:1417–1426. doi: 10.1242/dev.122.5.1417. [DOI] [PubMed] [Google Scholar]
- 43.Crump J.G., Swartz M.E., Kimmel C.B. An integrin-dependent role of pouch endoderm in hyoid cartilage development. PLoS Biol. 2004;2:E244. doi: 10.1371/journal.pbio.0020244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miller C.T., Schilling T.F., Lee K., Parker J., Kimmel C.B. sucker encodes a zebrafish Endothelin-1 required for ventral pharyngeal arch development. Development. 2000;127:3815–3828. doi: 10.1242/dev.127.17.3815. [DOI] [PubMed] [Google Scholar]
- 45.Maschhoff K.L., Baldwin H.S. Molecular determinants of neural crest migration. Am. J. Med. Genet. 2000;97:280–288. doi: 10.1002/1096-8628(200024)97:4<280::aid-ajmg1278>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 46.Clouthier D.E., Schilling T.F. Understanding endothelin-1 function during craniofacial development in the mouse and zebrafish. Birth Defects Res. C Embryo Today. 2004;72:190–199. doi: 10.1002/bdrc.20007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.