

Abstract
Nearly every ciliated organism possesses three B9 domain-containing proteins: MKS1, B9D1, and B9D2. Mutations in human MKS1 cause Meckel syndrome (MKS), a severe ciliopathy characterized by occipital encephalocele, liver ductal plate malformations, polydactyly, and kidney cysts. Mouse mutations in either Mks1 or B9d2 compromise ciliogenesis and result in phenotypes similar to those of MKS. Given the importance of these two B9 proteins to ciliogenesis, we examined the role of the third B9 protein, B9d1. Mice lacking B9d1 displayed polydactyly, kidney cysts, ductal plate malformations, and abnormal patterning of the neural tube, concomitant with compromised ciliogenesis, ciliary protein localization, and Hedgehog (Hh) signal transduction. These data prompted us to screen MKS patients for mutations in B9D1 and B9D2. We identified a homozygous c.301A>C (p.Ser101Arg) B9D2 mutation that segregates with MKS, affects an evolutionarily conserved residue, and is absent from controls. Unlike wild-type B9D2 mRNA, the p.Ser101Arg mutation failed to rescue zebrafish phenotypes induced by the suppression of b9d2. With coimmunoprecipitation and mass spectrometric analyses, we found that Mks1, B9d1, and B9d2 interact physically, but that the p.Ser101Arg mutation abrogates the ability of B9d2 to interact with Mks1, further suggesting that the mutation compromises B9d2 function. Our data indicate that B9d1 is required for normal Hh signaling, ciliogenesis, and ciliary protein localization and that B9d1 and B9d2 are essential components of a B9 protein complex, disruption of which causes MKS.
Introduction
Meckel syndrome (MKS [MIM 249000]) is a rare autosomal-recessive disorder resulting in perinatal lethality and characterized by renal cysts, hepatic ductal plate malformation, and central nervous system defects, such as occipital encephalocele.1 Mutations in six genes are known to cause MKS: MKS1 (MIM 609883), TMEM216 (MKS2 [MIM 613277]), TMEM67 (MKS3 [MIM 609884]), CEP290 (MKS4 [MIM 610142]), RPGRIP1L (MKS5 [MIM 610937]), and CC2D2A (MKS6 [MIM 612013]).2–7 In addition, mutations in NPHP3 (MKS7 [MIM 608002]) and TCTN2 (MKS8) cause MKS-like phenotypes.8,9 The products of these genes localize to cilia and/or basal bodies, supporting the hypothesis that ciliary dysfunction underlies MKS.
MKS1, a variant of which was the first gene linked to MKS, encodes a protein containing a B9 domain, structurally similar to the lipid binding domains of the C2 family.10 Although none of the other previously recognized MKS proteins contain B9 domains, bilaterians including mammals, Drosophila, and C. elegans possess two other B9 domain-containing proteins, B9d1 and B9d2 (also known as Stumpy [MIM 611951]). B9 domain-containing proteins are also present in diverse ciliated protists, including Trypanosomes, Naegleria, and Batrachochytrium, but are absent from nonciliated eukaryotes, including most yeast and higher plants (see Figure S1 available online). Consistent with a role in cilia function, all three B9 domain proteins localize to the transition zone of C. elegans cilia or the basal body and axoneme of mammalian cilia.11–14 Mouse embryos homozygous for a loss-of-function mutation in Mks1 display hallmarks of MKS, such as renal cysts, ductal plate malformation, and brain malformations similar to occipital meningoencephalocele. In addition, these mutants displayed cleft palate and polydactyly, phenotypes associated commonly with MKS1 (MIM 249000).13,15–18 Conditional inactivation of B9d2 in the brain and kidney cause hydrocephalus and renal cysts, accompanied by defects in ependymal and kidney cilia.14
Primary cilia are required for mammalian development, in part because of their roles in mediating Hh signal transduction.19 Hh signals participate in the patterning of a large number of tissues, including the neural tube and limb buds. Components of the Hh pathway, including Patched1 (Ptc1), Smoothened (Smo), and the Gli transcriptional effectors, localize to primary cilia, suggesting that critical Hh signal transduction events occur within cilia.20–22 Consistent with this hypothesis, vertebrate mutations that disrupt ciliogenesis cause defects in Hh signaling and concomitant developmental phenotypes.23
To investigate further the role of B9 proteins in ciliogenesis and in the pathogenesis of MKS, we analyzed mice mutant for the third member of the B9 genes, B9d1. Strikingly, loss of B9d1 resulted in MKS-like phenotypes in mouse embryos which, taken together with the mouse mutant data for B9d2, suggested that these two loci are candidates for causing severe ciliopathies. Therefore, we screened MKS fetuses for mutations in B9D1 and B9D2 and identified a homozygous mutation in B9D2. We also found that all three B9 proteins interact physically with each other but that the MKS-associated disease allele of B9d2 exhibited decreased affinity for Mks1. These findings reveal that a complex comprised of the three B9 proteins support mammalian ciliogenesis, disruption of which results in Hh signaling defects and MKS.
Material and Methods
B9 Protein Conservation
We identified MKS1, B9D1, and B9D2 orthologs by using reciprocal best match by BLAST. MKS1 orthologs include Tetrahymena TTHERM_00630490, Batrachochytrium BDEG_00264, Tricoplax JGI 61316, and Naegleria JGI 29577. B9D1 orthologs include Batrachochytrium BDEG_03036, Chlamydomonas Au9.Cre01.g066000, Paramecia GSPATP00029931001, Tetrahymena TTHERM_00219110, Leishmania major LmjF32.0280, Trypanosoma brucei Tb10.61.2290, Trichoplax JGI 52971, Monosiga JGI 24999, Naegleria JGI 52666, Thalassiosira JGI 264691, and Phytophthora JGI 71279. B9D2 orthologs include Batrachochytrium BDEG_03725, Chlamydomonas Au9.Cre03.g196050, Naegleria JGI 30379, Trypanosoma brucei Tb11.03.0750, Leishmania major LmjF25.0320, Tetrahymena TTHERM_00633350, Paramecium ICIS-1, Trichoplax JGI 52819, Monosiga JGI 17074, and Thalassiosira JGI 38153. Protein sequences were aligned with the ID weight matrix of ClustalW, and the alignment was used to create an unrooted nearest-joining tree.
Mouse Strains
All mouse protocols were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of California, San Francisco. B9d1+/− mice (B9d1tm1a(EUCOMM)Wtsi) were obtained from the European Conditional Mouse Mutagenesis Program (EUCOMM). The targeted mutation was made in C57BL/6N ES cells, which were injected into B6(Cg)-Tyrc-2J/J blastocysts. The mice were maintained by crossing to C57BL/6J mice (Jackson Laboratory). The mixed background strain was created by outcrossing B9d1+/− mice with CD1 mice (Charles River Laboratory) for two generations. The B9d1 conditional allele was created by crossing B9d1tm1a(EUCOMM)Wtsi mice to mice expressing FLPe recombinase (Tg(ACTFLPe)9205Dym/J) to remove the En2 splice acceptor and βGeo cassette (B9d1flox/flox). Subsequently, B9d1flox/flox mice were crossed to mice expressing Cre recombinase (Tg(ACTB-cre)2Mrt/J) to delete exon 3. We considered noon on the day a copulation plug was detected to be 0.5 days postcoitus.
Immunohistochemistry, Histology, and In Situ Hybridization
For whole-mount staining of nodes, E8.0 embryos were dissected in PBS + 10% FBS, rinsed in PBS, and fixed in 4% paraformaldehyde (PFA) in PBS for 30 min on ice, blocked for 30 min in 2% BSA + 5% donkey serum in PBS + 0.1% Triton X-100 (PBT), and incubated with primary antibodies overnight at 4°C in 2% BSA + 1% donkey serum in PBT. For neural tube analysis, E9.5 embryos were harvested in PBS + 10% FBS, rinsed in PBS, and fixed for 1 hr in 4% PFA in PBS on ice. Embryos were equilibrated in 30% sucrose overnight and mounted in O.C.T. mounting media (Fisher). 12 μm sections were stained as previously described.24 For liver and kidney analysis, organs were fixed in 10% formalin overnight at 4°C, sectioned, and stained with hematoxylin and eosin according to standard protocols. Immunofluorescent staining of liver and kidney sections was performed as described.25 Biotinylated lectin staining of kidney sections was performed as described,26 with the exception that avidin/biotin blocking solution (Vector Labs) was applied according to the manufacturer's instructions after sections were dewaxed.
The antibodies employed were mouse α-acetylated tubulin 6-11B-1 (1:1,000, Sigma), rabbit α-Ninein (1:20,000, gift of James Silbourne), mouse α-E-cadherin (1:100, BD), mouse α-Sox9 (1:100), goat α-γ tubulin (1:100, Santa Cruz Biotechnology), mouse α-Cytokeratin-19 (1:130, Novacastra), rabbit α-Pax6 (1:100, Covance), mouse α-Nkx2.2 (1:20), mouse α-FoxA2 (1:20), and mouse α-Islet1/2 (1:20, Developmental Studies Hybridoma Bank). These last three antibodies were developed under the auspices of the NICHD and maintained by the Department of Biological Sciences, The University of Iowa (Iowa City, IA). Biotinylated lectins employed were Dolichos biflorus agglutinin (10 μg/ml, Vector Labs B-1035) and Lotus tetragonolobus lectin (10 μg/ml, Vector Lab B-1325). Subsequent to secondary antibody staining with Avidin-conjugated Alexa 488 (1:300, Invitrogen), nodal and neural tube cilia were observed with a Nikon C1 spectral confocal microscope. Neural tube pattern as well as cilia staining of liver and kidney sections were observed with a Leica SP5 confocal microscope. Kidney and liver H&E as well as Sox9 and E-cadherin staining of liver sections were observed with a Zeiss Observer D1 inverted epifluorescent microscope. Postacquisition image processing was performed with Metamorph imaging software (Molecular Devices) or Image J (NIH).
To visualize cartilage, E13.5 embryos were harvested in PBS + 10% FBS, frozen at −20°, and stained with alcian blue as described.27 In situ hybridization was performed as described.27,28 Cartilage preparations and whole-mount embryos were observed with a Zeiss Discovery V12 steREO microscope.
Electron Microscopy
For SEM analysis, E8.0 (2–3 somite) embryos were fixed for 1 hr in 2% sodium cacodylate buffer, postfixed in 1% osmium tetroxide in sodium cacodylate buffer, and dehydrated through a graded ethanol series. Samples were mounted on aluminum stubs with carbon dots, critical point dried in a Tousimis AutoSamdri 815, and sputter coated with gold in a Tousimis chamber. Samples were visualized on a Hitachi S-5000 scanning electron microscope. For TEM analysis, E9.5 embryos were fixed for 24 hr at RT in 0.1 M phosphate buffer (pH 7.4, PB) containing 2% paraformaldehyde and 2.5% glutaraldehyde. Embryos were rinsed in PB and postfixed for 2 hr with 2% osmium tetroxide. After a graded ethanol dehydration series and propylene oxide infiltration, embryos were embedded in Araldite resin (Durcupan, Fluka). To select the area of interest, we cut semithin sections (1.5 μm) with a diamond knife and stained with 1% toluidine blue. Subsequently, we cut ultrathin sections (60–90 nm) from the area of interest with a diamond knife, stained them with lead citrate, and examined them with a Fei Tecnai Spirit electron microscope.
Plasmid Construction
pGLAP5-Rer1, pGLAP5-Mks1, pGLAP5-B9d1, pGLAP5-B9d2, pEF/V5/Frt-Rer1, pEF/V5/Frt-Mks1, pEF/V5/Frt-B9d1, and pEF/V5/Frt-B9d2 were created by polymerase chain reaction (PCR) amplification of the coding sequences from verified mouse cDNA clones (Open Biosystems). PCR products were TOPO cloned into the pCR8/GW/TOPO Gateway entry vector (Invitrogen) and recombined subsequently into either pGLAP5 (Addgene plasmid 19706)29 or pEF/V5/Frt (Invitrogen) destination vectors with Clonase II (Invitrogen). The B9d2 p.Ser101Arg mutation was introduced into pCR8/GW/-B9d2 via site directed mutagenesis with Quikchange II XL (Agilent). pCR8/GW-B9d2 S101R was used to create pGLAP5-B9d2 S101R and pEF/V5/Frt-B9d2 S101R via recombination, as described above. The p.Ser101Arg mutation was introduced into human B9D2 cDNA clone IOH4997 of the Invitrogen ORF collection with QuickChange site-directed mutagenesis (Agilent). Wild-type and p.Ser101Arg B9D2 cDNAs were cloned into pEGFP-N1 (Clontech) to generate pGFP-wtB9D2 or pGFP-B9D2-S101R. B9D2 and B9D2-S101R were subcloned into the pCS2+ Gateway destination vector (Invitrogen) from pENTR221 (Invitrogen) with Clonase II (Invitrogen) to generate pCS2-wtB9D2 or pCS2-B9D2-S101R.
Cell Culture, Immunofluorescence, and Microscopy
hTERT-RPE1 cells were grown to 70% confluency in DMEM/F12 media supplemented with 10% FBS and transfected with pGFP-wtB9D2 or pGFP-B9D2-S101R with FUGENE6 (Invitrogen). 18 hr posttransfection, cells were switched to low-serum (0.5% FBS) medium; 48 hr after serum starvation, the cells were washed with PBS and fixed in 4% PFA, permeabilized, and blocked with PBS-T (0.1% Triton X-100) containing 10% goat serum, stained with primary antibodies as indicated, and observed with a Zeiss LSM 510 META confocal microscope.
B9d1 wild-type and mutant MEFs were derived from E12.5 mouse embryos, as described, and grown in DMEM + 15% FBS.27 For Smo localization analysis, MEFs were grown to confluency, starved in Opti-MEM (Invitrogen) for 24 hr, and treated for 6 hr with 1 μM of the small molecule Smoothened AGonist (SAG) (Enzo Life Sciences) in dimethyl sulfoxide (DMSO, Sigma). MEFs were then fixed in 4% PFA for 5 min at 37°C and −20°C methanol for 3 min. For analysis of Hh pathway activation, MEFs were treated for 20 hr with 1 μM SAG.
Antibodies employed were rabbit α-Smo (1:100), rabbit α-Ift88 (1:100, Proteintech), rabbit α-Arl13b (1: 1,5000, gift of Tamara Caspary), rabbit α-ACIII (1:100, Santa Cruz Biotechnology), mouse α-acetylated tubulin 6-11B-1 (1:1,000, Sigma), and goat α-γ tubulin (1:100, Santa Cruz Biotechnology). Smo antibodies were generated by injection of a Smo-MBP fusion protein comprising the C-terminal tail of mouse Smo into rabbits (Pacific Immunology) and affinity purified with recombinant GST-Smo C terminus.
qRT-PCR
For analysis of Hh pathway activation, RNA was extracted from MEFs with an RNAeasy kit (QIAGEN), and cDNA synthesis was performed with a Maxima first-strand cDNA synthesis kit (Fermentas). qRT-PCR was performed with Maxima SYBR Green qPCR 2x master mix with ROX (Fermentas) and primers homologous to Ptch1, Gli1, and β-actin (Table S2) on an ABI 7900HT real-time PCR machine. Levels of Ptch1 and Gli were normalized to levels of β-actin. The qRT-PCR of each RNA sample was performed in quadruplicate and the experiment was replicated. The fold increase is the average of the fold increase of the Ptch1 or Gli1 mRNA levels (normalized to β-actin) in the experimental treatments compared to the average of the Ptch1 or Gli1 mRNA levels (normalized to β-actin) in the vehicle-only control. Error bars represent the standard deviation. Statistical significance was assessed with Student's t test.
Human Genetics
The study was approved by the review board and ethics committee of the RWTH Aachen University and the participants gave informed consent.
Linkage Analysis
DNA samples from the index family were used for linkage analysis of genes, mutant versions of which cause MKS, and related disease loci. A minimum of two informative flanking markers were typed.5 Primers for PCR amplification were as published by the Genome Data Base. Microsatellite markers were analyzed on an ABI PRISM 3130 genetic analyzer (Applied Biosystems) with a universal fluoresceinated primer ComF.30 Each marker was amplified in a three-primer PCR reaction in which the forward primer of the two specific primers was tailed with the sequence corresponding to ComF (specFcom). A 10:1 molar ratio of primers ComF to specFcom ensured that specFcom was exhausted during the early amplification cycles.
Mutation Analysis
We sequenced the exons encoding B9D2 (MIM 611951, Table S2) on chromosome 19q13 (GenBank NM_030578.3, NP_085055; mutation numbering +1 corresponds to the A of the initiation codon in exon 2). Genomic DNA from a parent and an affected individual was amplified by PCR with oligonucleotide primers complementary to flanking intronic sequences (available upon request). PCR products were purified with the DNA Clean & ConcentratorTM-5 kit (Zymo Research) and sequenced with ABI BigDye chemistry (Applied Biosystems) with the PCR primers. Samples were run and analyzed on an ABI PRISM 3130 (Applied Biosystems). Prediction of the functionality of the identified nonconservative missense mutation c.301A>C (p.Ser101Arg) was performed as described with a multisequence alignment of orthologs and several bioinformatic algorithms.31,32
Morpholino and mRNA Injection of Zebrafish Embryos
Spice-blocking morpholino (MO) against zebrafish b9d2 and a control MO (Gene Tools) were injected into 1- or 2-cell stage zebrafish embryos. After testing 2 ng, 4 ng, and 6 ng of b9d2 MO, we selected 2 ng for subsequent experiments. We observed similar, but more severe, phenotypes with a translation-blocking MO directed against the first ATG of b9d2. However, because we were unable to fully rescue the phenotype, we considered the MO unreliable and generated a splice-blocking MO. We transcribed mRNA encoding human B9D2 WT and p.Ser101Arg from pCS2-wtB9D2 and pCS2-B9D2-S101R (SP6 Message Machine, Ambion). For functional assays in zebrafish, we coinjected 60 pg wild-type or mutant B9D2 mRNA with 2 ng b9d2 MO.
Injected zebrafish embryos were classified based on phenotype. Embryos similar to age-matched controls from the same clutch were classified as normal. Embryos displaying mildly shortened body axes, mediolaterally elongated somites, and notochord imperfections were categorized as Class I. Embryos displaying severely shortened body axes, mediolaterally elongated somites, and widened and kinked notochords were categorized as Class II. Embryo classifications were based on the generalized classification scheme that has been developed for other ciliopathy loci.33,34 We compared the proportions of normal and affected embryos resulting from injections of morpholinos or mRNAs, by means of the chi-square test.
Immunoprecipitation and Immunoblot
COS1 cells were grown on 10 cm diameter plates, transfected with pGLAP5- or pEF/V5/Frt-Rer1, Mks1, B9d1, B9d2, or B9d2 p.Ser101Arg plasmids via JetPrime (Polyplus-Transfection), and lysed after 24 hr in a buffer of 50 mM HEPES (pH 7.4), 300 mM KCl, 1 mM EGTA, 1 mM MgCl2, 10% glycerol, 0.3% Nonidet P-40, 0.5 mM DTT, and protease and phosphatase inhibitors. Lysates were cleared by centrifugation at 13,000 × g for 10 min, and then incubated with GFP-Trap beads (Chromotek) overnight at 4°C. Beads were washed three times in lysis buffer, resuspended in 3× SDS-PAGE loading buffer, then denatured at 100°C for 5 min. Immunocomplexes were resolved on 4%–15% TGX gradient gels (Bio-Rad) and transferred to PVDF membrane for immunoblotting (Bio-Rad).
For Gli3 analysis, E10.5 embryos were lysed in RIPA with protease and phosphatase inhibitors. Lysates were cleared at 13,000 × g for 10 min, and protein concentration was assessed by Bradford assay (Bio-Rad). Proteins were resolved on 7% SDS-PAGE gels and transferred to PVDF membranes.
For immunoblotting, membranes were blocked with 5% nonfat dry milk (NFDM) in TBS with 0.1% Tween (TBST), then incubated with primary antibodies in 5% NFDM-TBST followed by secondary antibodies in 5% NFDM-TBST. Blots were developed with either Pico enhanced chemiluminescence (ECL, Pierce) or Western lightning ECL (Perkin Elmer). Apparent molecular weights were determined via full-range rainbow prestained protein standards (Amersham).
Primary antibodies employed were mouse α-GFP (1:1,000, Santa Cruz Biotechnology), chicken α-GFP (1:1000, Aves), goat α-V5 (1:2,000, AbCam), mouse α-Gli3 (1:5,000, gift from Suzie Scales, Genentech), and mouse α-tubulin (1:1,000, Sigma). Horseradish peroxidase conjugated secondary antibodies were donkey α-mouse (1:20,000, Jackson Labs), goat α-chicken (1:5,000, Jackson Labs), and rabbit α-goat (1:5,000, Santa Cruz Biotechnology).
Mass Spectrometry
NIH 3T3 cell lines harboring stable, single-integrations of pGLAP5 vector or pGLAP5-B9d1 were created with NIH 3T3 Flp-In cells according to the manufacturer's instructions (Invitrogen). Fifty 15 cm diameter plates each of pGLAP5 (control) and pGLAP5-B9d1 stable cells were used for LAP purification as described,35 with the exception that GFP trap beads were used in the purification (Chromotek). Purified LAP-complexes were denatured with 3× SDS PAGE sample buffer at 100°C for 10 min, and were then resolved on a 4%–15% Tris-glycine gradient gel (Bio-Rad).
SYPRO ruby (Invitrogen) stained gel bands were excised and digested with trypsin as reported,36 with the additional use of Protease Max (Promega) for increased peptide recovery and shorter digestion times. The alkylation reagent used was acrylamide, providing a propanamide modification.
Nano reversed phase HPLC was done with an Eksigent 2D nanoLC (Eksigent) with buffer A consisting of 0.1% formic acid in water and buffer B 0.1% formic acid in acetonitrile. A fused silica column self packed with duragel C18 matrix (Peeke) was used with a linear gradient from 5% B to 40% B over 60 min at a flow rate of 600 nL/minute. The nanoHPLC was interfaced with a Michrom Advance source. The mass spectrometer was an LTQ-Orbitrap Velos (Thermo Scientific), which was set in data-dependent acquisition mode to perform MS/MS in the ion trap of the top eight most intense ions. A full scan in the Orbitrap of the precursor ion was done at a resolution of 60,000 at 400 m/z. The .RAW files were then converted to mzXML format with msconvert script and searched with a Sorcerer (SageN) processor with Sequest. The database was searched with a precursor mass tolerance of 50 ppm.
All hits with three or more identified peptides in the B9d1LAP purification and zero or one peptide identified in the control purification were reported.
Modeling of the B9D2 C2 Domain
A model of the B9D2 C2 domain, based on the calcium-free piccolo C2a domain, was used as a template for electrostatic surface modeling with adaptive poisson-boltzmann solver (APBS) in the visual molecular dynamics software (VMD).37,38 A protein databank (PDB) file of the modeled B9D2 C2 domain produced by ModBase was converted to a PQR file with PDB2PQR before applying APBS in VMD.39–41 Software defaults were used in the electrostatic surface modeling to color positive residues blue and negative residues red.
Results
Mouse B9d1 Mutants Display Developmental Defects Similar to Those of MKS
Given the importance of Mks1 and B9d2, two genes encoding B9 domain proteins, to ciliary structure, we investigated the function of the third member of this family, B9d1.13,14,42 We obtained a targeted allele of B9d1 (B9d1tm1a(EUCOMM)Wtsi) from the European Conditional Mouse Mutagenesis Program (EUCOMM) that contains a gene trap in the second of six introns (Figure S2A). The resultant mutant allele is predicted to encode a fusion between the first 44 amino acids and a βgeo reporter. Given that the wild-type transcript is almost completely abrogated in homozygous B9d1 mutant embryos (Figure S2B), the B9d1 gene trap is likely to create a null or near null allele. In support of this conclusion, we also analyzed mice homozygous for a deletion of B9d1 exon 3, which encodes a large portion of the B9 domain, and these mutants displayed phenotypes indistinguishable from those homozygous for the gene trap allele (data not shown).
B9d1 mutants die at approximately E14.5 (Table 1). Mutant phenotypes were assessed in the strain of derivation, C57/Bl6, where possible, and in a mixed C57/Bl6-CD1 strain to observe later stages of development, because homozygous mutant embryos on the mixed background die between E17.5 and P1. At E18.5, these B9d1 mutant mice displayed kidneys that were larger than wild-type and contained multiple cystic lesions in all of the mutants examined (n = 5/5; Figure 1A). Dilation and cystic pathology was apparent in multiple nephron segments (Figure S3). Kidney tubules were well ciliated in wild-type animals, whereas the tubules were enlarged and cilia were reduced in number and length in B9d1 mutants (Figure 1B). During liver development, hepatic ductal plates form as rings around portal veins and are subsequently remodeled, positioning bile ducts centrally adjacent to portal veins by birth.43 In contrast, ductal plates persisted as peripheral rings in B9d1 mutant livers at P1, and hyperplastic portal mesenchyme separated enlarged bile ducts from portal veins (Figures 1C and 1D). Cilia were present and similar in length in wild-type and mutant bile ducts despite the occurrence of ductal plate malformation (Figure 1E).
Table 1.
Survival of B9d1 Embryos
|
Genotype |
|||
|---|---|---|---|
| Age | B9d1+/+ | B9d1+/− | B9d1−/− |
| E8.0 | 10/47 (21) | 24/47 (51) | 13/47 (28) |
| E8.5 | 2/15 (13) | 9/15 (60) | 4/15 (27) |
| E9.5 | 22/95 (23) | 48/95 (51) | 25/95 (26) |
| E10.5 | 35/112 (31) | 57/112 (51) | 20/112 (18) |
| E11.5 | 15/55 (27) | 28/55 (51) | 12/55 (22) |
| E12.5 | 19/67 (28) | 33/67 (49) | 15/67 (23) |
| E13.5 | 28/93 (30) | 41/93 (44) | 24/93 (26) |
| E14.5 | 5/12 (42) | 7/12 (58) | 0/12 (0) |
Embryonic survival scored at the ages listed. Shown is the number of surviving embryos among the total examined, with percentages in parentheses.
Figure 1.
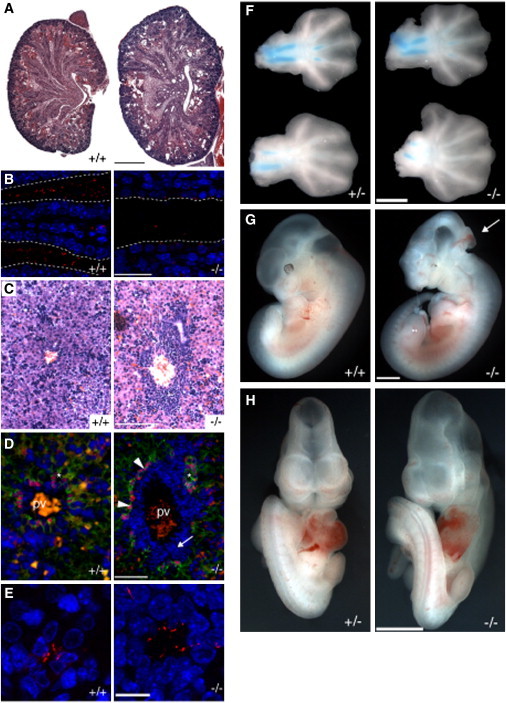
B9d1 Mutants Display Defects Similar to Those of MKS
(A) Coronal sections of E18.5 kidneys stained with H&E. Mutant kidneys are larger and contain multiple cysts. Scale bar represents 500 μm.
(B) P1 kidney tubules stained with a marker of cilia, acetylated tubulin (AcTub, red), and nuclei (DAPI, blue). B9d1 mutant tubules possess fewer cilia than wild-type tubules. Dotted lines mark the borders of tubules. Scale bar represents 25 μm.
(C) P1 liver sections stained with H&E. Scale bar represents 100 μm.
(D) Liver sections stained with a marker of bile ducts, Sox9 (red), E-cadherin (green), and nuclei (DAPI, blue). The ductal plate (arrowheads) fails to remodel in mutant livers, resulting in an abundance of portal mesenchyme (arrow) and separation of bile ducts (asterisks) from the central portal vein (pv). Scale bar represents 50 μm.
(E) Bile ducts stained for cilia (AcTub, red) and nuclei (DAPI, blue). Cilia are present in mutant bile ducts. Scale bar represents 10 μm.
(F) E13.5 forelimbs (top) and hindlimbs (bottom) stained with alcian blue, showing preaxial polydactyly.
(G) Left lateral view of E11.5 embryos. B9d1 mutants exhibit reduced telencephalons, microphthalmia, and occasional exencephaly (arrow).
(H) Frontal view of E10.5 embryos. B9d1 mutants display midline facial defects, randomized heart looping, and holoprosencephaly.
Scale bars for (F)–(H) represent 1 mm.
B9d1 mutants also developed preaxial polydactyly with either one or two extra digits, with 92% (n = 23/25) penetrance in hindlimbs and 24% (n = 6/25) penetrance in forelimbs (Figure 1F). We also observed occasional exencephaly (n = 3/49; Figure 1G). Thus, B9d1 mouse mutants display renal cystic dysplasia, the defect most commonly associated with MKS, as well as ductal plate malformation of the liver and polydactyly, other prominent characteristics of MKS.
In addition to displaying hallmarks of MKS, B9d1 mutants display dextrocardia with a frequency of 44% (n = 20/45), suggesting that heart looping is randomized in the mutant embryos (Figure 1H). We observed additional phenotypes associated with human ciliopathies in B9d1 mutants at varying frequencies, including holoprosencephaly (Figure 1H), microphthalmia (n = 32/37; Figure 1G), cleft palate (n = 9/19; Figure S4), ventricular septal defect (VSD), and thinning of the myocardial wall (Figure S5).
Randomized heart looping can be due to disruptions in fluid flow in the embryonic node.44 Because beating cilia propel nodal flow leftward, mutations in ciliogenic genes such as Kif3a and Ift88 disrupt left-right patterning.45,46 To assess whether defects in ciliogenesis might underlie the randomized heart looping observed in B9d1 mutant mice, we examined nodes with both immunofluorescence and scanning electron microscopy (SEM). B9d1 mutants exhibited normal node morphology, but lacked almost all nodal cilia (Figures 2A–2D); the few cilia that did form were short and displayed swollen tips (Figure 2D). In addition to analyzing nodal cilia, we examined the expression pattern of Lefty genes, early molecular markers, and genes implicated in the establishment of left-right patterning.28,47–49 In response to nodal flow, Lefty1/2 are expressed in the floorplate and left lateral plate mesoderm (LPM) of E8.0 embryos. Expression of Lefty1/2 was absent in B9d1 mutant embryos at E8.0 (n = 2/2; Figure 2E), which is similar to other mutants in which L-R patterning is disrupted at an early stage of axis specification.44,50–54 We surmise that the absence of nodal cilia is the likely cause of the laterality and heart looping defects of B9d1 mutants.
Figure 2.
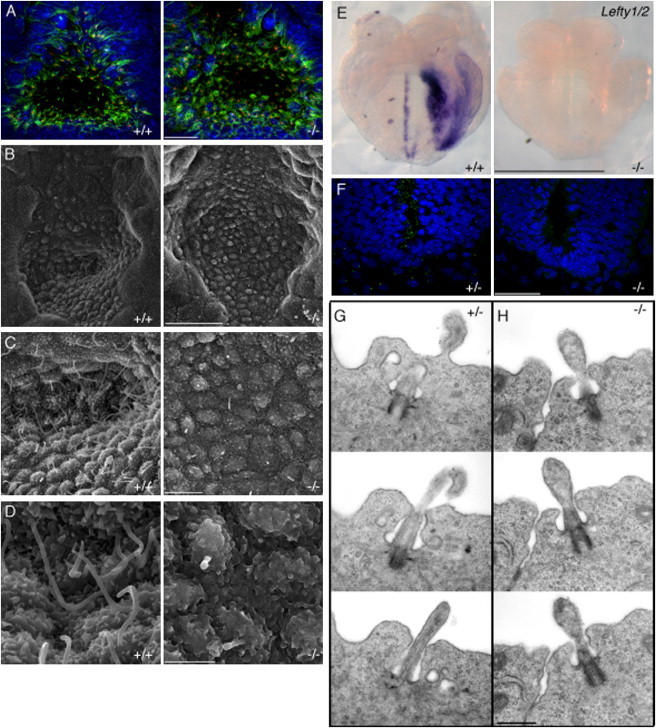
B9d1 Mutant Nodes and Neural Tubes Have Few and Abnormal Cilia and Mutants Do Not Express an Early Marker of LR Patterning
(A) Ventral view of E8.0 nodes with anterior oriented upward, depicting cilia (AcTub, green), basal bodies (Ninein, red), and nuclei (DAPI, blue). Scale bar represents 50 μm. Few cilia are present in B9d1 mutant nodes, whereas the number of basal bodies is unaffected.
(B–D) Scanning electron micrographs of E8.0 nodes reveal a reduction in the number of cilia in B9d1 mutants.
(B) The morphology of the node is normal in B9d1 mutants. Scale bar represents 20 μm.
(C) There is a dramatic reduction in the number of cilia in B9d1 mutant nodes. Scale bar represents 5 μm.
(D) The few cilia that form in B9d1 mutant nodes are short and can display bulges at their distal tips. Scale bar represents 2 μm.
(E) Frontal view of E8.0 embryos stained for expression of Lefty1/2. Expression is absent in the midline and left lateral plate mesoderm of B9d1 mutant embryos. Scale bar represents 500 μm.
(F) Transverse sections of E10.5 embryos showing the ventral neural tube and perineural mesenchyme stained with a marker of cilia (Arl13b, green) and nuclei (DAPI, blue). Scale bar represents 50 μm. B9d1 mutants show a reduction in the number of cilia in both tissues.
(G and H) Transmission electron micrographs of cilia in E9.5 neural tubes. Scale bar represents 500 nm.
(G) Serial sections through a B9d1 heterozygous E9.5 neural tube cilium.
(H) Serial sections through a B9d1 mutant E9.5 neural tube cilium reveal a docked basal body, a disrupted axoneme, and a bulging ciliary membrane.
Mouse B9d1 Is Required for Hh Signaling and Ciliary Protein Localization
Cilia are essential for the transduction of several intercellular signals, most notably Hh during neural tube development.23,24,55–59 B9d1 mutants lack cilia in the neural tube (Figure 2F) and exhibit holoprosencephaly (Figure 1H), a phenotype associated with abrogated Hh signaling.60 Further analysis of the neural tube by transmission electron microscopy (TEM) revealed that basal bodies in B9d1 mutant embryos dock properly at the apical surface of the cell yet do not form axonemes (Figures 2G and 2H).
To test whether Hh signaling is affected in B9d1 mutants, we examined patterning of the ventral neural tube.61 During neural tube development, high levels and durations of Hh signaling specify the ventral-most cell fates, including the floor plate, whereas moderate Hh signaling specifies more dorsal neural subtypes, including motor neurons.62,63
Molecular analysis of FoxA2 expression, a marker of the floor plate, indicated that the floor plate is reduced in B9d1 mutants (Figure 3A). Similar to the floor plate, high levels of Hh signaling are required for the induction of adjoining V3 interneuron precursors.64,65 Analysis of Nkx2.2, a marker of V3 interneuron precursors, revealed reduced expression in B9d1 mutants (Figure 3B). Additionally, B9d1 mutants showed a variable reduction in the number of motor neurons, as indicated by Islet1/2 expression (Figure 3C). The loss or reduction of ventral neural marker expression in B9d1 mutants was accompanied by the ventral expansion of Pax6 expression (Figures 3A–3C), normally restricted to the dorsal neural tube by Hh signaling.66 Together, these results indicate that B9d1 is required for high and moderate level Hh signaling in the neural tube.
Figure 3.
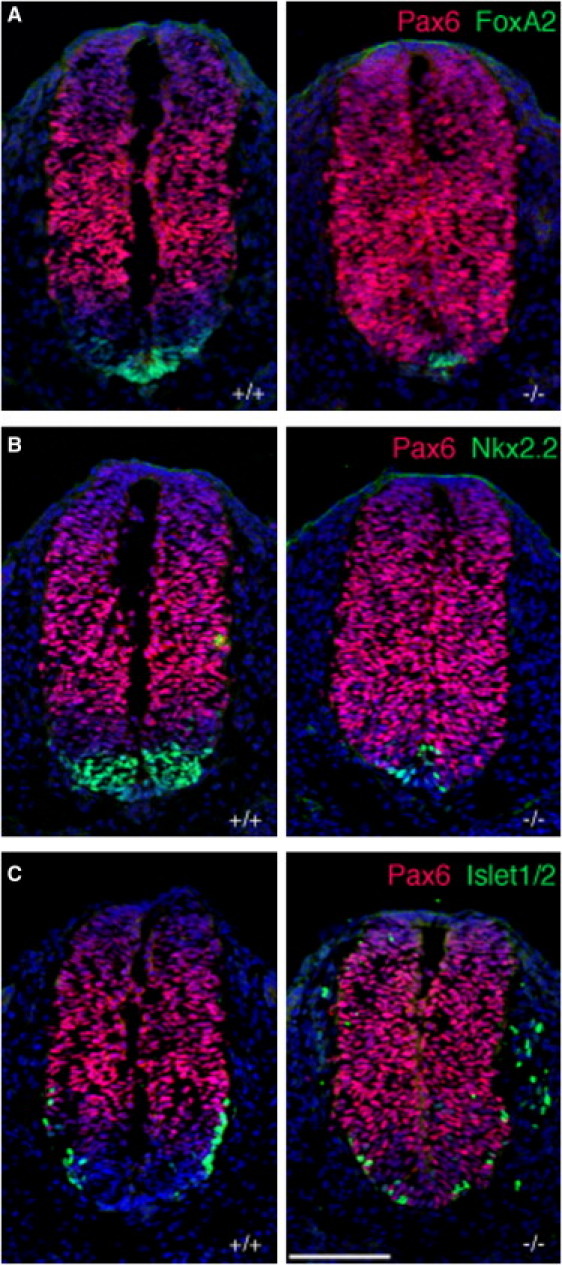
B9d1 Is Required for Hh-Dependent Neural Tube Patterning
Transverse sections of E9.5 embryos stained for expression of Pax6 (red) and, in green, FoxA2 (A), Nkx2.2 (B), or Islet1/2 (C). Nuclei are visualized with DAPI (blue).
(A) B9d1 mutants show reduction of the floorplate marker FoxA2 and ventrally expanded Pax6 expression.
(B) B9d1 mutants have reduced expression of the V3 interneuron precursor marker Nkx2.2.
(C) Expression of motor neuron marker Islet1/2 is reduced and ventrally shifted in B9d1 mutants.
Scale bar represents 100 μm.
In addition to their roles in promoting activation of the Hh transcriptional program, cilia are essential for the formation of the transcriptional repressor form of Gli3 (Gli3R).22,59 Unlike the neural tube, Shh controls anteroposterior limb bud patterning principally by inhibiting the formation of Gli3R. Shh is secreted from the zone of polarizing activity in the posterior of the limb bud, creating an opposing gradient of Gli3R decreasing from anterior to posterior.67 Loss of Gli3 results in polydactyly, similar to Shh gain of function.68,69 Mice with defects in ciliogenesis can display preaxial polydactyly, attributable to defects in Gli3R formation.22,59,70,71
The presence of preaxial polydactyly in B9d1 mutants (Figure 1F) was consistent with defective Gli3 processing. B9d1 mutants typically developed one or two supernumary preaxial digits on hindlimbs and occasionally on forelimbs, a limb phenotype more pronounced than that reported for Mks1 mutants.13,15 To assess whether B9d1 is required for Gli3 processing, we analyzed Gli3 in B9d1 heterozygote and mutant mouse embryo lysates. Full-length Gli3 was stabilized in mutant embryos compared to wild-type (Figure 4A), consistent with reduced Hh responsiveness.
Figure 4.
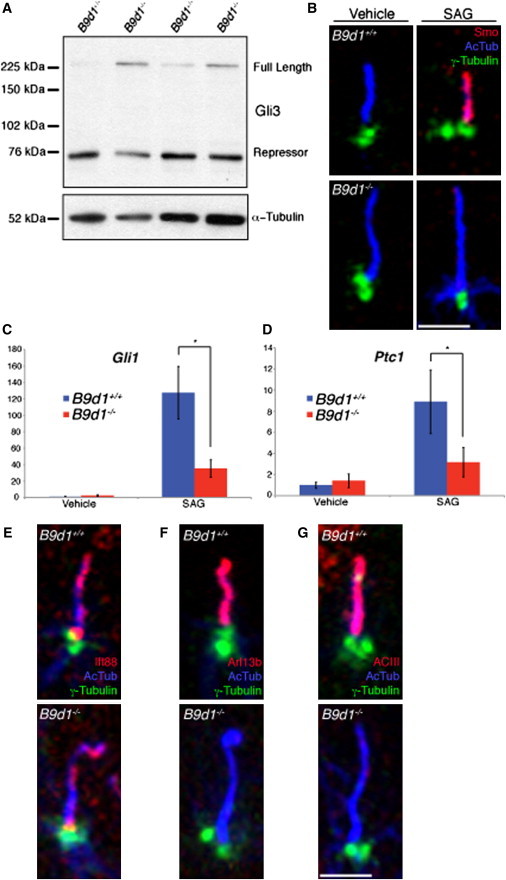
Hh Signaling and Protein Localization Defects in B9d1 Mutants
(A) Gli3 processing was assessed in E10.5 B9d1 heterozygote or mutant embryo lysates. Full-length Gli3 is stabilized in mutant lysates compared to the heterozygotes.
(B) B9d1 wild-type and mutant MEFs stained for cilia (AcTub, blue), basal bodies (γ-Tubulin, green), and Smo (red) in response to SAG treatment. Smo fails to localize to B9d1 mutant MEFs treated with SAG. Scale bar represents 2.5 μm.
(C and D) Gli1 and Ptc1 mRNA levels of B9d1 wild-type and mutant MEFs stimulated with SAG. The transcriptional response to SAG treatment is abrogated in B9d1 mutant MEFs. y axis values represent the fold change of mRNA compared to wild-type MEFs treated with vehicle only. Error bars represent the standard deviation. Asterisk indicates p < 0.0001.
(E–G) B9d1 wild-type and mutant MEFs stained for cilia (AcTub, blue), basal bodies (γ-Tubulin, green), and the ciliary proteins (red) Ift88 (E), Arl13b (F), and ACIII (G). Arl13b and ACIII fail to localize to B9d1 mutant cilia.
Scale bar represents 2.5 μm.
In response to Hh ligand, Smoothened (Smo), a positive regulator of Hh signaling, translocates into primary cilia and activates the pathway.20 To examine further the role of B9d1 in Hh signal transduction, we analyzed the localization of Smo to cilia in response to Smo AGonist (SAG). Similar to the cilia status of bile ducts, mouse embryonic fibroblasts (MEFs) derived from B9d1 mutant embryos were ciliated (Figure 4B). In response to SAG treatment, Smo localized to cilia in WT MEFs but not B9d1 mutant MEFs (Figure 4B; Figure S6), consistent with reduced Hh responsiveness. We also quantified the change in mRNA levels of Gli1 and Ptc1, general transcriptional targets of Hh signaling, in MEFs treated with SAG. B9d1 mutant MEFs treated with SAG showed blunted increases in Gli1 and Ptc1 activation as compared to WT SAG-treated MEFs (Figures 4C and 4D). Together, these results indicate that B9d1 is required for robust responsiveness to Hh signals even in cells that do not require B9d1 for ciliogenesis.
Given the failure of Smo to move to MEF cilia in response to SAG, we asked whether other ciliary proteins might fail to localize properly to the cilia of B9d1 mutant MEFs. Whereas the intraflagellar transport protein Ift88 localized to both WT and B9d1 mutant MEF cilia, the small GTPase Arl13b and the adenylate cyclase ACIII were found only in WT MEF cilia (Figures 4E–4G; Figure S6).22,55,72 This protein localization defect is similar to that seen upon disruption of Tctn1, Tctn2, Tmem67, or Cc2d2a, components of a larger transition zone complex that also contains B9 proteins (Table S3).73 Thus, B9d1 is required for the ciliary localization of a discrete subset of proteins, including Smo, Arl13b, and ACIII.
B9D2 Is a Candidate MKS Locus
B9d2, a B9 protein related to B9d1, localizes to the basal body and the ciliary axoneme, and loss of B9d2, either in vivo or in cultured cells, results in ciliogenic defects.11,14,42 In addition, conditional inactivation of B9d2 in the kidney produces cysts similar to those of Mks1 and B9d1 mutant mice.13–15 The phenotypic overlap observed between Mks1, B9d1, and B9d2 mutant mice, and the evolutionarily co-occurrence of these genes, suggested that human B9D1 and B9D2 variants could cause MKS.
To test this hypothesis, we used a combination of homozygosity mapping and candidate gene sequencing to screen B9D1 and B9D2 in 96 unrelated MKS patients. No regions of homozygosity overlapping with B9D1 segregated with MKS phenotypes in our patient cohort, and sequencing analysis without bias for linkage or homozygosity did not identify any nonsynonymous changes that would be consistent with a pathogenic role of this gene in our cohort of MKS patients. We note, however, that a recent study reported a B9D1 mutation in an individual with MKS.74 We identified a fifth-degree consanguineous multiplex family from Surinam with Indian-Pakistani background (Figure 5A), in which two affected fetuses displayed overlapping phenotypes including cystic kidneys, ductal plate malformation, polydactyly, and occipital encephalocele (Figures 5B–5M). Linkage analysis demonstrated identical homozygous haplotypes in both patients (Figure 5A) that were distinct from the haplotypes of the unaffected son (Figure 5A), but no regions of homozygosity in other known MKS disease genes. Sequencing revealed a homozygous, nonconservative missense mutation (c.301A>C) in B9D2 resulting in a serine to arginine substitution (p.Ser101Arg) in both affected individuals (Figure 5N). This change segregated with the MKS phenotype in this family, has not been described in all accessible databases of human genome variation, and was not present among 300 ethnically matched control chromosomes or 388 control chromosomes from other ethnic backgrounds.
Figure 5.
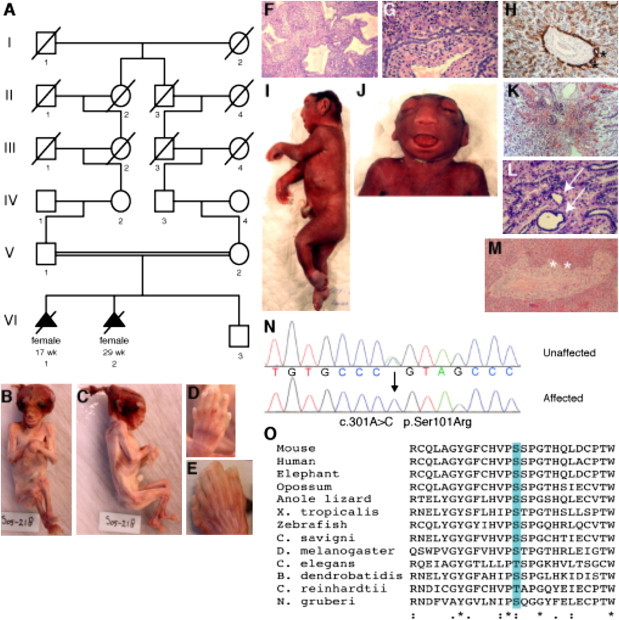
A B9D2 Mutation Is Associated with MKS
(A) Pedigree structure of a fifth-degree consanguineous multiplex family, which includes two affected MKS individuals harboring a homozygous point mutation in B9D2 (c.301A>C).
(B and C) Frontal and lateral views of an affected fetus aborted at the 17th gestational week because of occipital encephalocele.
(D and E) Polydactyly of the hands and feet.
(F) H&E-stained kidney section shows renal cysts.
(G) H&E-stained liver section reveals ductal plate malformation.
(H) Cytokeratin-19 staining shows defective modeling and persistence of bile duct remnants, confirming the ductal plate malformation (asterisks).
(I and J) Affected fetus aborted at the 29th gestational week. Postmortem analysis confirmed anencephaly, with occipital encephalocele, sacral dimple, and postaxial polydactyly of both hands and feet.
(K) H&E-stained cystic kidney section shows reduction of glomeruli and tubuli.
(L) H&E-stained kidney section at the level of the medulla showing some small cysts (arrows). Compared to normal fetal kidney control samples, the affected renal tissue showed less glomeruli. The Bowman's capsule was not clearly dilated and the proximal renal tubuli seemed normally developed. Immunohistochemistry with epithelial membrane antigen (EMA) and CD15 antibodies (data not shown) demonstrated that cyst formations mainly affected the distal tubuli, although some small renal cysts were also seen at the medullary level.
(M) H&E-stained liver section reveals fetal hematopoesis and ductal plate malformation (asterisks).
(N) B9D2 sequence traces are shown for an affected fetus (bottom) and one of the parents (top). Arrow indicates position of homozygous mutation (c.301A>C).
(O) ClustalW alignment of B9D2 protein sequences from diverse ciliated eukaryotes reveals conservation of either serine or threonine at the position corresponding to Ser101 of the human sequence (highlighted). Asterisks indicate identical residues, colons indicate conserved residues, and periods indicate semiconserved residues.
Residue 101 is invariantly serine in chordate B9D2 proteins and is either a serine or threonine in the B9D2 orthologs of ciliated protists Chlamydomonas, Naegleria, and Batrachochytrium (Figure 5O). This high degree of conservation suggests that serine 101 is important for B9D2 function and that substituting this small, polar, nonionizable residue with a large, basic residue such as arginine may not be tolerated (Figure S7). Similarly, in silico analysis predicted that this mutation is pathogenic (Table S1).
The Candidate MKS p.Ser101Arg Mutation Causes Loss of B9D2 Function
We have shown previously that morpholino (MO) suppression of mks1 in zebrafish results in developmental defects that include shortened body axis, broad notochords, long, thin somites, and failure of tail extension.34 Further, we and others have demonstrated the tractability of in vivo rescue in this system to determine the pathogenic effect of alleles found in ciliary genes with high specificity and sensitivity.75–77 To test the pathogenicity of the b9d2 MKS allele, we generated a splice-blocking MO against zebrafish b9d2 and attempted rescue with control or mutant human B9D2 mRNA. Injection of increasing amounts (2, 4, 6 ng) of b9d2 MO resulted in dosage-dependent phenotypes that can be grouped into three classes based on previously reported criteria (normal, class I, and class II).34 Class I embryos displayed mildly shortened body axes, mediolaterally elongated somites, and notochord imperfections; class II embryos displayed severely shortened body axes, mediolaterally elongated somites, and widened and kinked notochords (Figure 6A; Figure S8).
Figure 6.
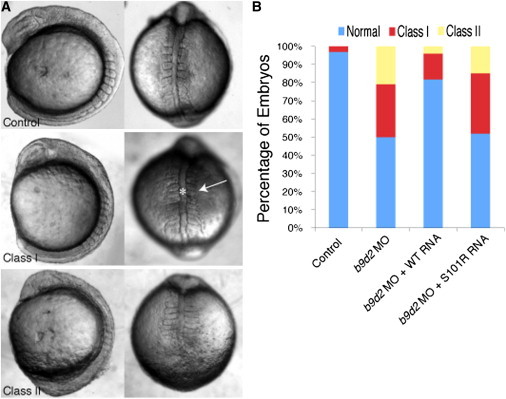
B9D2 p.Ser101Arg Fails to Rescue Zebrafish Phenotypes Caused by Suppression of b9d2
(A) Injection of MOs against b9d2 produces defects in zebrafish development including shortened body axis, widened and kinked notochord (asterisk), and broadened somites (arrow).
(B) Graph of the frequency of class I and II phenotypes among zebrafish embryos. Whereas coinjection of wild-type human B9D2 RNA (WT RNA) reduces the frequency of developmental defects caused by 2 ng b9d2 MO, B9D2 S101R RNA does not rescue the phenotype (b9d2 MO + S101R).
The specificity of the MO-induced phenotypes was confirmed by coinjection of human B9D2 mRNA. Upon masked scoring of 50–150 embryos per injection, we observed that whereas 29% of the b9d2 morphants were designated as class I and 21% were class II, coinjection of b9d2 morpholino with 60 pg human B9D2 mRNA reduced the prevalence of class I to 14% and of class II to 4%. In contrast, coinjection of b9d2 morpholino with 60 pg of human B9D2 c.301A>C mRNA yielded morphants of which 33% were scored as class I and 14% were class II. The failure of B9D2 c.301A>C to reduce the frequency of b9d2 morpholino phenotypes suggests that the MKS-associated mutation is a strong hypomorphic or functional null allele (Figure 6B).
Effect of the B9D2 Mutation on Protein Function
To investigate the pathogenicity of the B9D2 mutant allele and the mechanisms by which it may result in ciliary dysfunction and human disease, we explored the potential consequences of the p.Ser101Arg amino acid substitution in mammalian cells. B9D2 has been shown to localize primarily to the basal body and, under certain conditions, to the axoneme of the primary cilium.11 We therefore tested whether introduction of the Arg101 residue affects B9D2 localization. Upon expression of GFP-B9D2 and GFP-B9D2 S101R in hTERT-RPE1 cells, we found that both localize to the basal body (Figure 7), rendering mislocalization an unlikely cause of disease and pointing instead to potential biochemical causes.
Figure 7.
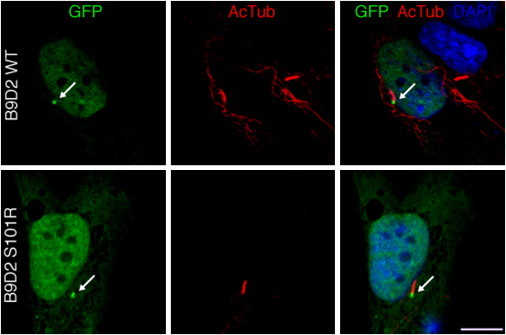
Localization of Wild-Type B9D2 and B9D2 S101R
hTERT-RPE1 cells expressing GFP-tagged wild-type B9D2 or B9D2 S101R (green). Expressions of both are enriched at the base of cilia (AcTub, red, arrows). Nuclei are visualized with DAPI (blue). Scale bar represents 10 μm.
The phenotypic overlap caused by loss of any of the three mouse B9 proteins raised the possibility that the B9 proteins promote ciliogenesis by acting together in a nonredundant fashion. Similarly, the presence of MKS-associated mutations in B9D1, B9D2, and MKS1 suggests that these proteins may function cooperatively in humans, reminiscent of the related ciliopathy, Bardet-Biedl syndrome (BBS [MIM 209900]), in which the products of several causative genes can form a complex.78 To investigate whether B9 proteins might similarly form a MKS-related complex, we assessed whether tagged B9 proteins coimmunoprecipitated. GFP-tagged versions of Mks1, B9d1, and B9d2 coimmunoprecipitated V5-tagged versions of the other B9 proteins (Figure 8). GFP-tagged B9 proteins did not coimmunoprecipitate with the V5 vector or a V5-tagged control protein, Rer1, indicating that the interaction of the B9 proteins with each other is specific.
Figure 8.
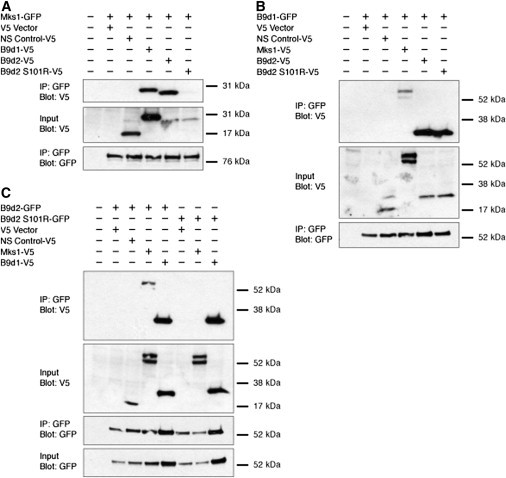
B9 Proteins Form a Complex and B9d2 S101R Fails to Interact with Mks1
Coimmunoprecipitation of epitope-tagged B9 proteins. Immunoprecipitation of GFP-tagged Mks1 (A), B9d1 (B), B9d2 and B9d2 S101R (C) revealed interactions with V5-tagged Mks1, B9d1, B9d2, and B9d2S101R but not vector alone or a nonspecific (NS) control protein, Rer1. In contrast to all other B9 protein combinations, B9d2 S101R did not interact with Mks1.
To confirm further the observed interactions, we created NIH 3T3 cell lines harboring stable, single integrations of a control plasmid expressing the localization and affinity purification (LAP) tag (pGLAP5) or a plasmid expressing B9d1 with a carboxy-terminally fused LAP (pGLAP5-B9d1). We tandem affinity purified LAP proteins from the control and B9d1-LAP-expressing cells, separated them by gel electrophoresis, sectioned the gel lanes, and subjected them to mass spectrometry.29,35 Mass spectrometric analysis detected abundant peptides of Mks1 (50 peptides with 37% coverage) and B9d2 (32 peptides with 42% coverage) among the proteins that copurified with B9d1-LAP. We also identified a number of interacting proteins that have been suggested recently to be part of a larger transition zone complex functioning within the cilium (Table S3).73
To determine how the p.Ser101Arg mutation affects B9D2 activity, we asked whether this mutation alters B9 protein complex formation. Reciprocal coimmunoprecipitations indicated that B9d2 p.Ser101Arg failed to interact with Mks1, without compromising its interaction with B9d1 (Figure 8). These results indicate that the B9 proteins interact physically and that the MKS-associated mutant form of B9d2 disrupts its interaction with Mks1, thus compromising the integrity of this ciliogenic complex. Therefore, we propose that mutations that disrupt the interactions among components of the B9 complex result in human MKS.
Discussion
In the last decade, our understanding of the role of primary cilia in human disorders has benefited from the identification of a burgeoning group of ciliopathy-associated genes. In this study, we showed that disruption of B9d1 in the mouse perturbs ciliogenesis and ciliary protein localization, abrogates Hh signaling, and results in phenotypes that fall on the severe end of the ciliopathy clinical spectrum. In addition, we identified a nonconservative missense mutation in B9D2 in individuals affected with MKS. Finally, we demonstrated that the B9 domain containing proteins physically interact and that the MKS-associated mutant form of B9d2 has diminished function and reduced capacity to interact with Mks1, previously implicated in MKS.2,79 Together, these results suggest that a complex of B9 proteins cooperatively supports mammalian ciliogenesis and ciliary protein localization, and abrogated complex function results in MKS.
B9d1 Is Essential for Ciliogenesis, Ciliary Protein Localization, and Hh Signal Transduction
Ciliary defects compromise the response to Hh signals in vertebrates by abrogating the ability of Smo to activate both Gli2 and Gli3R formation.19,59 The Hh-related phenotypes of B9d1 mutants include dorsalization of the neural tube consistent with decreased Gli2 activity, as well as limb bud patterning defects associated with disrupted Gli3 processing. In MEFs, B9d1 is dispensible for ciliogenesis but is required for robust response to the Hh pathway activator SAG. This probably reflects the requirement for B9d1 for the movement of Smo in response to pathway activation and supports the contention that some MKS phenotypes, such as polydactyly, are due to disruptions in Hh signal transduction.15
Because cilia may participate in multiple signaling pathways, other manifestations of MKS may be due to defects in intercellular communication apart from Hh signaling.33,80–82 In addition to Smo, B9d1 is essential for the ciliary localization of Arl13b and ACIII. Similar defects in ciliary membrane composition are observed in mouse mutants affecting B9d1-interacting proteins.73 It is likely that alterations in ciliary membrane composition affect diverse ciliary functions, resulting in some of the pleiotropic phenotypes associated with MKS.
Mutation of B9D2 Is Associated with MKS
We detected a homozygous missense mutation affecting a highly conserved residue of B9D2 in the affected MKS individuals of a single pedigree, suggesting that B9D2 is essential for human development. In addition to the human genetic data, the failure of B9D2 bearing the p.Ser101Arg mutation to rescue zebrafish b9d2 morphants or to interact with Mks1 indicates that this mutation abrogates function.
A significant fraction of cases of other genetically heterogeneous ciliopathies, such as nephronophthisis and BBS, are caused by private mutations.83–85 Our work indicates that this may be true for MKS as well. Human genetic studies with limited pedigrees are insufficient to prove the causality of alleles and, by extension, the causal role of specific genes in the phenotype. However, testing the allele in a physiologically relevant system provides insight into its function. In the case of the MKS-associated allele of B9D2, we used three approaches to provide evidence of causality: human genetics, in vivo rescue, and biochemical interaction data.
Although we did not detect B9D1 mutations in our cohort of 96 MKS patients, a recent study identified B9D1 mutations as a cause of MKS.74 If B9D1 mutations result in early gestational lethality, as exhibited by the B9d1 mutant mice, B9D1 mutations detected in postnatal MKS cases may not reflect the prevalence of the alleles.
The B9 Domain of B9D2 Promotes Interaction with MKS1
It is striking that the B9 domain-containing proteins are found only in organisms that form cilia and that all B9 proteins are involved in ciliary functions in both C. elegans and vertebrates.11–15 The p.Ser101Arg mutation of B9D2 identified in MKS individuals occurs within the B9 domain, suggesting that it is critical to protein function. The B9 domain is related to the C2 domain involved in calcium-dependent lipid binding present in several other MKS proteins, including MKS5 (RPGRIP1L) and MKS6 (CC2D2A).86 Despite the possibility that MKS1, B9D1, and B9D2 suggests shared biochemical activities with C2 domain proteins, the overlapping ciliary defects exhibited by the mouse B9 mutants suggest that they are not functionally redundant either with each other or with C2 domain proteins.13,15
The MKS-associated mutation in the B9 domain of B9D2 does not destabilize the protein, interfere with its localization to the ciliary base, or compromise its interaction with B9d1. Instead, the inability of the mutant form of B9d2 to physically interact with Mks1 reveals that the mutation compromises formation of the B9 protein complex. These results suggest that B9 domains may support protein-protein interactions and that disruption of the B9 protein complex may underlie some forms of MKS.
B9 Proteins Have Nonredundant Functions in Development
In support of the contention that B9 proteins function together in a complex, B9d1 mutant mice display phenotypes associated with the MKS1 variant of MKS, including cleft palate, holoprosencephaly, microphthalmia, and polydactyly.2,16–18 Furthermore, the two MKS fetuses harboring B9D2 mutations displayed polydactyly. The late gestational MKS fetus, in addition to occipital encephalocele, exhibited anencephaly and a sacral dimple, reflecting neural tube closure defects.
Mutations in each B9 gene in humans can be associated with MKS, and loss of each B9 protein in mice causes phenotypes consistent with MKS.2,13–15,74 However, the descriptions of the three B9 mouse mutant phenotypes are not identical. The B9d2-null phenotype has not been described, but conditional deletion of B9d2 in the nervous system and kidneys caused kidney cysts, hydrocephalus, and a loss or stunting of cilia in the CNS.14
Two distinct Mks1 mutations cause cystic kidneys, polydactyly, neural tube defects, reduced skull ossification, other skeletal abnormalities, and hepatic defects. Mks1krc is a splice-site mutation that results in deletion of the B9 domain, and Mks1del64-323 is an in-frame deletion of exons 3–10 that removes the first nine amino acids of the B9 domain.13,15 Despite extensive phenotypic similarities, the two mutants are not indistinguishable. At E10.5, Mks1krc mutants did not display gross abnormalities, whereas Mks1del64-323 mutants displayed expansion of the mesencephalic flexure, reduced telencephalon size, and microphthalmia, similar to B9d1 mutants.13,15 Moreover, Mks1del64-323 mutants exhibited neural tube patterning defects similar to those of B9d1 mutants and more profound than those of Mks1krc mutants.13,15 Because Mks1krc phenotypes were assessed on a C3H or mixed C3H/CD1 background and Mks1del64-323 phenotypes were assessed on a C3H or C57Bl/6 background (C. Lo, personal communication), the phenotypic differences may reflect differences in allele strength or modification by background-dependent loci.13,15
The Relationship of Ciliary Dysfunction and Ciliopathies
In addition to MKS, defects in the function of primary cilia are thought to underlie related disorders including BBS, Leber congenital amaurosis (LCA [MIM 204000]), and Senior-Løken syndrome (SLSN [MIM 266900]). Several of these disorders share retinal degeneration as a feature but include other characteristics such as obesity, polydactyly, and renal abnormalities (BBS), midface hypoplasia (LCA), or nephronophthisis, a cystic kidney disorder (SLSN). Further examples of ciliopathies include Joubert syndrome (JBTS [MIM 213300]), characterized by hypoplasia of the cerebellar vermis, and Orofaciodigital syndrome (OFD [MIM 311200]), characterized by malformation of the face, mouth, and digits. It is not clear how ciliary dysfunction results in such diverse phenotypes.87
Moreover, some ciliopathy genes display extensive allelism. For example, mutations in CEP290 are associated with MKS, JBTS, SLSN, LCA, and BBS.88 Although some CEP290 mutations may affect discrete functions or compromise activity to different degrees, other CEP290 mutations are associated with more than one ciliopathy, suggesting that modifier effects may account for the distinct resultant phenotypes.88,89
In support of the possibility that ciliary modifier loci have strong effects on expressivity, we observed that mouse B9d1 mutants with C57/Bl6 or mixed CD1-C57/Bl6 backgrounds displayed distinct phenotypes. It will be interesting to assess whether other B9D1 or B9D2 mutations underlie ciliopathies related to MKS, such as JBTS, and whether weaker alleles of B9 genes modify other ciliopathies.
A second possible explanation for the origins of different ciliopathy phenotypes is that different ciliogenic protein complexes have distinct functions, affecting ciliogenesis to different extents or in different tissues. In support of this possibility, B9 mutants display tissue-specific defects in ciliogenesis and ciliary protein mislocalization. For example, in mouse B9d1 mutants, cilia are absent in the node and neural tube, reduced in number and length in cystic kidneys, and present in bile ducts and MEFs. Similarly, in mouse Mks1 mutants, cilia are reduced in number and length in the node, limb buds, and neural tube, but not in lung airways and bile ducts.15
As in the mammalian bile duct, B9 proteins are not required for ciliogenesis in C. elegans, as shown by the fact that nematodes mutant for the orthologs of Mks1, B9d1, B9d2, or a combination of all three do not display overt structural ciliary defects.11,12 The C. elegans orthologs of B9 proteins are, however, required for cilia-dependent sensory functions.12
Thus, B9 proteins are required for the formation of cilia in some tissues and organisms and are required for ciliary function in others. B9d1 is essential for the localization of a subset of ciliary proteins, so defects in ciliary protein composition are likely to underlie some intercellular signaling abnormalities in mouse B9 mutants. The similarity of mouse Mks1 and B9d1 mutant phenotypes to human MKS suggests that MKS may be caused not by the absence of cilia, but by tissue-specific ciliary loss and dysfunction.
Acknowledgments
We thank Caroline Miller and JoDee Fish at the Gladstone Institute Histology Core, the Berkeley EM lab, Kurt Thorn and the Nikon Imaging Center at UCSF, William Wallantus and the Institute for Regenerative Medicine Imaging Core at UCSF, Chris Adams and the Stanford University Mass Spectrometry Bio-X core, and members of the J.F.R. lab for discussions and critical reading of this manuscript. This work was funded by grants from the NSF to W.E.D., the NIH (R01DK072301, R01DK075972, and R01HD04260 to N.K. and R01AR054396 to J.F.R.), the Burroughs Wellcome Fund, the Packard Foundation, and the Sandler Family Supporting Foundation to J.F.R., the PKD Foundation and the Deutsche Nierenstiftung to C.B., and the Deutsche Forschungsgemeinschaft (DFG BE 3910/4-1 and SFB/TRR57) to K.Z. and C.B. N.K. is a Distinguished Brumley Professor.
Contributor Information
Nicholas Katsanis, Email: katsanis@cellbio.duke.edu.
Carsten Bergmann, Email: carsten.bergmann@bioscientia.de.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
AlignGVGD, http://agvgd.iarc.fr/index.php
European Conditional Mouse Mutagenesis Program (EUCOMM), http://www.eucomm.org/
Genome Database (GDB), http://www.gdb.org/
HUGO Gene Nomenclature Committee (HGNC), http://www.genenames.org/
ModBase, http://modbase.compbio.ucsf.edu
Mutation Taster, http://www.mutationtaster.org
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
PDB2PQR Program, http://kryptonite.nbcr.net/pdb2pqr/
References
- 1.Badano J.L., Mitsuma N., Beales P.L., Katsanis N. The ciliopathies: An emerging class of human genetic disorders. Annu. Rev. Genomics Hum. Genet. 2006;7:125–148. doi: 10.1146/annurev.genom.7.080505.115610. [DOI] [PubMed] [Google Scholar]
- 2.Kyttälä M., Tallila J., Salonen R., Kopra O., Kohlschmidt N., Paavola-Sakki P., Peltonen L., Kestilä M. MKS1, encoding a component of the flagellar apparatus basal body proteome, is mutated in Meckel syndrome. Nat. Genet. 2006;38:155–157. doi: 10.1038/ng1714. [DOI] [PubMed] [Google Scholar]
- 3.Valente E.M., Logan C.V., Mougou-Zerelli S., Lee J.H., Silhavy J.L., Brancati F., Iannicelli M., Travaglini L., Romani S., Illi B. Mutations in TMEM216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes. Nat. Genet. 2010;42:619–625. doi: 10.1038/ng.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith U.M., Consugar M., Tee L.J., McKee B.M., Maina E.N., Whelan S., Morgan N.V., Goranson E., Gissen P., Lilliquist S. The transmembrane protein meckelin (MKS3) is mutated in Meckel-Gruber syndrome and the wpk rat. Nat. Genet. 2006;38:191–196. doi: 10.1038/ng1713. [DOI] [PubMed] [Google Scholar]
- 5.Frank V., den Hollander A.I., Brüchle N.O., Zonneveld M.N., Nürnberg G., Becker C., Du Bois G., Kendziorra H., Roosing S., Senderek J. Mutations of the CEP290 gene encoding a centrosomal protein cause Meckel-Gruber syndrome. Hum. Mutat. 2008;29:45–52. doi: 10.1002/humu.20614. [DOI] [PubMed] [Google Scholar]
- 6.Delous M., Baala L., Salomon R., Laclef C., Vierkotten J., Tory K., Golzio C., Lacoste T., Besse L., Ozilou C. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat. Genet. 2007;39:875–881. doi: 10.1038/ng2039. [DOI] [PubMed] [Google Scholar]
- 7.Tallila J., Jakkula E., Peltonen L., Salonen R., Kestilä M. Identification of CC2D2A as a Meckel syndrome gene adds an important piece to the ciliopathy puzzle. Am. J. Hum. Genet. 2008;82:1361–1367. doi: 10.1016/j.ajhg.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bergmann C., Fliegauf M., Brüchle N.O., Frank V., Olbrich H., Kirschner J., Schermer B., Schmedding I., Kispert A., Kränzlin B. Loss of nephrocystin-3 function can cause embryonic lethality, Meckel-Gruber-like syndrome, situs inversus, and renal-hepatic-pancreatic dysplasia. Am. J. Hum. Genet. 2008;82:959–970. doi: 10.1016/j.ajhg.2008.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shaheen R., Faqeih E., Seidahmed M.Z., Sunker A., Alali F.E., Khadijah A., Alkuraya F.S.A. A TCTN2 mutation defines a novel Meckel Gruber syndrome locus. Hum. Mutat. 2011;32:573–578. doi: 10.1002/humu.21507. [DOI] [PubMed] [Google Scholar]
- 10.Zhang D., Aravind L. Identification of novel families and classification of the C2 domain superfamily elucidate the origin and evolution of membrane targeting activities in eukaryotes. Gene. 2010;469:18–30. doi: 10.1016/j.gene.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bialas N.J., Inglis P.N., Li C., Robinson J.F., Parker J.D., Healey M.P., Davis E.E., Inglis C.D., Toivonen T., Cottell D.C. Functional interactions between the ciliopathy-associated Meckel syndrome 1 (MKS1) protein and two novel MKS1-related (MKSR) proteins. J. Cell Sci. 2009;122:611–624. doi: 10.1242/jcs.028621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Williams C.L., Winkelbauer M.E., Schafer J.C., Michaud E.J., Yoder B.K. Functional redundancy of the B9 proteins and nephrocystins in Caenorhabditis elegans ciliogenesis. Mol. Biol. Cell. 2008;19:2154–2168. doi: 10.1091/mbc.E07-10-1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cui C., Chatterjee B., Francis D., Yu Q., SanAgustin J.T., Francis R., Tansey T., Henry C., Wang B., Lemley B. Disruption of Mks1 localization to the mother centriole causes cilia defects and developmental malformations in Meckel-Gruber syndrome. Dis Model Mech. 2011;4:43–56. doi: 10.1242/dmm.006262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Town T., Breunig J.J., Sarkisian M.R., Spilianakis C., Ayoub A.E., Liu X., Ferrandino A.F., Gallagher A.R., Li M.O., Rakic P., Flavell R.A. The stumpy gene is required for mammalian ciliogenesis. Proc. Natl. Acad. Sci. USA. 2008;105:2853–2858. doi: 10.1073/pnas.0712385105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weatherbee S.D., Niswander L.A., Anderson K.V. A mouse model for Meckel syndrome reveals Mks1 is required for ciliogenesis and Hedgehog signaling. Hum. Mol. Genet. 2009;18:4565–4575. doi: 10.1093/hmg/ddp422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Consugar M.B., Kubly V.J., Lager D.J., Hommerding C.J., Wong W.C., Bakker E., Gattone V.H., 2nd, Torres V.E., Breuning M.H., Harris P.C. Molecular diagnostics of Meckel-Gruber syndrome highlights phenotypic differences between MKS1 and MKS3. Hum. Genet. 2007;121:591–599. doi: 10.1007/s00439-007-0341-3. [DOI] [PubMed] [Google Scholar]
- 17.Khaddour R., Smith U., Baala L., Martinovic J., Clavering D., Shaffiq R., Ozilou C., Cullinane A., Kyttälä M., Shalev S., SOFFOET (Société Française de Foetopathologie) Spectrum of MKS1 and MKS3 mutations in Meckel syndrome: A genotype-phenotype correlation. Mutation in brief #960. Online. Hum. Mutat. 2007;28:523–524. doi: 10.1002/humu.9489. [DOI] [PubMed] [Google Scholar]
- 18.Tallila J., Salonen R., Kohlschmidt N., Peltonen L., Kestilä M. Mutation spectrum of Meckel syndrome genes: One group of syndromes or several distinct groups? Hum. Mutat. 2009;30:E813–E830. doi: 10.1002/humu.21057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goetz S.C., Anderson K.V. The primary cilium: A signalling centre during vertebrate development. Nat. Rev. Genet. 2010;11:331–344. doi: 10.1038/nrg2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Corbit K.C., Aanstad P., Singla V., Norman A.R., Stainier D.Y., Reiter J.F. Vertebrate Smoothened functions at the primary cilium. Nature. 2005;437:1018–1021. doi: 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- 21.Rohatgi R., Milenkovic L., Scott M.P. Patched1 regulates hedgehog signaling at the primary cilium. Science. 2007;317:372–376. doi: 10.1126/science.1139740. [DOI] [PubMed] [Google Scholar]
- 22.Haycraft C.J., Banizs B., Aydin-Son Y., Zhang Q., Michaud E.J., Yoder B.K. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 2005;1:e53. doi: 10.1371/journal.pgen.0010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huangfu D., Liu A., Rakeman A.S., Murcia N.S., Niswander L., Anderson K.V. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature. 2003;426:83–87. doi: 10.1038/nature02061. [DOI] [PubMed] [Google Scholar]
- 24.Reiter J.F., Skarnes W.C. Tectonic, a novel regulator of the Hedgehog pathway required for both activation and inhibition. Genes Dev. 2006;20:22–27. doi: 10.1101/gad.1363606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Eyll J.M., Pierreux C.E., Lemaigre F.P., Rousseau G.G. Shh-dependent differentiation of intestinal tissue from embryonic pancreas by activin A. J. Cell Sci. 2004;117:2077–2086. doi: 10.1242/jcs.01067. [DOI] [PubMed] [Google Scholar]
- 26.Leathem A., Atkins N. Lectin binding to formalin-fixed paraffin sections. J. Clin. Pathol. 1983;36:747–750. doi: 10.1136/jcp.36.7.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nagy A. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2003. Manipulating the Mouse Embryo: A Laboratory Manual. [Google Scholar]
- 28.Meno C., Ito Y., Saijoh Y., Matsuda Y., Tashiro K., Kuhara S., Hamada H. Two closely-related left-right asymmetrically expressed genes, lefty-1 and lefty-2: Their distinct expression domains, chromosomal linkage and direct neuralizing activity in Xenopus embryos. Genes Cells. 1997;2:513–524. doi: 10.1046/j.1365-2443.1997.1400338.x. [DOI] [PubMed] [Google Scholar]
- 29.Torres J.Z., Miller J.J., Jackson P.K. High-throughput generation of tagged stable cell lines for proteomic analysis. Proteomics. 2009;9:2888–2891. doi: 10.1002/pmic.200800873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Warner J.P., Barron L.H., Goudie D., Kelly K., Dow D., Fitzpatrick D.R., Brock D.J. A general method for the detection of large CAG repeat expansions by fluorescent PCR. J. Med. Genet. 1996;33:1022–1026. doi: 10.1136/jmg.33.12.1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mathe E., Olivier M., Kato S., Ishioka C., Hainaut P., Tavtigian S.V. Computational approaches for predicting the biological effect of p53 missense mutations: A comparison of three sequence analysis based methods. Nucleic Acids Res. 2006;34:1317–1325. doi: 10.1093/nar/gkj518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schwarz J.M., Rödelsperger C., Schuelke M., Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 33.Gerdes J.M., Liu Y., Zaghloul N.A., Leitch C.C., Lawson S.S., Kato M., Beachy P.A., Beales P.L., DeMartino G.N., Fisher S. Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nat. Genet. 2007;39:1350–1360. doi: 10.1038/ng.2007.12. [DOI] [PubMed] [Google Scholar]
- 34.Leitch C.C., Zaghloul N.A., Davis E.E., Stoetzel C., Diaz-Font A., Rix S., Alfadhel M., Lewis R.A., Eyaid W., Banin E. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat. Genet. 2008;40:443–448. doi: 10.1038/ng.97. [DOI] [PubMed] [Google Scholar]
- 35.Cheeseman I.M., Desai A. A combined approach for the localization and tandem affinity purification of protein complexes from metazoans. Sci. STKE. 2005;2005:pl1. doi: 10.1126/stke.2662005pl1. [DOI] [PubMed] [Google Scholar]
- 36.Shevchenko A., Tomas H., Havlis J., Olsen J.V., Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2006;1:2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- 37.Baker N.A., Sept D., Joseph S., Holst M.J., McCammon J.A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14 doi: 10.1016/0263-7855(96)00018-5. 33-8, 27-8. [DOI] [PubMed] [Google Scholar]
- 39.Pieper U., Webb B.M., Barkan D.T., Schneidman-Duhovny D., Schlessinger A., Braberg H., Yang Z., Meng E.C., Pettersen E.F., Huang C.C. ModBase, a database of annotated comparative protein structure models, and associated resources. Nucleic Acids Res. 2011;39(Database issue):D465–D474. doi: 10.1093/nar/gkq1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dolinsky T.J., Nielsen J.E., McCammon J.A., Baker N.A. PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32(Web Server issue) doi: 10.1093/nar/gkh381. W665-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dolinsky T.J., Czodrowski P., Li H., Nielsen J.E., Jensen J.H., Klebe G., Baker N.A. PDB2PQR: Expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007;35(Web Server issue) doi: 10.1093/nar/gkm276. W522-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ponsard C., Skowron-Zwarg M., Seltzer V., Perret E., Gallinger J., Fisch C., Dupuis-Williams P., Caruso N., Middendorp S., Tournier F. Identification of ICIS-1, a new protein involved in cilia stability. Front. Biosci. 2007;12:1661–1669. doi: 10.2741/2178. [DOI] [PubMed] [Google Scholar]
- 43.Gunay-Aygun M. Liver and kidney disease in ciliopathies. Am. J. Med. Genet. C. Semin. Med. Genet. 2009;151C:296–306. doi: 10.1002/ajmg.c.30225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nonaka S., Tanaka Y., Okada Y., Takeda S., Harada A., Kanai Y., Kido M., Hirokawa N. Randomization of left-right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell. 1998;95:829–837. doi: 10.1016/s0092-8674(00)81705-5. [DOI] [PubMed] [Google Scholar]
- 45.Marszalek J.R., Ruiz-Lozano P., Roberts E., Chien K.R., Goldstein L.S. Situs inversus and embryonic ciliary morphogenesis defects in mouse mutants lacking the KIF3A subunit of kinesin-II. Proc. Natl. Acad. Sci. USA. 1999;96:5043–5048. doi: 10.1073/pnas.96.9.5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takeda S., Yonekawa Y., Tanaka Y., Okada Y., Nonaka S., Hirokawa N. Left-right asymmetry and kinesin superfamily protein KIF3A: New insights in determination of laterality and mesoderm induction by kif3A−/− mice analysis. J. Cell Biol. 1999;145:825–836. doi: 10.1083/jcb.145.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meno C., Saijoh Y., Fujii H., Ikeda M., Yokoyama T., Yokoyama M., Toyoda Y., Hamada H. Left-right asymmetric expression of the TGF beta-family member lefty in mouse embryos. Nature. 1996;381:151–155. doi: 10.1038/381151a0. [DOI] [PubMed] [Google Scholar]
- 48.Meno C., Shimono A., Saijoh Y., Yashiro K., Mochida K., Ohishi S., Noji S., Kondoh H., Hamada H. lefty-1 is required for left-right determination as a regulator of lefty-2 and nodal. Cell. 1998;94:287–297. doi: 10.1016/s0092-8674(00)81472-5. [DOI] [PubMed] [Google Scholar]
- 49.Izraeli S., Lowe L.A., Bertness V.L., Good D.J., Dorward D.W., Kirsch I.R., Kuehn M.R. The SIL gene is required for mouse embryonic axial development and left-right specification. Nature. 1999;399:691–694. doi: 10.1038/21429. [DOI] [PubMed] [Google Scholar]
- 50.Rana A.A., Barbera J.P., Rodriguez T.A., Lynch D., Hirst E., Smith J.C., Beddington R.S. Targeted deletion of the novel cytoplasmic dynein mD2LIC disrupts the embryonic organiser, formation of the body axes and specification of ventral cell fates. Development. 2004;131:4999–5007. doi: 10.1242/dev.01389. [DOI] [PubMed] [Google Scholar]
- 51.Kamura K., Kobayashi D., Uehara Y., Koshida S., Iijima N., Kudo A., Yokoyama T., Takeda H. Pkd1l1 complexes with Pkd2 on motile cilia and functions to establish the left-right axis. Development. 2011;138:1121–1129. doi: 10.1242/dev.058271. [DOI] [PubMed] [Google Scholar]
- 52.Pennekamp P., Karcher C., Fischer A., Schweickert A., Skryabin B., Horst J., Blum M., Dworniczak B. The ion channel polycystin-2 is required for left-right axis determination in mice. Curr. Biol. 2002;12:938–943. doi: 10.1016/s0960-9822(02)00869-2. [DOI] [PubMed] [Google Scholar]
- 53.Zhang M., Bolfing M.F., Knowles H.J., Karnes H., Hackett B.P. Foxj1 regulates asymmetric gene expression during left-right axis patterning in mice. Biochem. Biophys. Res. Commun. 2004;324:1413–1420. doi: 10.1016/j.bbrc.2004.09.207. [DOI] [PubMed] [Google Scholar]
- 54.Hadjantonakis A.K., Pisano E., Papaioannou V.E. Tbx6 regulates left/right patterning in mouse embryos through effects on nodal cilia and perinodal signaling. PLoS ONE. 2008;3:e2511. doi: 10.1371/journal.pone.0002511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Caspary T., Larkins C.E., Anderson K.V. The graded response to Sonic Hedgehog depends on cilia architecture. Dev. Cell. 2007;12:767–778. doi: 10.1016/j.devcel.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 56.Eggenschwiler J.T., Bulgakov O.V., Qin J., Li T., Anderson K.V. Mouse Rab23 regulates hedgehog signaling from smoothened to Gli proteins. Dev. Biol. 2006;290:1–12. doi: 10.1016/j.ydbio.2005.09.022. [DOI] [PubMed] [Google Scholar]
- 57.Huangfu D., Anderson K.V. Cilia and Hedgehog responsiveness in the mouse. Proc. Natl. Acad. Sci. USA. 2005;102:11325–11330. doi: 10.1073/pnas.0505328102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.May S.R., Ashique A.M., Karlen M., Wang B., Shen Y., Zarbalis K., Reiter J., Ericson J., Peterson A.S. Loss of the retrograde motor for IFT disrupts localization of Smo to cilia and prevents the expression of both activator and repressor functions of Gli. Dev. Biol. 2005;287:378–389. doi: 10.1016/j.ydbio.2005.08.050. [DOI] [PubMed] [Google Scholar]
- 59.Liu A., Wang B., Niswander L.A. Mouse intraflagellar transport proteins regulate both the activator and repressor functions of Gli transcription factors. Development. 2005;132:3103–3111. doi: 10.1242/dev.01894. [DOI] [PubMed] [Google Scholar]
- 60.Chiang C., Litingtung Y., Lee E., Young K.E., Corden J.L., Westphal H., Beachy P.A. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature. 1996;383:407–413. doi: 10.1038/383407a0. [DOI] [PubMed] [Google Scholar]
- 61.Stamataki D., Ulloa F., Tsoni S.V., Mynett A., Briscoe J. A gradient of Gli activity mediates graded Sonic Hedgehog signaling in the neural tube. Genes Dev. 2005;19:626–641. doi: 10.1101/gad.325905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Echelard Y., Epstein D.J., St-Jacques B., Shen L., Mohler J., McMahon J.A., McMahon A.P. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell. 1993;75:1417–1430. doi: 10.1016/0092-8674(93)90627-3. [DOI] [PubMed] [Google Scholar]
- 63.Roelink H., Porter J.A., Chiang C., Tanabe Y., Chang D.T., Beachy P.A., Jessell T.M. Floor plate and motor neuron induction by different concentrations of the amino-terminal cleavage product of sonic hedgehog autoproteolysis. Cell. 1995;81:445–455. doi: 10.1016/0092-8674(95)90397-6. [DOI] [PubMed] [Google Scholar]
- 64.Litingtung Y., Chiang C. Specification of ventral neuron types is mediated by an antagonistic interaction between Shh and Gli3. Nat. Neurosci. 2000;3:979–985. doi: 10.1038/79916. [DOI] [PubMed] [Google Scholar]
- 65.Wijgerde M., McMahon J.A., Rule M., McMahon A.P. A direct requirement for Hedgehog signaling for normal specification of all ventral progenitor domains in the presumptive mammalian spinal cord. Genes Dev. 2002;16:2849–2864. doi: 10.1101/gad.1025702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ericson J., Rashbass P., Schedl A., Brenner-Morton S., Kawakami A., van Heyningen V., Jessell T.M., Briscoe J. Pax6 controls progenitor cell identity and neuronal fate in response to graded Shh signaling. Cell. 1997;90:169–180. doi: 10.1016/s0092-8674(00)80323-2. [DOI] [PubMed] [Google Scholar]
- 67.Wang B., Fallon J.F., Beachy P.A. Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell. 2000;100:423–434. doi: 10.1016/s0092-8674(00)80678-9. [DOI] [PubMed] [Google Scholar]
- 68.Hui C.C., Joyner A.L. A mouse model of greig cephalopolysyndactyly syndrome: the extra-toesJ mutation contains an intragenic deletion of the Gli3 gene. Nat. Genet. 1993;3:241–246. doi: 10.1038/ng0393-241. [DOI] [PubMed] [Google Scholar]
- 69.Riddle R.D., Johnson R.L., Laufer E., Tabin C. Sonic hedgehog mediates the polarizing activity of the ZPA. Cell. 1993;75:1401–1416. doi: 10.1016/0092-8674(93)90626-2. [DOI] [PubMed] [Google Scholar]
- 70.May S.R., Ashique A.M., Karlen M., Wang B., Shen Y., Zarbalis K., Reiter J., Ericson J., Peterson A.S. Loss of the retrograde motor for IFT disrupts localization of Smo to cilia and prevents the expression of both activator and repressor functions of Gli. Dev. Biol. 2005;287:378–389. doi: 10.1016/j.ydbio.2005.08.050. [DOI] [PubMed] [Google Scholar]
- 71.Tran P.V., Haycraft C.J., Besschetnova T.Y., Turbe-Doan A., Stottmann R.W., Herron B.J., Chesebro A.L., Qiu H., Scherz P.J., Shah J.V. THM1 negatively modulates mouse sonic hedgehog signal transduction and affects retrograde intraflagellar transport in cilia. Nat. Genet. 2008;40:403–410. doi: 10.1038/ng.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang Z., Li V., Chan G.C., Phan T., Nudelman A.S., Xia Z., Storm D.R. Adult type 3 adenylyl cyclase-deficient mice are obese. PLoS ONE. 2009;4:e6979. doi: 10.1371/journal.pone.0006979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Garcia-Gonzalo F.R., Corbit K., Sirerol-Piquer M.S., Ramaswami G., Otto E., Noriega T.R., Seol A.D., Bennett C.L., Robinson J.F., Josifova D.J. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat. Genet. 2011 doi: 10.1038/ng.891. in press. Published online July 3, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hopp K., Heyer C.M., Hommerding C.J., Henke S.A., Sundsbak J.L., Patel S., Patel P., Consugar M.B., Czarnecki P.G., Gliem T.J. B9D1 is revealed as a novel Meckel syndrome (MKS) gene by targeted exon-enriched next-generation sequencing and deletion analysis. Hum. Mol. Genet. 2011;20:2524–2534. doi: 10.1093/hmg/ddr151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zaghloul N.A., Liu Y., Gerdes J.M., Gascue C., Oh E.C., Leitch C.C., Bromberg Y., Binkley J., Leibel R.L., Sidow A. Functional analyses of variants reveal a significant role for dominant negative and common alleles in oligogenic Bardet-Biedl syndrome. Proc. Natl. Acad. Sci. USA. 2010;107:10602–10607. doi: 10.1073/pnas.1000219107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Davis E.E., Zhang Q., Liu Q., Diplas B.H., Davey L.M., Hartley J., Stoetzel C., Szymanska K., Ramaswami G., Logan C.V., NISC Comparative Sequencing Program TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat. Genet. 2011;43:189–196. doi: 10.1038/ng.756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chiang A.P., Beck J.S., Yen H.J., Tayeh M.K., Scheetz T.E., Swiderski R.E., Nishimura D.Y., Braun T.A., Kim K.Y., Huang J. Homozygosity mapping with SNP arrays identifies TRIM32, an E3 ubiquitin ligase, as a Bardet-Biedl syndrome gene (BBS11) Proc. Natl. Acad. Sci. USA. 2006;103:6287–6292. doi: 10.1073/pnas.0600158103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nachury M.V., Loktev A.V., Zhang Q., Westlake C.J., Peränen J., Merdes A., Slusarski D.C., Scheller R.H., Bazan J.F., Sheffield V.C., Jackson P.K. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell. 2007;129:1201–1213. doi: 10.1016/j.cell.2007.03.053. [DOI] [PubMed] [Google Scholar]
- 79.Chen C.P. Meckel syndrome: Genetics, perinatal findings, and differential diagnosis. Taiwan J. Obstet. Gynecol. 2007;46:9–14. doi: 10.1016/S1028-4559(08)60100-X. [DOI] [PubMed] [Google Scholar]
- 80.Corbit K.C., Shyer A.E., Dowdle W.E., Gaulden J., Singla V., Chen M.H., Chuang P.T., Reiter J.F. Kif3a constrains beta-catenin-dependent Wnt signalling through dual ciliary and non-ciliary mechanisms. Nat. Cell Biol. 2008;10:70–76. doi: 10.1038/ncb1670. [DOI] [PubMed] [Google Scholar]
- 81.Schneider L., Clement C.A., Teilmann S.C., Pazour G.J., Hoffmann E.K., Satir P., Christensen S.T. PDGFRalphaalpha signaling is regulated through the primary cilium in fibroblasts. Curr. Biol. 2005;15:1861–1866. doi: 10.1016/j.cub.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 82.Boehlke C., Kotsis F., Patel V., Braeg S., Voelker H., Bredt S., Beyer T., Janusch H., Hamann C., Gödel M. Primary cilia regulate mTORC1 activity and cell size through Lkb1. Nat. Cell Biol. 2010;12:1115–1122. doi: 10.1038/ncb2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hurd T.W., Hildebrandt F. Mechanisms of nephronophthisis and related ciliopathies. Nephron, Exp. Nephrol. 2011;118:e9–e14. doi: 10.1159/000320888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Otto E.A., Ramaswami G., Janssen S., Chaki M., Allen S.J., Zhou W., Airik R., Hurd T.W., Ghosh A.K., Wolf M.T., GPN Study Group Mutation analysis of 18 nephronophthisis associated ciliopathy disease genes using a DNA pooling and next generation sequencing strategy. J. Med. Genet. 2011;48:105–116. doi: 10.1136/jmg.2010.082552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Janssen S., Ramaswami G., Davis E.E., Hurd T., Airik R., Kasanuki J.M., Van Der Kraak L., Allen S.J., Beales P.L., Katsanis N. Mutation analysis in Bardet-Biedl syndrome by DNA pooling and massively parallel resequencing in 105 individuals. Hum. Genet. 2011;129:79–90. doi: 10.1007/s00439-010-0902-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vierkotten J., Dildrop R., Peters T., Wang B., Rüther U. Ftm is a novel basal body protein of cilia involved in Shh signalling. Development. 2007;134:2569–2577. doi: 10.1242/dev.003715. [DOI] [PubMed] [Google Scholar]
- 87.Lancaster M.A., Gleeson J.G. The primary cilium as a cellular signaling center: Lessons from disease. Curr. Opin. Genet. Dev. 2009;19:220–229. doi: 10.1016/j.gde.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Coppieters F., Lefever S., Leroy B.P., De Baere E. CEP290, a gene with many faces: Mutation overview and presentation of CEP290base. Hum. Mutat. 2010;31:1097–1108. doi: 10.1002/humu.21337. [DOI] [PubMed] [Google Scholar]
- 89.Waters A.M., Beales P.L. Ciliopathies: An expanding disease spectrum. Pediatr. Nephrol. 2011;26:1039–1056. doi: 10.1007/s00467-010-1731-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.