

Abstract
An effective, general protocol for the Diversity-Oriented Synthesis (DOS) of 2,4,6-trisubstituted piperidine congeners has been designed and validated. The successful strategy entails a modular approach to all possible stereoisomers of the selected piperidine scaffold, exploiting Type II Anion Relay Chemistry (ARC), followed in turn by intramolecular SN2 cyclization, chemoselective removal of the dithiane moieties and carbonyl reductions.
Nature’s biosynthesis of architecturally complex molecules often comprises iterative reaction sequences utilizing complex molecular machines, such as polyketide synthases and the ribosome, to unite activated, stereochemically pure building blocks.1 In an attempt to mimic Nature’s iterative biosynthesis of complex molecules, we developed and validated Type I and Type II Anion Relay Chemistry (ARC) (Scheme 1),2 two closely related synthetic methods comprising multicomponent union protocols. In addition to providing access to specific architectures, the ARC tactic also holds considerable potential for Diversity-Oriented Synthesis (DOS).3 Many DOS programs, however, suffer from the inability to provide access to all possible stereoisomers of a selected scaffold. We have therefore set as a goal for our DOS programs, the construction of all possible stereoisomers of the selected scaffold. Such a goal, if widely adopted by the DOS community, will require, and in many cases demand the development of new, innovative synthetic methods to access the targeted congeners in an efficient fashion, an outcome not dissimilar to one of the core goals of natural product total synthesis.
Scheme 1.

Type I and Type II ARC
Having achieved the development and application of Type I Anion Relay chemistry (Scheme 1), initially as a tri-component coupling protocol, which we employed to great advantage in a number of complex molecule synthetic programs,4 we subsequently devised the Type II ARC tactic, also an iterative multi-component union strategy, which like the Type I ARC process exploits bifunctional linchpins. The Type II ARC protocol holds, we believe, even more potential for the design and synthesis of complex molecular structures. The development of the Type II ARC process however required the design, synthesis and validation of a series of effective bifunctional linchpins.2
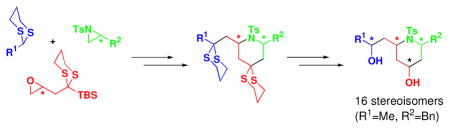
To illustrate the utility of the Type II ARC tactic in the area of DOS, we report here the synthesis of all possible stereoisomers of a family of 2,4,6-trisubstituted piperidines (Scheme 2: VI), utilizing this union tactic, followed in turn by an intramolecular SN2 cyclization, and further elaboration.
Scheme 2.

General Synthetic Route to Access Diverse Piperidine Analogues via Type II ARC.
From the medicinal perspective, the piperidine scaffold has attracted considerable interest in synthetic5 and biological6 communities. However, notwithstanding the availability of numerous methods to access individual members of the 2,4,6-trisubstituted piperidine family in a stereocontrolled fashion, there are few general methods that can provide access to all stereoisomers. 7
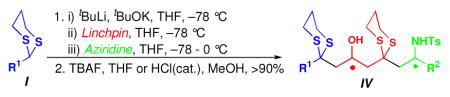
The Type II ARC tactic, as illustrated in Scheme 2, not only would provide a convergent route to 2,4,6- trisubstituted piperidines, but also enables chemical and stereochemical diversification at the C(2) and C(6) stereogenic centers, depending on the components I–III employed. In addition, the two dithiane groups provide synthetic handles for further chemoselective diversification. To initiate this program, the three requisite components for the Type II ARC reaction were prepared: initiating nucleophiles I (dithianes 1a–1d), bifunctional linchpins II [(+)-2, (−)-2], and aziridines III [(+)-3a, (+)-3b, and (−)-3a, (−)-3c)], the latter readily accessible from enantiomerically pure amino acids.8
With these components in hand, reaction conditions for the Type II ARC protocol were optimized based on our earlier studies.4 Conditions employing the modified Schlosser base9 proved highly effective without the use of co-solvents such as HMPA or DMPU to enhance the nucleophilicity of dithiane anion.10 The initial multicomponent adducts were subjected to removal of the TBS group with TBAF (Table 1).
Table 1.
Multi-component reaction (Type II ARC).
 | |||||
|---|---|---|---|---|---|
| entry | dithiane | linchpin | aziridine | confign (*,*)a | yieldb (%) |
| 1 | 1a | (+)-2 | (+)-3a | (S,S)-4 | 74 |
| 2 | 1a | (+)-2 | (−)-3a | (S,R)-4 | 69 |
| 3 | 1a | (−)-2 | (+)-3a | (R,S)-4 | 69 |
| 4 | 1a | (−)-2 | (−)-3a | (R,R)-4 | 74 |
| 5 | 1a | (+)-2 | (+)-3b | (S,S)-5 | 61 |
| 6 | 1a | (+)-2 | (−)-3c | (S,S)-6 | 41 |
| 7 | 1b | (+)-2 | (+)-3a | (S,S)-7 | 65 |
| 8 | 1b | (−)-2 | (+)-3a | (R,S)-7 | 59 |
| 9 | 1b | (+)-2 | (+)-3b | (S,S)-8 | 55 |
| 10 | 1c | (+)-2 | (+)-3a | (R,S)-9 | 56 |
| 11 | 1d | (+)-2 | (+)-3a | (R,S)-10 | 52 |
| 12 | 1d | (−)-2 | (+)-3a | (S,S)-10 | 55 |
Absolute configuration of the corresponding stereocenters.
Isolated yield for ARC.
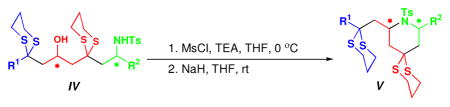
Mesylation of the hydroxy group then furnished the substrates for the subsequent intramolecular SN2 cyclizations. Examination of a variety of conditions, including solvents, bases, and leaving groups to suppress potential elimination reactions11 revealed that treatment of the mesylates in dilute THF solution with NaH effectively provided both 2,6-cis and 2,6-trans-piperidines, again in preparatively useful yields (Table 2).
Table 2.
Intramolecular SN2 Cyclization.
 | |||||
|---|---|---|---|---|---|
| entry | substrate (*,*)a | R1/R2 | product (*,*)a | ring substitution | yield (%)b |
| 1 | (S,S)-4 | Me/Bn | (R,S)-11 | cis | 87 |
| 2 | (R,R)-4 | Me/Bn | (S,R)-11 | cis | 85 |
| 3 | (S,R)-4 | Me/Bn | (R,R)-11 | trans | 55 |
| 4 | (R,S)-4 | Me/Bn | (S,S)-11 | trans | 58 |
| 5 | (S,S)-5 | Me/nPr | (R,S)-12 | cis | 89 |
| 6 | (R,S)-10 | Ph/Bn | (S,S)-13 | cis | 81 |
Absolute stereochemistry of the chiral center.
Yield over two steps.
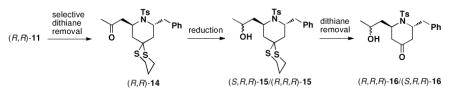
Next, the utility of the two dithiane groups was explored (Scheme 3). Treatment of (R,S)-11 with Hg(ClO4)2 and 2,6-lutidine in wet THF led to regioselective removal of the more accessible side chain dithiane moiety to furnish ketone (R,S)-14, which in turn was subjected to various reduction conditions (Table 3A; Entry 1–5). Use of the Corey (R)-CBS reagent12 (Table 3A; Entry 4) and Al(OiPr)3 (Table 3A; Entry 5) proved optimal. The resultant diastereomeric alcohols (S,R,S)-15 and (R,R,S)- 15, readily separable by column chromatograpy, were then subjected to removal of the remaining dithiane moiety under the Stork conditions13 to provide hydroxy ketones (S,R,S)-16 and (R,R,S)-16.
Scheme 3.

Functional and Stereochemical Diversification of 2,6-cis-disubstituted Piperidine (R,S)-11.
Table 3.
Screening Conditions for Reduction of Ketones.
|
A. Reduction of (R,S)-14 | |||
|---|---|---|---|
| entry | condition | product ratioa (S,R,S)-14: (R,R,S)-14 | yieldb (%) |
| 1 | A | 2:1 | 93 |
| 2 | B | 3:1 | 97 |
| 3 | C | 4:1 | 92 |
| 4 | D | 1:20 | 93 |
| 5 | E | 5:1 | 88 |
| B. Reduction of (S,R,S)-16 (entry 1–4) and (R,R,S)-16 (entry 5)
| |||
|---|---|---|---|
| entry | condition | product ratioa (S,R,S,R)-17: (S,R,S,S)-17 | yieldb (%) |
| 1 | A | 5:1 | 93 |
| 2 | B | 20:1 | 97 |
| 3 | F | 2:1 | 95 |
| 4 | G | 1:1.5 | 89 |
|
| |||
| 5 | G | 1:1.3c | 91 |
Ratio of diastereomers was determined by 1H-NMR.
Combined yield of diastereomers.
The ratio of (R,R,S,R)-17: (R,R,S,S)-17, Conditions: A: NaBH4, MeOH, 0 °C; B: L-Selectride, THF, −78–0 °C; C: (R)-CBS reagent, BH3•THF, THF, 0 °C; D: (S)-CBS reagent, BH3•THF, THF, 0 °C; E: Al(OiPr)3, iPrOH, reflux; F: BH3•THF, THF, −78–0 °C; G: SmI2, H2O, THF, −78–0 °C.
Ketones 16 were also subjected to various reduction conditions. Regardless of steric encumberance of the hydride reducing agent, (S,R,S)-16 led to +-hydroxy isomer (S,R,S,R)-17 as the major diastereomer (Table 3B; Entry 1–3). Molecular mechanics calculations (MMFF94) revealed that the 2,6-diaxial chair-like conformer C possesses a lower energy, by ca. 16 kcal/mol than the 2,6- diequatorial chair-like conformer D, due to pseudo A1,3- strain14 between the substituents at the 2- and 6-positions and the tosyl group, thus leading to hydride attack from the more accessible α-face of C (Figure 1). In accordance with this reasoning, an increase in the bulkiness of the hydride reagent (L-Selectride) led to excellent selectivity (ca. 20:1) to provide (S,R,S,R)-17 (Table 3B; Entry 4).
Figure 1.

Proposed Conformational Analysis for the Reduction of (S,R,S)-16 and (R,R,S)-16.
At this juncture, we presumed that the diastereoselectivity could be reversed under dissolving metal conditions15 to obtain diastereomer (S,R,S,S)-17. Treatment of (S,R,S)-16 with SmI2 (4.0 equiv) and H2O (6.0 equiv) in THF furnished the desired α-hydroxy isomer as the major product, albeit with poor selectivity (Table 3B; Entry 4). The lack of diastereoselectivity in the dissolving metal reductions presumably arises from competition of the two possible chelated intermediates (Figure 1; E and F). The structures and the relative configurations of (S,R,S,R)-17 and (R,R,S,R)-17 were confirmed by X-ray crystallographic analysis. Under the same reduction conditions, (R,R,S,R)-17 and (R,R,S,S)-17 were obtained from (R,R,S)-16 (Table 3B; Entry 5).
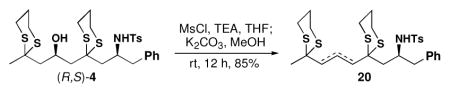
Based on the successful elaboration of the 2,6-cispiperidine congeners from (R,S)-11, the 2,6-trans congener (R,R)-11 was subjected to the same procedure.16 Following selective removal of the less-hindered dithiane moiety of (R,R)-11, all attempts to arrive at a single diastereomeric alcohol employing a wide variety of reducing agents proved unsuccessful. Equally disappointing, separations of the two diastereomeric alcohols (S,R,R)-15/(R,R,R)-15, as well as the hydroxy ketones (S,R,S)-16/(R,R,S)-16 could not be achieved.
To solve this issue, we explored the regioselective reduction of dione (R,R)-18 (Scheme 4), which was generated by removal of the both dithiane moieties in (R,R)-11 employing the Corey-Erickson protocol.17 Pleasingly, the internal ketone of (R,R)-18 was reduced regioselectively upon treatment with one equivalent of the bulky reducing agent [LiAlH(OCEt3)3] to provide a mixture of (S,R,R)-19 and (S,R,S)-19, readily separable by flash column chromatography. Reduction of the remaining side chain ketone employing BH3•THF furnished mixtures (ca. 1:1) of diastereomeric diols [(S,R,R,R)- 17/(R,R,R,R)-17 and (S,R,R,S)-17/(R,R,R,S)- 17], which were separated by SFC, thereby providing access to all possible stereoisomers obtainable from (R,R)-11.
Scheme 4.

Functional and Stereochemical Diversification of 2,6-trans-Disubstituted Piperidine (R,R)-11
Identical synthetic steps were followed with (S,R)-11 and (S,S)-11 to prepare the enantiomeric library ent-A.
In summary, an effective DOS strategy has be designed and validated to access the complete matrix of stereoisomers of the targeted 2,4,6-trisubsituted piperidine scaffold, exploiting our modular Type II ARC protocol, followed in turn by intramolecular SN2 cyclization. Regioselective dithiane removal and reduction conditions were then examined and optimized. In the context of complex molecule synthesis, the reported non-selective reductions would be viewed as a shortcoming. However for Diversity Oriented Synthesis (DOS) directed at the construction of a complete matrix of congeners, non-selective reactions in conjunction with effective chromatographic separation has considerable advantage. Nonetheless, the lack of observed selectivity, serves to reveal a continuing need to develop new, selective reactivity to enhance the synthetic arsenal.
Supplementary Material
Figure 2.

The Complete Piperidne Library
Acknowledgments
Financial support was provided by the NIH through Grant GM-29028. We thank Drs G. Furst, P. J. Carroll and R. Kohli at the University of Pennsylvania for assistance in obtaining the NMR, X-ray diffraction and high-resolution mass spectra, respectively.
Footnotes
Supporting Information Available Experimental procedures and spectroscopic and analytical data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Shen B. Curr Opin Chem Biol. 2003;7:285. doi: 10.1016/s1367-5931(03)00020-6. [DOI] [PubMed] [Google Scholar]
- 2.(a) Smith AB, III, Wuest WM. Chem Commun. 2008:5883. doi: 10.1039/b810394a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Smith AB, III, Kim W-S, Wuest WM. Angew Chem Int Ed. 2008;47:7082. doi: 10.1002/anie.200802301. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Smith AB, III, Kim WS, Tong R. Org Lett. 2010;12:588. doi: 10.1021/ol902784q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Smith AB, III, Tong R. Org Lett. 2010;12:1260. doi: 10.1021/ol100130x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Smith AB, III, Kim WS. Proc Natl Acad Sci USA. 2011;108:6787. doi: 10.1073/pnas.1015265108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schreiber SL. Science. 2000;287:1964. doi: 10.1126/science.287.5460.1964. [DOI] [PubMed] [Google Scholar]
- 4.(a) Smith AB, III, Doughty VA, Lin Q, Zhuang L, McBriar MD, Boldi AM, Moser WH, Murase N, Nakayama K, Sobukawa M. Angew Chem, Int Ed. 2001;40:191. [PubMed] [Google Scholar]; (b) Smith AB, III, Lin Q, Doughty VA, Zhuang L, McBriar MD, Kerns JK, Brook CS, Murase N, Nakayama K. Angew Chem, Int Ed. 2001;40:196. [PubMed] [Google Scholar]; (c) Smith AB, III, Pitram SM, Fuertes MJ. Org Lett. 2003;5:2751. doi: 10.1021/ol034989g. [DOI] [PubMed] [Google Scholar]; (d) Smith AB, III, Kim DS. Org Lett. 2004;6:1493. doi: 10.1021/ol049601b. [DOI] [PubMed] [Google Scholar]; (e) Smith AB, III, Kim DS. Org Lett. 2005;7:3247. doi: 10.1021/ol0510264. [DOI] [PubMed] [Google Scholar]; (f) Smith AB, III, Kim DS. J Org Chem. 2006;71:2547. doi: 10.1021/jo052314g. [DOI] [PubMed] [Google Scholar]
- 5.Recent examples, see: Krishna PR, Sreeshailam A. Tetrahedron Lett. 2007;48:6924.Chandraskhar S, Babu GSK, Reddy ChR. Tetrahedron Asymmetry. 2009;20:2216.Gnamm C, Krauter CM, Brödner K, Helmchen G. Chem Eur J. 2009;15:2050. doi: 10.1002/chem.200802525.Kumar RSC, Reddy GV, Shankaraiah G, Babu KS, Rao JM. Tetrahedron Lett. 2010;51:1114.Cui L, Li C, Zhang L. Angew Chem Int Ed. 2010;49:9178. doi: 10.1002/anie.201004712.
- 6.(a) Daly J. J Nat Prod. 1998;61:162. doi: 10.1021/np970460e. [DOI] [PubMed] [Google Scholar]; (b) Kuznetsov VV. Khim-Farm Zh. 1991;25:61. [Google Scholar]; Chem Abst. 1991;115:158846. [Google Scholar]; J Enzym Inhib Med Ch. 2005;20:551. [Google Scholar]; (c) Li H, Asberom T, Bara TA, Clader JW, Greenlee WJ, Josien HB, Mcbriar MD, Nomeir A, Pissarnitski DA, Rajagopalan M, Xu R, Zhao Z, Song L, Zhang L. Bioog Med Chem Lett. 2007;17:6290. doi: 10.1016/j.bmcl.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 7.Recent reviews, see: Laschat S, Dickner T. Synthesis. 2000:1781.Weintraub JS, Sabol PM, Kane JM, Borcherding DR. Tetrahedron. 2003;59:2953.Buffat MGP. Tetrahedron. 2004;60:1701.Cossy J Chem Rec. 2005;5:70. doi: 10.1002/tcr.20035.
- 8.Alonso DA, Andersson PG. J Org Chem. 1998;63:9455. doi: 10.1021/jo972082o. [DOI] [PubMed] [Google Scholar]
- 9.Schlosser M, Strunk S. Tetrahedron Lett. 1984;25:741. [Google Scholar]
- 10.Reich HJ, Sanders AW, Fielder AT, Bevan MJ. J Am Chem Soc. 2002;124:13386. doi: 10.1021/ja026915q. [DOI] [PubMed] [Google Scholar]
-
11.Elimination reaction: see supporting information for details.
- 12.Corey EJ, Shibata S, Bakshi RK. J Org Chem. 1988;53:2861. [Google Scholar]
- 13.(a) Fleming FF, Funk L, Altundas R, Tu Y. J Org Chem. 2001;66:6502. doi: 10.1021/jo0157829. [DOI] [PubMed] [Google Scholar]; (b) Stork G, Zhao K. Tetrahedron. 1989;30:287. [Google Scholar]
- 14.(a) Beak P, Zajdel WJ. J Am Chem Soc. 1984;106:1010. [Google Scholar]; (b) Brown JD, Foley MA, Comins DL. J Am Chem Soc. 1988;110:7445. [Google Scholar]; (c) Heintzelman GR, Weinreb SM, Parvez M. J Org Chem. 1996;61:4594. doi: 10.1021/jo960035a. [DOI] [PubMed] [Google Scholar]; (d) Neipp CE, Martin SF. J Org Chem. 2003;68:8867. doi: 10.1021/jo0349936. [DOI] [PubMed] [Google Scholar]; (e) Lemire A, Charette AB. Org Lett. 2005;7:2747. doi: 10.1021/ol051022z. [DOI] [PubMed] [Google Scholar]; (f) Cariou CAM, Snaith JS. Org Biomol Chem. 2006;4:51–53. doi: 10.1039/b515547a. [DOI] [PubMed] [Google Scholar]
- 15.(a) Singh AK, Bakshi RK, Corey EJ. J Am Chem Soc. 1987;109:6187. [Google Scholar]; (b) Keck GE, Wager CA, Seel T, Wager TT. J Org Chem. 1999;64:2172. [Google Scholar]; (d) Keck GE, Wager CA. Org Lett. 2000;2:2307. doi: 10.1021/ol006072c. [DOI] [PubMed] [Google Scholar]
-
16.See the Supporting information for details.
- 17.Corey EJ, Erickson BW. J Org Chem. 1971;36:3553. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.