

Abstract
The cis-regulatory regions on DNA serve as binding sites for proteins such as transcription factors and RNA polymerase. The combinatorial interaction of these proteins plays a crucial role in transcription initiation, which is an important point of control in the regulation of gene expression. We present here an analysis of the performance of an in silico method for predicting cis-regulatory regions in the plant genomes of Arabidopsis (Arabidopsis thaliana) and rice (Oryza sativa) on the basis of free energy of DNA melting. For protein-coding genes, we achieve recall and precision of 96% and 42% for Arabidopsis and 97% and 31% for rice, respectively. For noncoding RNA genes, the program gives recall and precision of 94% and 75% for Arabidopsis and 95% and 90% for rice, respectively. Moreover, 96% of the false-positive predictions were located in noncoding regions of primary transcripts, out of which 20% were found in the first intron alone, indicating possible regulatory roles. The predictions for orthologous genes from the two genomes showed a good correlation with respect to prediction scores and promoter organization. Comparison of our results with an existing program for promoter prediction in plant genomes indicates that our method shows improved prediction capability.
Sequencing and annotation of a large number of eukaryotic genomes has made available an enormous amount of information regarding genetic coding sequences (CDS). These data can be effectively utilized for studying and modifying the expression of genes if the location and contribution of cis-regulatory regions, which control spatial and temporal regulation of gene expression, are available. However, the precise annotation of regulatory regions is difficult as compared with the identification of genes, primarily because regulatory regions do not code for an identifiable product. In fact, regulatory regions are bound by proteins such as transcription factors, which bring about transcription and its regulation. Determining transcription factor-binding sites (TFBSs) from chromatin immunoprecipitation methods has limitations and requires a lot of downstream data processing (Farnham, 2009). Moreover, the mere binding of a transcription factor at a particular site does not warrant its involvement in the regulation of a gene. Development of computational approaches that enable accurate prediction of cis-regulatory sites could thus greatly aid in deciphering the regulatory mechanisms at the genome level.
The preponderance of noncoding DNA in the eukaryotic genome makes it difficult to identify promoter regions. Most efforts toward the prediction of regulatory regions have traditionally focused on the detection of consensus sequences for the TATA box, Initiator elements, TFBSs, etc. Such sequence-based prediction of short motifs might be inadequate because a large number of false hits are possible by chance. Moreover, there is increasing evidence to suggest that consensus sequences vary greatly and are even absent in many cases. The TATA box, which is considered as the signature sequence for promoters, is not found in a majority of core promoters in eukaryotes (Cooper et al., 2006; ENCODE Project Consortium, 2007), and TATA-binding protein can recognize the core promoter irrespective of the underlying sequence with the help of additional factors (Pugh, 2000). A recent study on nucleosomal positioning in Schizosaccharomyces pombe shows that nucleosome-depleted regions at promoters do not show the sequence characteristics (poly[A+T] tracts) that are crucial for nucleosome depletion in Saccharomyces cerevisiae, thus raising questions about sequence conservation at these sites (Lantermann et al., 2010).
The view that structural features of DNA (rather than sequence) might be able to give a better understanding of the regulatory landscape was first suggested by Pedersen et al. (1999) and is slowly gaining ground. DNA at promoter sites may have special features that play a major role in transcription by allowing protein-DNA interactions and communication between factors bound at distal promoters. Structural features of DNA, such as GC skew, bendability, topography, free energy, curvature, nucleosome positioning, base stacking, relative entropy of nucleotides, etc., have been shown to give characteristic patterns at the transcription start site (TSS) and functional noncoding regions such as promoters (Florquin et al., 2005; Fujimori et al., 2005; Kanhere and Bansal, 2005a, 2005b; Alexandrov et al., 2006; Lee et al., 2007; Abeel et al., 2008a; Cao et al., 2009; Parker et al., 2009; Rangannan and Bansal, 2009; Tanaka et al., 2009). Although these properties are inherently sequence dependent, they give additional insight into long-range interactions that might not be evident from sequence alone. Moreover, the structural features found at promoter regions are sometimes conserved across species (Fujimori et al., 2005; Kanhere and Bansal, 2005a; Abeel et al., 2008a). Thus, a prediction program that effectively captures the structural patterns at promoters could help in predicting regulatory regions across genomes irrespective of the availability of training data.
The majority of the currently available promoter prediction programs (PPPs) such as ARTS, Eponine, and ProSOM focus on promoter prediction in the human genome or related genomes (Down and Hubbard, 2002; Sonnenburg et al., 2006; Abeel et al., 2008b, 2009) for which processed experimental data and detailed annotation, such as from deepCAGE sequencing, are already available. Since these programs require pretraining on the genome, they cannot be readily applied to other genomes, such as plants. CpG island predictors cannot be used for plants, since a suitable prediction criterion is unavailable (Rombauts et al., 2003) and they are purported to be absent in plant genomes (Yamamoto et al., 2007b). Sequence-based PPPs for plants are either repositories of TFBSs and cis-regulatory elements reported in individual studies, such as PLACE (Higo et al., 1999), Osiris (Morris et al., 2008), and AGRIS (Davuluri et al., 2003), or in silico analysis of overrepresented k-mers at promoters (Molina and Grotewold, 2005; Yamamoto et al., 2007a; Lichtenberg et al., 2009). EP3 (Abeel et al., 2008a) is the only PPP available currently that predicts extended promoter regions in plant genomes. However, the promoter prediction property (base stacking) used in EP3 is selected based on analysis in the human genome only. Also, some minimal training is apparently involved in the EP3 program as well, since different thresholds are used for different organisms (Arabidopsis [Arabidopsis thaliana], 0.0583; rice [Oryza sativa], 0.1394). Many plant genomes have been recently sequenced (Ming et al., 2008; Rensing et al., 2008; International Brachypodium Initiative, 2010; Schmutz et al., 2010), and a large number of genomes are in the sequencing pipeline, such as the multinational Brassica rapa sequencing project (for a full list, see National Center for Biotechnology Information Genome Projects). A promoter prediction tool suited for plant genomes could help in the annotation of putative cis-regulatory regions as well as in finding new genes for these newly sequenced genomes.
We present here a detailed analysis of the performance of the program PromPredict, a simple program that captures the free energy pattern at promoter regions from DNA sequence information without requiring any pretraining, for the model monocot and eudicot plant genomes of rice (cv Nipponbare; Rice Annotation Project, 2008) and Arabidopsis (Arabidopsis Genome Initiative, 2000). PromPredict was originally developed to predict putative promoters using the whole-genome percentage GC of select bacterial genomes to define the baseline cutoffs for relative free energy of promoter regions (Rangannan and Bansal, 2009). It has now been generalized for genome prediction using 1,000-nucleotide fragments with 20% to 80% GC (Rangannan and Bansal, 2010). It should be noted that the promoters are not predicted on the basis of motif composition or organization of cis-regulatory modules but solely on the basis of relative free energy of adjoining sequences. We compare and contrast the genomic features and prediction characteristics in the two plant genomes to highlight the similarities and differences in their genome architecture. Such a comparison can shed light on the evolution of monocot and dicot lineages of flowering plants. The predictions are assigned to five different score classes to indicate their relative strength (as discussed below). We also compare the performance of PromPredict with the EP3 program.
RESULTS
A comparison of the annotated genomes (excluding mitochondrial and chloroplast chromosomes) of rice and Arabidopsis gave some interesting insights into the genome composition of the two plants. Arabidopsis has a small and compact genome with gene density 1 order of magnitude higher than that for the rice genome (Table I). The length of the Arabidopsis genome is less than half the length of the rice genome, but it has 40% of its genome being transcribed as compared with approximately 22% in rice at the current state of annotation. However, the rice genome has longer primary transcripts, and introns contribute to a majority of the primary transcript length (Table I). Moreover, the rice genome has a higher average GC content and a greater GC variation, which is also reflected in the various regions of the gene (Table I; Supplemental Fig. S1).
Table I. Comparison of Arabidopsis and rice genomes.
The sequence data for five chromosomes of Arabidopsis (approximately 119 Mb) and 12 chromosomes of rice (approximately 382 Mb) were analyzed for their genome characteristics.
| Feature | Arabidopsis | Rice |
| Characteristica | ||
| Gene density (genes Mb−1) | Approximately 238.8 | Approximately 63.4 |
| Transcribed region (% of genome length) | 40.0% | 21.8% |
| Exon coverage | 20.3% | 6.3% |
| Protein-coding genes | 27,169 (28,289bc) | 23,057 |
| ncRNA genes | 1,243 (1,263b) | 1,527 |
| ncRNA genes (median length in nucleotides) | 82 | 74 |
| Average GC content | 36% | 42.4% |
| Median length (nucleotides) of various regions in protein-coding genes (% of primary transcript length) | ||
| Primary transcript | 2,095 | 3,163 |
| Intergenic | 924 | 5,300 |
| 5′ UTR | 105 (6.1%) | 106 (9.1%) |
| 3′ UTR | 208 (9.5%) | 260 (13.5%) |
| Intron | 100 (33.6%) | 96 (47.8%) |
| CDS | 129 (50.8%) | 132 (29.7%) |
| Average GC percentage of various regions in protein-coding genes | ||
| Primary transcript | 39.2 ± 3.0 | 47.5 ± 8.3 |
| Intergenic | 31.9 ± 4.9 | 41.9 ± 5.6 |
| 5′ UTR | 37.7 ± 8.0 | 55 ± 15.5 |
| 3′ UTR | 31.7 ± 4.8 | 40.6 ± 7.2 |
| Intron | 32.6 ± 4.2 | 38.6 ± 7.9 |
| CDS | 44.9 ± 3.2 | 51.1 ± 11.3 |
Excluding mitochondrial and chloroplast chromosomes, transposons, and pseudogenes.
Gene models from TAIR that were considered for analysis (see “Materials and Methods”).
Protein-coding gene models with TSS information considered for analysis: 20,094; non-protein-coding transcripts in rice were also considered for analysis: 1,152.
Average Free Energy Profile
Distortion of the DNA double helix, such as separation of strands and bending of DNA, is necessary for binding of RNA polymerase and transcription factors at the promoter site. The free energy of DNA melting is a dinucleotide sequence-dependent secondary structure property that comprises not only hydrogen bonding energy but also base stacking energy and hence is slightly different from mere AT or GC contents. Figure 1, A and B, shows the average free energy (AFE) profiles for Arabidopsis and rice genomes in the vicinity of the TSS. The details of calculating the AFE profile are mentioned in “Materials and Methods.” Both plants show similar free energy profiles with a significant difference between upstream and downstream regions and a peak just upstream of the TSS. Overall, the profiles show a less stable upstream region followed by a relatively stable downstream region. However, the difference in stability is much greater for rice (approximately 3.5 kcal mol−1) as compared with Arabidopsis (approximately 1.5 kcal mol−1), as seen in Figure 1C. It should be noted here that AT-rich sequences tend to be less stable, even though the correlation is not exact and depends on their dinucleotide frequencies as seen in the vicinity of the TSS. The Arabidopsis profile has a higher free energy (less stability) than rice owing to its AT-rich genome. Similarly, the AFE profiles shift with variation in GC content of the sequences (Supplemental Fig. S2). In conclusion, the promoter region is characterized by relative instability when compared with the downstream stable region for both the plant genomes. This characteristic can be used to identify promoter regions, as shown by Rangannan and Bansal (2007, 2009) for prokaryotes and by Abeel et al. (2008a) for eukaryotes.
Figure 1.

A and B, AFE profiles in the vicinity of the TSS for all five chromosomes of Arabidopsis (A) and six representative (even numbered) chromosomes of rice (B). The AFE values for upstream, downstream, and full-length shuffled sequences are shown as dashed lines. C, Comparison of free energy profiles (shown in red) and percentage AT occurrence (shown in green) over the region −500 to +500 bp with respect to (w.r.t.) TSS for chromosome 1 of Arabidopsis and rice.
The high stability trough in the profiles is found around 100 to 200 nucleotides downstream of the TSS. This region is beyond the 5′ untranslated region (5′ UTR) of most genes in both Arabidopsis and rice (Table I) and hence overlaps with the first CDS. Moreover, the 5′ UTR and CDS in rice also have higher average GC contents than those of the primary transcript (Table I). The GC richness of the region immediately downstream of the TSS is more pronounced in rice than in Arabidopsis (Supplemental Fig. S1). These observations could account for the presence of GC content gradients previously reported in monocots (Wong et al., 2002).
Performance of PromPredict on Plant Genomes
We tested the latest version of the program PromPredict (Rangannan and Bansal, 2010) on rice and Arabidopsis genomes in order to find cis-regulatory sites (see “Web Resources” below). The program detects relative differences in free energy and applies cutoffs based on the GC content of a sequence. It compares the free energy of two adjacent sequences and predicts a cis-regulatory region at the upstream sequence if the two criteria, (1) free energy of the upstream sequence (E1 value) and (2) the difference in free energy between the two sequences (D value), are greater than predetermined cutoff values (Supplemental Protocol S1; Supplemental Fig. S3; Supplemental Table S1; see “Materials and Methods”). The predictions are directional, depending on the orientation of the input sequence: forward or reverse strand.
The whole-genome prediction performance of the program is presented in Table II in terms of the recall and precision values for the various gene data sets (for precision and recall calculations, see “Materials and Methods”). The region −500 to +100 bp with respect to the TSS (true-positive region [TP region]) was considered for determining true positives in protein-coding genes, as this covers the upstream region as well as most 5′ UTRs. The region −1,000 to 0 bp was considered for noncoding RNA (ncRNA) genes and for protein-coding genes with only translation start site (TLS) information. The predictions obtained within the gene beyond the true-positive region are considered as false positives (FPpred.). Overall, more than 92% of genes (henceforth referred to as TPgenes) have a true-positive prediction (TPpred.) within −500 to +100 bp. Both the genomes have approximately 1.5 TPpred. per TPgene on average. As expected, the longer protein-coding genes have more FPpred., thus leading to lower values of precision as compared with that for ncRNA genes.
Table II. PromPredict performance on Arabidopsis and rice genomes.
For protein-coding genes, the region considered for determining true positives was −500 to +100 bp in the vicinity of the TSS. For ncRNA genes and protein-coding genes with only TLS information, the region considered for determining true positives was −1,000 to 0 bp of the start site.
| Gene Type | No. of Genes | Recall | Precision |
| Arabidopsis | |||
| Protein-coding genes | 20,094 | 0.92 | 0.33 |
| Protein-coding genes, TLSa | 8,195 | 0.96 | 0.51 |
| ncRNA genes | 1,263 | 0.93 | 0.76 |
| Rice | |||
| Protein-coding genes | 23,057 | 0.92 | 0.24 |
| ncRNA genes | 1,527 | 0.95 | 0.90 |
| Non-protein-coding transcripts | 1,152 | 0.96 | 0.47 |
Protein-coding genes with only TLS information.
Although a gene might have more than one prediction in the TP region, the prediction nearest to the TSS will correspond to the core promoter. A frequency plot for distance of the nearest prediction from the TSS (Fig. 2A) shows that the majority of the TPpred. obtained are proximal to the TSS: 70% of predictions for Arabidopsis and 63% of predictions for rice are within −200 to +100 bp of the TSS. For ncRNA genes, the region −500 to 0 bp of the TSS contains 92% to 93% of the predictions for both Arabidopsis and rice genomes (Fig. 2B). However, a significant number of predictions are also obtained farther away from the TSS, especially in rice. Interestingly, Arabidopsis predictions are clustered closer to the TSS (0 to −100 bp of the TSS), while rice predictions show an almost uniform distribution over 0 to −200 bp relative to the TSS in both protein-coding and ncRNA genes.
Figure 2.

Percentage frequency distribution plots showing the distance of promoter predictions from TSS in 50-nucleotide bins for protein-coding genes (A) and ncRNA genes (B). For protein-coding genes, the predictions within −500 to +100 bp with respect to (w.r.t.) TSS, and for ncRNA genes, predictions within −1,000 to 0 bp with respect to TSS, are considered where position 0 corresponds to the TSS. [See online article for color version of this figure.]
If the closest prediction for a gene is found near the TSS (−100 to +50 bp), it might correspond to the core promoter and hence have a stronger signal. The free energy difference with respect to the downstream sequence would be greater for these predictions as compared with those present distally. However, we found that this is not true. We calculated the AFE profiles for 1001-nucleotide sequences clustered according to the proximity of the closest prediction to the TSS (50-nucleotide bins from Fig. 2). The AFE plots in Figure 3 have a broad low-stability region corresponding to the 50-nucleotide bin where the closest prediction lies and another peak at the −35 region. The first peak is expected because the algorithm recognizes this feature for prediction. The difference in AFE for upstream and downstream regions is almost constant for all plots irrespective of the distance of the instability peak from the TSS. The AFE for the peaks is less (by 1.5–2 kcal mol−1) than the AFE observed at the same position in Figure 1, but the peaks follow the general trend of the overall profile. We propose that such a profile might be a characteristic of cis-regulatory sites spread over a longer region upstream of the TSS.
Figure 3.

AFE plots for sequences from each frequency class from Figure 2 for Arabidopsis (A) and rice (B). It is seen that the predictions occurring in each frequency class correspond to peaks in AFE profiles at a particular distance from the TSS. The plots depict the AFE for sequences with the closest prediction present at a given distance (−500 to +100 bp with respect to [w.r.t.]). The color code used to depict the AFE profile, for sequences with predictions in each 50-nucleotide bin, is indicated in the box at right.
The second sharper peak found ubiquitously at the −35 position might indicate the presence of a TATA box at this region. However, Web logos for this region did not show any strong consensus TATA sequence (data not shown). A comparison of tetramer frequencies in the −50- to −20-bp region and the −500- to +500-bp region shows a relatively high occurrence of TATA and AAAA tetramers in the core promoter region for both Arabidopsis and rice (Supplemental Fig. S5). Interestingly, while several AT-containing tetramers are preferentially located at the upstream −35 region in Arabidopsis, some C-rich sequences also show overrepresentation in this region for rice promoters.
An analysis for the overlap of predictions with 92 TFBSs in rice as obtained from Osiris (Morris et al., 2008) was carried out. Fifty-six percent of TPpred. contained within them entire TFBS motifs, while 98% of TPpred. overlapped with at least half of the TFBS sequence. Ninety-one percent of the reported TFBSs overlapped at least partially (half or more) with TPpred., out of which 58% were found to overlap completely. Although a substantial number of TPpred. were found to contain AT-rich TFBSs, a significant number of GC-rich TFBSs were also found to occur within the predictions.
Prediction Score
We categorized the predictions on the basis of the difference in free energy between a prediction and its downstream region, denoted as the D value. The score classes are formed on the basis of the maximum D value (Dmax) of predictions and the GC content range of the surrounding 1001-nucleotide sequence (for details of categorization, see “Materials and Methods” and Supplemental Fig. S4). Table III shows that most of the predictions in the higher score categories are TPpred., whereas the FPpred. show a preponderance toward the lower score categories. The D value cutoffs used for relative score categorization of predictions are similar to the cutoffs applied for promoter prediction. Both are dependent on GC content of the flanking 1,001-nucleotide sequence and the frequency of obtaining a prediction in a particular GC range. If we raise the cutoff for promoter prediction to the category classification cutoffs, the precision can be improved. However, the number of TPgenes is reduced as a consequence and recall decreases. A segregation of the predicted signals according to their score thus allows user-defined stringency settings. We suggest that the three highest classes should be considered where multiple predictions are obtained.
Table III. Variable prediction cutoffs.
If the cutoff values for prediction are increased to mean − sd, mean, mean + sd, and mean + 2 sd, the precision and recall values change as shown in the rows Medium to Highest from bottom to top. Hence, predictions can be chosen according to the precision and recall desired. The TPpred. and FPpred. are categorized according to their Dmax scores. The highest Dmax score for TPpred. of a TPgene is considered as the score for that gene and is used to categorize the TPgenes in the score classes.
| Score Class | TPpred. | TPpred. | FPpred. | FPpred. | TPgenes | TPgenes | Recall | Precision | F Value |
| % | % | % | |||||||
| Arabidopsis | |||||||||
| Highest | 1,100 | 3.98 | 772 | 1.38 | 1,076 | 5.84 | 0.05 | 0.59 | 0.09 |
| Very high | 3,738 | 13.53 | 2,974 | 5.32 | 3,497 | 18.99 | 0.23 | 0.56 | 0.33 |
| High | 8,723 | 31.57 | 13,160 | 23.52 | 7,073 | 38.4 | 0.58 | 0.45 | 0.51 |
| Medium | 12,075 | 43.7 | 31,944 | 57.09 | 6,168 | 33.49 | 0.89 | 0.34 | 0.49 |
| Low | 1,993 | 7.21 | 7,102 | 12.69 | 604 | 3.28 | 0.92 | 0.33 | 0.49 |
| Total | 27,629 | 100 | 55,952 | 100 | 18,418 | 100 | 0.92 | 0.33 | 0.49 |
| Rice | |||||||||
| Highest | 3,651 | 11.18 | 2,846 | 2.75 | 3,482 | 16.57 | 0.15 | 0.56 | 0.24 |
| Very high | 6,439 | 19.72 | 7,202 | 6.96 | 5,609 | 26.69 | 0.39 | 0.5 | 0.44 |
| High | 10,491 | 32.13 | 25,350 | 24.5 | 7,379 | 35.11 | 0.71 | 0.37 | 0.48 |
| Medium | 9,872 | 30.23 | 54,706 | 52.87 | 4,125 | 19.63 | 0.89 | 0.25 | 0.39 |
| Low | 2,198 | 6.73 | 13,365 | 12.92 | 424 | 2.02 | 0.91 | 0.24 | 0.38 |
| Total | 32,651 | 100 | 103,469 | 100 | 21,019 | 100 | 0.91 | 0.24 | 0.38 |
Distribution of False-Positive Predictions in Primary Transcript
The PromPredict program is able to predict cis-regulatory elements for more than 90% of the annotated genes (considering all score classes). However, on applying the lowest cutoffs, the precision of prediction is lower (i.e. a substantial number of predictions are found in the primary transcript region [FP region]). An analysis of the locations and relative scores of FPpred. (Table IV) showed that the majority of FPpred. were obtained in the noncoding regions of the primary transcript (i.e. introns and UTRs). We found that approximately 20% of FPpred. were found in the first intron alone, which could be a putative cis-regulatory region. If we consider the predictions in the first intron as TPpred., the precision increases to 0.47 for Arabidopsis and 0.40 for rice. Interestingly, the median length for the first intron (177 nucleotides for Arabidopsis and 1,176 nucleotides for rice) is greater than that for all introns (100 nucleotides for Arabidopsis and 96 nucleotides for rice). Only a few FPpred. were found in the CDS region (approximately 6%), although it constitutes a substantial length of the primary transcript (50.8% in Arabidopsis and 29.7% in rice). If these predictions are considered as FPpred., the precision increases to 0.96 for both Arabidopsis and rice.
Table IV. Percentage distribution of FPpred.
The location of FPpred. in coding and noncoding regions of primary transcripts as a percentage of the total FPpred. is shown.
| Region | Arabidopsis | Rice |
| 5′ UTR | 7.3% | 6.6% |
| 3′ UTR | 14.3% | 9.1% |
| Introns | 71.7% | 78.4% |
| First introna | 20.4% | 21% |
| CDS | 6.7% | 5.9% |
The nearest intron from the TSS that has length greater than 50 nucleotides is considered as the first intron irrespective of its location in the UTR or the coding region.
We also categorized FPpred. in score classes and according to their location in the primary transcript. The distribution is presented in Figure 4 as a percentage of FPpred. for each score class. Although introns dominate in all score classes, there is an increasing trend of predictions toward higher score categories (68% level of significance for the highest frequency). The same trend is observed in 5′ UTRs, although the number of FPpred. is low. On the other hand, in exons and 3′ UTRs, there is an increasing trend toward lower score categories (68% level of significance for the highest frequency).
Figure 4.

FP prediction distribution. The frequency distribution of FPpred. is shown from each score category found in various regions of the primary transcript as a percentage of the total FPpred. in each category for Arabidopsis (A) and rice (B) genomes. The majority of predictions for each category lie in the intronic region.
Analysis of False Negatives
There are very few genes (8%–9%) that do not have any predictions between −500 and +100 bp relative to the TSS, termed as false-negative genes (FNgenes). A comparison of the AFE profiles for these genes with the profiles for all TSSs from a representative chromosome (Fig. 5) showed that the distinct difference between upstream and downstream regions is absent in FNgenes. The immediate upstream region has higher stability while the immediate downstream region has lower stability in the FNgene profile than those observed for the corresponding regions in the profile of all TSSs. The Gene Ontology (GO) categorization of Arabidopsis and rice genes (Supplemental Protocol S2; Supplemental Table S3; Supplemental Fig. S6) does not show a preponderance of FNgenes in any particular GO category. However, slight differences are seen in the various categories, especially the presence of a greater percentage of FNgenes with unknown functions, processes, and cellular locations as well as the absence of FNgenes corresponding to vital processes such as DNA integration and chromatin assembly.
Figure 5.

Genes without a prediction in the TP region (FNgenes). AFE profile comparison is shown between FNgenes and all genes of chromosome 1 with respect to TSS for Arabidopsis (A) and rice (B). The number of genes considered in each case is indicated in parentheses. [See online article for color version of this figure.]
Correlation of Prediction Score with Gene Expression
Interspecies homology is routinely used for the characterization of gene functions. Thus, it would be interesting to see if orthologous genes from rice and Arabidopsis show similarity in their promoter organization as well. An analysis of the prediction score correlation in all orthologous gene pairs from rice and Arabidopsis was carried out. A total of 12,780 Arabidopsis orthologs and 12,615 rice orthologs were obtained (only protein-coding genes with TSS information was considered) using the g:Orth program of the g:profiler software (Reimand et al., 2007), which uses the Plant Ensembl database (Kersey et al., 2010). Of these, 11,941 (93.4%) and 11,554 (91.6%) genes were TPgenes in Arabidopsis and rice, respectively, and the remaining were FNgenes. Since there were multiple Arabidopsis orthologs for certain rice genes and vice versa, 12,359 pairs of orthologous genes were formed. The Dmax prediction scores for the orthologous gene pairs (see “Materials and Methods”) have been plotted in Figure 7 and give a correlation coefficient of 0.23. However, if only the ortholog pairs for which the prediction scores from the two genomes are from the same score class or differ by one level are considered (81% of total pairs; shown as blue +), the correlation coefficient is 0.51.
Figure 7.

Correlation between prediction scores for orthologous genes. The highest prediction scores corresponding to 11,941 TPgenes in Arabidopsis have been plotted against the scores for their 10,275 TPgene orthologs in rice. Since there is more than one Arabidopsis gene ortholog for some rice genes, 12,359 pairs of orthologous genes were formed. The 9,976 orthologous gene pairs with scores in the same class or differing by one level in the two genomes (crosses) give a Pearson correlation coefficient of 0.51 (dotted-dashed best fit line), while a value of 0.23 is obtained for all gene pairs (crosses and dots; solid best fit line). [See online article for color version of this figure.]
A comparison of the relative positions and scores of predictions in the promoter regions of certain orthologous gene pairs (gene IDs are given in Supplemental Table S4) showed that predictions of comparable strength and relative position are found in most cases (Fig. 8). Arabidopsis genes FAD2 (Kim et al., 2006) and PRF1 (Jeong et al., 2006), which have regulatory first introns, were also studied along with their rice orthologs. For FAD2 (Fig. 8E), intronic predictions were observed in the first intron of both Arabidopsis and rice. In addition, a ncRNA gene overlapping the first intron was found in the rice genome. The first intron of PRF1 (Fig. 8F) in Arabidopsis is long and covers the same length as two short introns in the rice homolog. The prediction for rice was found in the second short intron but at the same position from the TSS as the intronic prediction in Arabidopsis.
Figure 8.

Promoter predictions for six orthologous genes are shown for Arabidopsis (blue) and rice (red). The TSSs of the orthologs are aligned and correspond to nucleotide position 0 on the x axis. The orthologous genes are shown schematically at the bottom. Gray bars represent UTRs, thin black bars correspond to introns, and brown bars represent exons. The y axis indicates the Dmax score of the prediction. Only predictions within −500 to +100 bp of the TSS are true positives in each case. The six representative genes shown are Asp aminotransferase (A), copper/zinc (Cu/Zn) superoxide dismutase (B), Dof gene family (C), P-type ATPase (D), FAD2 (E), and PRF1 (F). The first intron for Arabidopsis genes in E and F has been shown to have regulatory functions. A ncRNA gene coincides with the first intron of the rice FAD2 gene as shown in E.
As mentioned earlier, the Dmax score of a prediction gives the relative difference in free energy between adjacent regions. The question then arises, can the score give an idea of the “strength” of the predicted promoter? For example, it has been shown that CpG islands are generally found upstream of housekeeping genes, whereas tissue-specific genes have strong promoters usually containing a TATA box. It would be interesting to see if the relative differences in DNA free energy can capture the promoter strength. We categorized the TPgenes from certain families, metabolic pathways (Mueller et al., 2003; Jaiswal et al., 2006), and GO terms (Gene Ontology Consortium, 2000) according to their score categories (Fig. 6). Most of the gene sets studied showed similar score distributions in the two genomes. For example, about 50% of genes involved in inflorescence had “very high” and “highest” scores in both genomes, which might indicate their tissue-specific roles. Also, 60% to 80% of predictions for heat shock proteins fall into the top three score categories. However, constitutively expressed genes such as ubiquitin and tubulin did not show such similarities, possibly owing to different expression rates for the protein isoforms.
Figure 6.

Classification of gene families, metabolic pathway genes, and genes from specific GO terms for Arabidopsis (A) and rice (B) according to the TP with the highest prediction score present within −500 to +100 bp of the TSS. The distribution of the score categories is presented as a percentage of the TPgenes present in that category. The number adjacent to each bar indicates the number of TPgenes. [See online article for color version of this figure.]
Comparison of PromPredict Performance with EP3
We compared the programs PromPredict and EP3 for whole-genome prediction in Arabidopsis and rice (Table V). For both programs, we considered the predictions within −500 to +500 bp for determining true positives, since that is the criterion used in EP3. The F value (harmonic mean of the recall and precision) calculated shows that PromPredict gives better prediction performance than EP3. However, the EP3 program gives slightly higher F values in the rice genome for protein-coding genes. Interestingly, PromPredict is able to predict cis-regulatory sites for ncRNA genes with much higher sensitivity and precision than EP3. It should be noted that all PromPredict predictions within −500 to +500 bp are considered irrespective of their score. If only the three higher score classes are considered, the F value would improve, as seen in Table III.
Table V. Comparison of PromPredict with EP3 (Abeel et al., 2008a).
In Arabidopsis, 20,094 (protein-coding) and 1,263 (RNA-coding) TSSs were considered for analysis. In rice, 23,057 (protein-coding) and 1,527 (RNA-coding) TSSs were considered for analysis. For Arabidopsis, PromPredict gave 386,264 predictions while EP3 gave 594,559 predictions. PromPredict predicted 1,284,547 signals and EP3 predicted 1,611,598 signals in the rice genome. The region considered for determining true positives is −500 to +500 bp of the TSS for protein-coding genes and −1,000 to 0 bp of the TSS for ncRNA genes.
| Feature | PromPredict |
EP3 |
||
| Protein | RNA | Protein | RNA | |
| Arabidopsis | ||||
| Recall | 0.96 | 0.94 | 0.48 | 0.28 |
| Precision | 0.42 | 0.75 | 0.49 | 0.51 |
| F value | 0.58 | 0.83 | 0.48 | 0.37 |
| TPpred. length (nucleotides)a | 71.6 ± 47.8 | 64.6 ± 45 | 400 | 400 |
| FPpred. length (nucleotides)a | 53.9 ± 38.9 | 61.6 ± 41.9 | 400 | 400 |
| TP coverage (%) | 13.9 | 12.7 | 50.7 | 46 |
| FP coverage (%) | 7.5 | 7.6 | 13.2 | 24.5 |
| Rice | ||||
| Recall | 0.97 | 0.95 | 0.77 | 0.15 |
| Precision | 0.31 | 0.9 | 0.62 | 0.86 |
| F value | 0.47 | 0.92 | 0.53 | 0.26 |
| TPpred. length (nucleotides)a | 94.6 ± 60.1 | 66.2 ± 48.8 | 400 | 400 |
| FPpred. length (nucleotides)a | 60.7 ± 44.5 | 45.8 ± 36.2 | 400 | 400 |
| TP coverage (%) | 17.9 | 14.2 | 51.8 | 7.6 |
| FP coverage (%) | 8.5 | 6 | 10.4 | 6.7 |
In PromPredict, the midpoint of a 100-nucleotide window is considered as a prediction if it satisfies the cutoffs. In EP3, the entire 400-nucleotide window is considered as a prediction if it satisfies the cutoffs.
We believe that the recall and precision values do not give a complete picture of the prediction quality. In order to be useful in guiding experimental analysis and annotation, it is important to have predictions of appropriate length. One of the major drawbacks of EP3 is that it uses nonoverlapping windows of fixed 400-nucleotide length for prediction. As a result, large chunks of the genome are predicted that might not be amenable to experimental validation. PromPredict, on the other hand, calculates free energy over overlapping windows of 100-nucleotide length and assigns it to the midpoint of the window. Thus, the prediction length varies (maximum of 300 nucleotides) depending on the local free energy in adjacent windows. The prediction coverage of the TP and FP regions indicates the percentage length of the region that is predicted to be “true.” The overall percentage of genome length covered by TP and FP predictions is significantly lower for PromPredict as compared with EP3. Overall, PromPredict gives a better performance for promoter prediction in plants than EP3.
DISCUSSION
The program PromPredict gives relatively good performance for the plant genomes of Arabidopsis and rice, although its cutoffs have been derived from prokaryotic analysis. The AFE profiles for plant genomes (Fig. 1) and prokaryotes (Rangannan and Bansal, 2009) are not identical, but the difference in free energy between upstream and downstream sequences is seen in both profiles, which is the prediction criterion used by PromPredict. Interestingly, the cutoffs derived by training on prokaryotes show good performance for eukaryotes (as shown here for plant genomes). However, slightly tweaking the cutoffs might give better predictions for each genome, and the prediction score classes outlined in Table III can serve as alternative cutoffs for achieving the required performance.
We compared the performance parameters of PromPredict with EP3, which is the only other program that predicts extended promoters in plant genomes (Table V). PromPredict gave better F values than EP3 except for protein-coding genes in rice. However, precision and recall parameters take only the number of predictions into consideration and ignore their length. Longer predictions could (wrongly) give better values for these parameters but would, in turn, increase the amount of experimental testing required. The EP3 algorithm gives longer and fixed length predictions that contribute significantly to the TP and FP regions as compared with PromPredict. PromPredict gives a much better performance for ncRNA genes than EP3 for both plant genomes, even though EP3 is based on similar parameters. Most motif searching and trained algorithms such as ARTS, Eponine, and ProSOM that look for consensus sequences or patterns are also expected to give a poor performance for ncRNA genes, because the organization of PolII promoters differs from PolI (Russell and Zomerdijk, 2005) and PolIII (Geiduschek and Kassavetis, 2001) promoters.
The two plant genomes were compared with respect to genome characteristics as well as prediction characteristics. The precision value obtained for rice was lower than for Arabidopsis due to the presence of higher FPpred.. However, this might be a result of longer primary transcripts in the rice genome, which have a preponderance of intronic regions. Predictions obtained in the primary transcript, especially in noncoding regions, cannot be ignored, as these might be alternative promoters or promoters for downstream genes. Yang (2009) has shown that broadly expressed genes in Arabidopsis and rice have longer noncoding regions, which might play a regulatory role. Carninci et al. (2006) have shown that alternative promoters present within primary transcripts are responsible for tissue-specific expression in humans. Forty-eight percent of oligo(dT)-primed CAGE libraries and 34% of random-primed CAGE libraries have at least one alternative promoter overlapping the sequence of known or predicted transcripts. Also, TSSs have been found in the 3′ UTR of certain protein-coding genes, which may code for transcripts that regulate downstream genes on the same or opposite strands. Moreover, regions located downstream of the TSS, such as introns (Rose, 2008; Rose et al., 2008) and 5′ UTRs (Lu et al., 2008), have been shown to be involved in the regulation of transcription by acting as enhancers or through mechanisms such as intron-mediated enhancement. Noncoding regions might also be involved in replication, transcription of regulatory ncRNAs, and transposition. Zhu et al. (2010) have shown that short conserved introns (50–150 nucleotides) in human and mouse show preferential location (3′ UTRs of universally expressed housekeeping genes) and nonrandom chromosomal distribution. They speculate that these introns might play regulatory roles in gene expression and nucleocytoplasmic transcript export.
Our analysis showed that about 95% to 96% of predictions were found in the noncoding region of the primary transcript, a majority of which were found in the introns and may be valid TSSs or cis-regulatory sites (Table IV). Also, we analyzed 24 introns that have been experimentally shown to regulate expression in Arabidopsis and rice (Table VI), out of which 21 were detected by PromPredict to contain a promoter signal. The introns closest to the TSS are suggested to be most important for the regulation of transcription, and interestingly, 20% of our FPpred. are found in the first intron alone. The remaining intronic predictions (50% of total FPpred.) might be signals for other processes, such as splicing in RNAs.
Table VI. Regulatory introns and predictions.
Some Arabidopsis and rice introns are known to regulate expression. The majority of these have an overlapping PromPredict prediction. The first intron in all the genes was involved in regulation, except for TWN2, where the first two introns were involved.
| Gene | Prediction Strength | Reference |
| Arabidopsis | ||
| RHD3 (At3g13870) | No prediction | Wang et al. (2002) |
| Histone H3 (At4g40040) | Medium | Chaubet-Gigot et al. (2001) |
| Histone H3 (At4g40030) | Low | Chaubet-Gigot et al. (2001) |
| EF-1α A1 (At1g07920) | Very high/medium | Curie et al. (1993) |
| EF-1α A3 (At1g07940) | High/medium | Chung et al. (2006) |
| eEF-1β (At2g18110) | Medium | Gidekel et al. (1996) |
| TWN2 intron 1 (At1g14610) | No prediction | Zhang and Somerville (1997) |
| TWN2 intron 2 (At1g14610) | High | Zhang and Somerville (1997) |
| Cox5c-1 (At2g47380) | Medium | Curi et al. (2005) |
| Cox5c-2 (At3g62400) | High/medium | Curi et al. (2005) |
| ACT1 (At2g37620) | High/medium | Vitale et al. (2003) |
| KC01 (At5g55630) | Medium | Czempinski et al. (2002) |
| PRF1 (At2g19760) | High | Jeong et al. (2006) |
| PRF2 (At4g29350) | Medium | Jeong et al. (2006) |
| ADF1 (At3g46010) | High | Jeong et al. (2006) |
| FAD2 (At3g12120) | High/medium | Kim et al. (2006) |
| SUVH3 (At1g73100) | Medium | Casas-Mollano et al. (2006) |
| ATMHX (At2g47600) | No prediction | David-Assael et al. (2006) |
| UBQ3 (At5g03240) | Medium | Norris et al. (1993) |
| UBQ10 (At4g05320) | High | Norris et al. (1993) |
| ATPK1 (At3g08730) | High/medium | Zhang et al. (1994) |
| Rice | ||
| TPI (Os01g0147900 | Medium | Xu et al. (1994), Snowden et al. (1996) |
| GAMyb (Os01g0812000) | Medium | Washio and Morikawa (2006) |
| RPBF (Os02g0252400) | Highest/very high/medium | Washio and Morikawa (2006) |
In order to determine the core promoters, the predictions closest to the TSS were considered for both protein-coding and ncRNA genes (Fig. 2). The predictions in Arabidopsis are concentrated closer to the TSS than in rice. However, the free energy of distal predictions is comparable to that of proximal predictions (Fig. 3), indicating that these might be putative core promoters and not prediction artifacts. Thus, it seems that free energy peaks might be present at different positions relative to the TSS for eukaryotic genes, in contrast to prokaryotic genes, where the peak is only localized close to the TSS (Rangannan and Bansal, 2009).
A genome-wide analysis of prediction scores in orthologous gene pairs gave a good correlation between the highest score TPpred. for each ortholog member (Fig. 7). A finer analysis of promoter organization in certain ortholog pairs further showed that the predictions in the vicinity of the TSS not only have comparable scores but also similar locations with respect to TSS (Fig. 8). Hence, there seems to be a good relationship between the promoter predictions in orthologous genes, and we propose that our predictions can also be used for studying promoter regions in these genes.
The AFE for FNgenes showed a different profile than that observed for all genes, especially in rice (Fig. 5). The presence of alternative structural profiles for bendability in human promoters has been reported (Florquin et al., 2005; Zeng et al., 2009). The presence of such alternative structural profiles might point toward different regulatory architectures that enable spatiotemporal expression specificity in eukaryotes. Hence, alternative free energy profiles (and other structural profiles) could be explored in plants to gain a better understanding of the promoter region and regulation.
CONCLUSION
We show here that the program PromPredict performs quite well in predicting cis-regulatory regions in plant genomes. This is indeed surprising, since the program has been trained on prokaryotes. It seems that the relative free energy difference criterion used in this program is a general property found in the vicinity of the TSS, as shown in the human genome by Abeel et al. (2008a). Hence, PromPredict might also be expected to perform well for other plants and eukaryotes.
The program is based on simple prediction criteria that are easy to program, and further enhancement of the program with other features might give better results. Since PromPredict predictions are biased toward unstable and hence AT-rich regions, complementing the program with other motifs like the Y patch or GA motif (Yamamoto et al., 2009) could be beneficial.
As our understanding of transcription develops, the actual complexity of the processes involved in gene regulation is revealed. Determination of putative regulatory sites where transcription factors could bind is but a small step in trying to understand the huge orchestra of regulatory mechanisms involved. It is difficult to make a one-to-one correlation between the cis-regulatory region and the corresponding regulated gene. In eukaryotic genomes, the sites involved in the regulation of a gene may vary in different tissues, adding to the complexity of the problem. This is further complicated by factors such as combinatorial regulation, nucleosome binding, and epigenetic modifications. Yet, common themes and patterns of regulation can be observed, as seen in this study. A combinatorial approach involving sequence and structural studies, both theoretical and experimental, would be most useful to further explore the mechanisms of transcription regulation.
MATERIALS AND METHODS
Data Sets
The Arabidopsis (Arabidopsis thaliana) genome and annotation data were extracted from the TAIR9 release of The Arabidopsis Information Resource (TAIR; Arabidopsis Genome Initiative, 2000; Rhee et al., 2003). The TAIR9 release contains multiple gene models for certain genes. Since we are considering the TSS positions in our analysis, gene models with overlapping or proximal TSSs will result in misrepresentation of the results. Hence, for TSSs (of the same gene) within 100 nucleotides of each other, only the most upstream TSS is considered. Applying this constraint, we have a data set of 20,094 protein-coding gene models with TSS information and 1,263 ncRNA gene models. Also, gene models with only TLS information were sorted so that gene models with TLSs at least 100 nucleotides apart are selected for a particular gene, to give the TLS data set of 8,195 genes. The Web browser for visualizing predictions shows all the gene models.
The rice (Oryza sativa ssp. japonica ‘Nipponbare’) genome was extracted from the Rice Annotation Project Database (RAPDB) Build 4 (Rice Annotation Project, 2007, 2008). The protein-coding genes with TSS information (23,057 genes) and ncRNA genes (1,527 genes) were considered for analysis. A total of 1,152 non-protein-coding primary transcripts were also considered for analysis. The latest build of RAPDB (Build 5) has 31,232 protein-coding genes and 1,515 noncoding primary transcripts.
AFE Profile
For calculating the AFE, dinucleotide parameters based on the model proposed by Allawi and Santalucia (1997) and Santalucia (1998) were used. Sequences of the same length were aligned with the TSS at the 0 position. An average profile is obtained by calculating the mean value of free energy at each position over all the sequences. The dinucleotide parameters averaged over a moving window of 15 nucleotides (frameshift of one nucleotide) were assigned to the midpoint of each window in order to reduce noise.
PromPredict Program
The PromPredict program was first written to predict promoter regions in prokaryotes (Kanhere and Bansal, 2005b; Rangannan and Bansal, 2007, 2009). The program is built to predict cis-regulatory regions in a given input sequence on the basis of relative free energy of neighboring regions in a 1,001-nucleotide-long fragment and does not require any genome-specific training. Hence, the program can be readily used for newly sequenced genomes for which gene and promoter information is scarce. Supplemental Figure S3A shows the AFE values in the −2,000 to +2,000 regions relative to TSS in Arabidopsis and rice. Supplemental Figure S3B shows the AFE values used as cutoffs to define the promoter regions in PromPredict and the AFE values in upstream (−500 to 0) and downstream (0 to +500) regions of Arabidopsis and rice. The values for Arabidopsis match well with the PromPredict cutoffs. The proximal downstream regions in rice are unusually GC rich, leading to lower AFE values, but in general, the AFE values for nonpromoter regions (+500 to +1,000) farther away from TSSs are similar to the cutoffs used in PromPredict.
PromPredict considers (1) the absolute free energy (E1) averaged over each overlapping window of a 100-nucleotide sequence (frameshift of one nucleotide) and (2) the relative free energy difference (D) of E1 with the free energy (E2) averaged over a downstream 100-nucleotide sequence separated by 50 nucleotides in the 5′→3′ direction. The free energy is calculated using the dinucleotide parameters based on the model proposed by Allawi and Santalucia (1997) and Santalucia (1998) as a sum over 15 nucleotides. The parameters E1 and D are then compared with predefined cutoff values (Supplemental Table S1). A sliding superwindow of 1,001 nucleotides (with a frameshift of 750 nucleotides) is used to determine the average GC content range used for cutoff values for each 100-nucleotide window within this superwindow. Further details of the algorithm are given in Supplemental Protocol S1. We have used the latest version of the program described by Rangannan and Bansal (2010).
Performance Evaluation and Comparison
The performance of PromPredict on rice and Arabidopsis genomes was evaluated using the distance-based cutoff as described by Bajic et al. (2004) and Abeel et al. (2009). It is obvious that the performance of a program would improve if the region considered for determining true positives was increased. However, the cis-regulatory regions are generally found within a particular distance upstream of the TSS/TLS and in some regions within the primary transcript, such as the 5′ UTR. The optimal length of this region largely depends on the organism under study. Previous analyses have considered a TP region of −150 to +50 bp for prokaryotes (Rangannan and Bansal, 2009), −500 to +500 bp for eukaryotes (Abeel et al., 2008a, 2009) with respect to TSS, and −500 to 0 bp for prokaryotes with respect to TLS (Rangannan and Bansal, 2009), where the 0 position corresponds to the TSS/TLS. For rice and Arabidopsis, we found that approximately 50% of the genes have a 5′ UTR length of 100 nucleotides or less (Table I). Hence, in our analysis, we considered the region −500 to +100 bp with respect to the TSS for determining true positives in protein-coding genes while the region −1,000 to 0 bp of the TSS/TLS was considered for ncRNA genes and for genes with only TLS information. For comparison of our results with EP3, we considered the region −500 to +500 bp with respect to the TSS for protein-coding genes and −1,000 to 0 bp of the TSS for ncRNA genes.
The least stable position of a prediction was considered as a single-nucleotide metric for defining true and false predictions to avoid ambiguity due to overlapping with the TP region. Therefore, any prediction with its least stable position lying within the TP region was considered as a TPpred., while any prediction (least stable position) lying within the transcribing region of a gene but not within the TP region (FP region) was considered as a FPpred.. The genes that have at least one TPpred. within the TP region of its TSS were considered as TPgenes. The genes that did not have any predictions within the TP region were considered as FNgenes. The performance parameters were defined as follows:
 |
 |
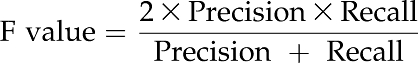 |
We also mention the average prediction length and the percentage prediction coverage of the TP region and FP region when comparing programs. The latter statistic indicates the percentage length of the TP or FP region that a program predicts to be true.
Prediction Score Categorization
The D value of a prediction reflects the difference in free energy of a prediction from its downstream region. Since an unstable region in a comparatively stable environment in DNA is a characteristic of promoter regions, we used the D value to determine the relative score of predictions. The Dmax for all predictions in Arabidopsis and rice were pooled, and the mean and sd for these data were calculated. Dmax cutoffs for categorizing predictions (Supplemental Table S2) were calculated on the basis of the GC content of a 1,001-nucleotide fragment (superwindow) containing the prediction. One of the following five score classes was assigned to each prediction depending on the score of the Dmax value of a prediction: (1) highest (Dmax > mean + 2 sd); (2) very high (mean + sd < Dmax < mean + 2 sd); (3) high (mean < Dmax < mean + sd); (4) medium (mean − sd < Dmax < mean); (5) low (cutoff < Dmax < mean − sd). Also, the genes were categorized according to the score class of the prediction with the highest score present within −500 to +100 bp of its TSS. All the predictions thus categorized according to their scores can be browsed online in the genome browser PlantcisProm constructed using Bioperl (Stein et al., 2002) along with annotated genes (see below).
Web Resources
The PlantcisProm genome browser (http://nucleix.mbu.iisc.ernet.in/plantcisprom) can be used to browse the whole-genome promoter predictions along with gene annotations for Arabidopsis and rice genomes. The PromPredict Web server (http://nucleix.mbu.iisc.ernet.in/prompredict/prompredict.html) can be used to predict promoter regions in the input sequence. Downloadable versions of the program are available for short (less than 10 Mb) and long (more than 10 Mb) genomic sequences.
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Figure S1. GC content distribution for Arabidopsis and rice sequences in the vicinity of the TSS.
Supplemental Figure S2. AFE profile variation with GC content for sequences in the vicinity of the TSS.
Supplemental Figure S3. Comparison of AFE values in the vicinity of the TSS with cutoff values used in PromPredict.
Supplemental Figure S4. Derivation of cutoff values for prediction score categories of Arabidopsis and rice predictions.
Supplemental Figure S5. Percentage frequency distribution of tetramers in the core promoter and the 1,001-nucleotide region surrounding the TSS.
Supplemental Figure S6. GO categorization of all genes and TPgenes from rice chromosome 1.
Supplemental Table S1. Cutoff values for PromPredict.
Supplemental Table S2. Cutoff values for prediction score categories.
Supplemental Table S3. GO categorization of TPgenes and FNgenes from Arabidopsis.
Supplemental Table S4. Gene IDs for orthologous genes.
Supplemental Protocol S1. Details of the PromPredict algorithm.
Supplemental Protocol S2. GO SLIM categories
Supplementary Material
Acknowledgments
We thank the RAPDB and TAIR teams for providing information on annotation queries.
References
- Abeel T, Saeys Y, Bonnet E, Rouzé P, Van De Peer Y. (2008a) Generic eukaryotic core promoter prediction using structural features of DNA. Genome Res 18: 310–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abeel T, Saeys Y, Rouzé P, Van De Peer Y. (2008b) ProSOM: core promoter prediction based on unsupervised clustering of DNA physical profiles. Bioinformatics 24: i24–i31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abeel T, Van De Peer Y, Saeys Y. (2009) Toward a gold standard for promoter prediction evaluation. Bioinformatics 25: i313–i320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov N, Troukhan M, Brover V, Tatarinova T, Flavell R, Feldmann K. (2006) Features of Arabidopsis genes and genome discovered using full-length cDNAs. Plant Mol Biol 60: 69–85 [DOI] [PubMed] [Google Scholar]
- Allawi HT, Santalucia J. (1997) Thermodynamics and NMR of internal G-T mismatches in DNA. Biochemistry 36: 10581–10594 [DOI] [PubMed] [Google Scholar]
- Arabidopsis Genome Initiative (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408: 796–815 [DOI] [PubMed] [Google Scholar]
- Bajic VB, Tan SL, Suzuki Y, Sugano S. (2004) Promoter prediction analysis on the whole human genome. Nat Biotechnol 22: 1467–1473 [DOI] [PubMed] [Google Scholar]
- Cao X, Zeng J, Yan H. (2009) Physical signals for protein-DNA recognition. Phys Biol 6: 036012–036021 [DOI] [PubMed] [Google Scholar]
- Carninci P, Sandelin A, Lenhard B, Katayama S, Shimokawa K, Frith MC, Ponjavic J, Semple CAM, Taylor MS, Forrest ARR, et al. (2006) Genome-wide analysis of mammalian promoter architecture and evolution. Nat Genet 38: 626–635 [DOI] [PubMed] [Google Scholar]
- Casas-Mollano J, Lao N, Kavanagh T. (2006) Intron-regulated expression of SUVH3, an Arabidopsis Su(var)3-9 homologue. J Exp Bot 57: 3301–3311 [DOI] [PubMed] [Google Scholar]
- Chaubet-Gigot N, Kapros T, Flenet M, Kahn K, Gigot C, Waterborg J. (2001) Tissue-dependent enhancement of transgene expression by introns of replacement histone H3 genes of Arabidopsis. Plant Mol Biol 45: 17–30 [DOI] [PubMed] [Google Scholar]
- Chung B, Simons C, Firth A, Brown C, Hellens R. (2006) Effect of 5′UTR introns on gene expression in Arabidopsis thaliana. BMC Genomics 7: 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper SJ, Trinklein ND, Anton ED, Nguyen L, Myers RM. (2006) Comprehensive analysis of transcriptional promoter structure and function in 1% of the human genome. Genome Res 16: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curi GC, Chan RL, Gonzalez DH. (2005) The leader intron of Arabidopsis thaliana genes encoding cytochrome c oxidase subunit 5c promotes high-level expression by increasing transcript abundance and translation efficiency. J Exp Bot 56: 2563–2571 [DOI] [PubMed] [Google Scholar]
- Curie C, Axelos M, Bardet C, Atanassova R, Chaubet N, Lescure B. (1993) Modular organization and development activity of an Arabidopsis thaliana EF-1α gene promoter. Mol Genet Genomics 238: 428–436 [DOI] [PubMed] [Google Scholar]
- Czempinski K, Frachisse J, Maurel C, Barbier-Brygoo H, Mueller-Roeber B. (2002) Vacuolar membrane localization of the Arabidopsis ‘two-pore’ K+ channel KCO1. Plant J 29: 809–820 [DOI] [PubMed] [Google Scholar]
- David-Assael O, Berezin I, Shoshani-Knaai N, Saul H, Mizrachy-Dagri T, Chen J, Brook E, Shaul O. (2006) AtMHX is an auxin and ABA-regulated transporter whose expression pattern suggests a role in metal homeostatis in tissues with photosynthetic potential. Funct Plant Biol 33: 661–672 [DOI] [PubMed] [Google Scholar]
- Davuluri R, Sun H, Palaniswamy S, Matthews N, Molina C, Kurtz M, Grotewold E. (2003) AGRIS: Arabidopsis Gene Regulatory Information Server, an information resource of Arabidopsis cis-regulatory elements and transcription factors. BMC Bioinformatics 4: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Down TA, Hubbard TJP. (2002) Computational detection and location of transcription start sites in mammalian genomic DNA. Genome Res 12: 458–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium (2007) Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447: 799–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnham PJ. (2009) Insights from genomic profiling of transcription factors. Nat Rev Genet 10: 605–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florquin K, Saeys Y, Degroeve S, Rouzé P, Van de Peer Y. (2005) Large-scale structural analysis of the core promoter in mammalian and plant genomes. Nucleic Acids Res 33: 4255–4264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimori S, Washio T, Tomita M. (2005) GC-compositional strand bias around transcription start sites in plants and fungi. BMC Genomics 6: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiduschek EP, Kassavetis GA. (2001) The RNA polymerase III transcription apparatus. J Mol Biol 310: 1–26 [DOI] [PubMed] [Google Scholar]
- Gene Ontology Consortium (2000) Gene Ontology: tool for the unification of biology. Nat Genet 25: 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gidekel M, Jimenez B, Herrera-estrella L. (1996) The first intron of the Arabidopsis thaliana gene coding for elongation factor 1-β contains an enhancer-like element. Gene 170: 201–206 [DOI] [PubMed] [Google Scholar]
- Higo K, Ugawa Y, Iwamoto M, Korenaga T. (1999) Plant cis-acting regulatory DNA elements (PLACE) database: 1999. Nucleic Acids Res 27: 297–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Brachypodium Initiative (2010) Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 463: 763–768 [DOI] [PubMed] [Google Scholar]
- Jaiswal P, Ni J, Yap I, Ware D, Spooner W, Youens-Clark K, Ren L, Liang C, Zhao W, Ratnapu K, et al. (2006) Gramene: a bird’s eye view of cereal genomes. Nucleic Acids Res 34: D717–D723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong Y, Mun J, Lee I, Woo J, Hong C, Kim S. (2006) Distinct roles of the first introns on the expression of Arabidopsis profilin gene family members. Plant Physiol 140: 196–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanhere A, Bansal M. (2005a) A novel method for prokaryotic promoter prediction based on DNA stability. BMC Bioinformatics 6: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanhere A, Bansal M. (2005b) Structural properties of promoters: similarities and differences between prokaryotes and eukaryotes. Nucleic Acids Res 33: 3165–3175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersey PJ, Lawson D, Birney E, Derwent PS, Haimel M, Herrero J, Keenan S, Kerhornou A, Koscielny G, Kähäri A, et al. (2010) Ensembl Genomes: extending Ensembl across the taxonomic space. Nucleic Acids Res 38: D563–D569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Kim H, Shin J, Chung C, Ohlrogge J, Suh M. (2006) Seed-specific expression of sesame microsomal oleic acid desaturase is controlled by combinatorial properties between negative cis-regulatory elements in the SeFAD2 promoter and enhancers in the 5′-UTR intron. Mol Genet Genomics 276: 351–368 [DOI] [PubMed] [Google Scholar]
- Lantermann AB, Straub T, Stralfors A, Yuan G-C, Ekwallkarl K, Korber P. (2010) Schizosaccharomyces pombe genome-wide nucleosome mapping reveals positioning mechanisms distinct from those of Saccharomyces cerevisiae. Nat Struct Mol Biol 17: 251–257 [DOI] [PubMed] [Google Scholar]
- Lee W, Tillo D, Bray N, Morse RH, Davis RW, Hughes TR, Nislow C. (2007) A high-resolution atlas of nucleosome occupancy in yeast. Nat Genet 39: 1235–1244 [DOI] [PubMed] [Google Scholar]
- Lichtenberg J, Yilmaz A, Welch J, Kurz K, Liang X, Drews F, Ecker K, Lee S, Geisler M, Grotewold E, et al. (2009) The word landscape of the non-coding segments of the Arabidopsis thaliana genome. BMC Genomics 10: 463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Sivamani E, Azhakanandam K, Samadder P, Li X, Qu R. (2008) Gene expression enhancement mediated by the 5′ UTR intron of the rice rubi3 gene varied remarkably among tissues in transgenic rice plants. Mol Genet Genomics 279: 563–572 [DOI] [PubMed] [Google Scholar]
- Ming R, Hou S, Feng Y, Yu Q, Dionne-Laporte A, Saw JH, Senin P, Wang W, Ly BV, Lewis KLT, et al. (2008) The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus). Nature 452: 991–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina C, Grotewold E. (2005) Genome wide analysis of Arabidopsis core promoters. BMC Genomics 6: 25–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RT, O’Connor TR, Wyrick JJ. (2008) Osiris: an integrated promoter database for Oryza sativa L. Bioinformatics 24: 2915–2917 [DOI] [PubMed] [Google Scholar]
- Mueller LA, Zhang P, Rhee SY. (2003) AraCyc: a biochemical pathway database for Arabidopsis. Plant Physiol 132: 453–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris S, Meyer S, Callis J. (1993) The intron of Arabidopsis thaliana polyubiquitin genes is conserved in location and is a quantitative determinant of chimeric gene expression. Plant Mol Biol 21: 895–906 [DOI] [PubMed] [Google Scholar]
- Parker SCJ, Hansen L, Abaan HO, Tullius TD, Margulies EH. (2009) Local DNA topography correlates with functional noncoding regions of the human genome. Science 324: 389–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen AG, Baldi P, Chauvin Y, Brunak S. (1999) The biology of eukaryotic promoter prediction: a review. Comput Chem 23: 191–207 [DOI] [PubMed] [Google Scholar]
- Pugh BF. (2000) Control of gene expression through regulation of the TATA-binding protein. Gene 255: 1–14 [DOI] [PubMed] [Google Scholar]
- Rangannan V, Bansal M. (2007) Identification and annotation of promoter regions in microbial genome sequences on the basis of DNA stability. J Biosci 32: 851–862 [DOI] [PubMed] [Google Scholar]
- Rangannan V, Bansal M. (2009) Relative stability of DNA as a generic criterion for promoter prediction: whole genome annotation of microbial genomes with varying nucleotide base composition. Mol Biosyst 5: 1758–1769 [DOI] [PubMed] [Google Scholar]
- Rangannan V, Bansal M. (2010) High quality annotation of promoter regions for 913 bacterial genomes. Bioinformatics 26: 3043–3050 [DOI] [PubMed] [Google Scholar]
- Reimand J, Kull M, Peterson H, Hansen J, Vilo J. (2007) g:Profiler: a Web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res 35: W193–W200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rensing SA, Lang D, Zimmer AD, Terry A, Salamov A, Shapiro H, Nishiyama T, Perroud P-F, Lindquist EA, Kamisugi Y, et al. (2008) The Physcomitrella genome reveals evolutionary insights into the conquest of land by plants. Science 319: 64–69 [DOI] [PubMed] [Google Scholar]
- Rhee S, Beavis W, Berardini TZ, Chen G, Dixon D, Doyle A, Garcia-Hernandez M, Huala E, Lander G, Montoya M, et al. (2003) The Arabidopsis Information Resource (TAIR): a model organism database providing a centralized, curated gateway to Arabidopsis biology, research materials and community. Nucleic Acids Res 31: 224–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice Annotation Project (2007) Curated genome annotation of Oryza sativa ssp. japonica and comparative genome analysis with Arabidopsis thaliana. Genome Res 17: 175–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice Annotation Project (2008) The Rice Annotation Project Database (RAP-DB): 2008 update. Nucleic Acids Res 36: D1028–D1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rombauts S, Florquin K, Lescot M, Marchal K, Rouzé P, Peer YVD. (2003) Computational approaches to identify promoters and cis-regulatory elements in plant genomes. Plant Physiol 132: 1162–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose AB. (2008) Intron-mediated regulation of gene expression. Curr Top Microbiol Immunol 326: 277–290 [DOI] [PubMed] [Google Scholar]
- Rose AB, Elfersi T, Parra G, Korf I. (2008) Promoter-proximal introns in Arabidopsis thaliana are enriched in dispersed signals that elevate gene expression. Plant Cell 20: 543–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell J, Zomerdijk JCBM. (2005) RNA-polymerase-I-directed rDNA transcription, life and works. Trends Biochem Sci 30: 87–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santalucia J. (1998) A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc Natl Acad Sci USA 95: 1460–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, Nelson W, Hyten DL, Song Q, Thelen JJ, Cheng J, et al. (2010) Genome sequence of the palaeopolyploid soybean. Nature 463: 178–183 [DOI] [PubMed] [Google Scholar]
- Snowden KC, Buchhholz WG, Hall TC. (1996) Intron position affects expression from the tpi promoter in rice. Plant Mol Biol 31: 689–692 [DOI] [PubMed] [Google Scholar]
- Sonnenburg S, Zien A, Ratsch G. (2006) ARTS: accurate recognition of transcription starts in human. Bioinformatics 22: e472–e480 [DOI] [PubMed] [Google Scholar]
- Stein LD, Mungall C, Shu S, Caudy M, Mangone M, Day A, Nickerson E, Stajich JE, Harris TW, Arva A, et al. (2002) The Generic Genome Browser: a building block for a model organism system database. Genome Res 12: 1599–1610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Koyanagi KO, Itoh T. (2009) Highly diversified molecular evolution of downstream transcription start sites in rice and Arabidopsis. Plant Physiol 149: 1316–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale A, We RJ, Cheng Z, Meagher RB. (2003) Multiple conserved 5′ elements are required for high-level pollen expression of the Arabidopsis reproductive actin ACT1. Plant Mol Biol 52: 1135–1151 [DOI] [PubMed] [Google Scholar]
- Wang H, Lee MM, Schiefelbein JW. (2002) Regulation of the cell expansion gene RHD3 during Arabidopsis development. Plant Physiol 129: 638–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washio K, Morikawa M. (2006) Common mechanisms regulating expression of rice aleurone genes that contribute to the primary response for gibberellin. Biochim Biophys Acta 1759: 478–490 [DOI] [PubMed] [Google Scholar]
- Wong GK-S, Wang J, Tao L, Tan J, Zhang J, Passey DA, Yu J. (2002) Compositional gradients in Gramineae genes. Genome Res 12: 851–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Yu H, Hall TC. (1994) Rice triosephosphate isomerase gene 5′ sequence directs β-glucuronidase activity in transgenic tobacco but requires an intron for expression in rice. Plant Physiol 106: 459–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y, Ichida H, Matsui M, Obokata J, Sakurai T, Satou M, Seki M, Shinozaki K, Abe T. (2007a) Identification of plant promoter constituents by analysis of local distribution of short sequences. BMC Genomics 8: 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto YY, Ichida H, Abe T, Suzuki Y, Sugano S, Obokata J. (2007b) Differentiation of core promoter architecture between plants and mammals revealed by LDSS analysis. Nucleic Acids Res 35: 6219–6226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto YY, Yoshitsugu T, Sakurai T, Seki M, Shinozaki K, Obokata J. (2009) Heterogeneity of Arabidopsis core promoters revealed by high-density TSS analysis. Plant J 60: 350–362 [DOI] [PubMed] [Google Scholar]
- Yang H. (2009) In plants, expression breadth and expression level distinctly and nonlinearly correlate with gene structure. Biol Direct 4: 45–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng J, Cao X-Q, Zhao H, Yan H. (2009) Finding human promoter groups based on DNA physical properties. Phys Rev E Stat Nonlin Soft Matter Phys 80: 041917. [DOI] [PubMed] [Google Scholar]
- Zhang JZ, Somerville CR. (1997) Suspensor-derived poly-embryony caused by altered expression of valyl-tRNA synthetase in the twn2 mutant of Arabidopsis. Proc Natl Acad Sci USA 94: 7349–7355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S-h, Lawton MA, Hunter T, Lamb CJ. (1994) atpk1, a novel ribosomal protein kinase gene from Arabidopsis. J Biol Chem 269: 17586–17592 [PubMed] [Google Scholar]
- Zhu J, He F, Wang D, Liu K, Huang D, Xiao J, Wu J, Hu S, Yu J. (2010) A novel role for minimal introns: routing mRNAs to the cytosol. PLoS ONE 5: e10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.