

Abstract
Cases of progressive multifocal leukoencephalopathy can occur in patients treated with the B cell depleting anti-CD20 antibody, rituximab, highlighting the importance of B cell surveillance of the central nervous system (CNS). The lymphoid chemokine, CXCL13, is critical for B cell recruitment and functional organization of peripheral lymphoid tissues, and CXCL13 levels are often elevated in the inflamed CNS. To more directly investigate the role of CXCL13 in CNS B cell migration, its role in animal models of infectious and inflammatory demyelinating disease was examined. During acute alphavirus encephalitis where viral clearance depends on the local actions of anti-viral antibodies, CXCL13 levels and B cell numbers increased in brain tissue over time. Surprisingly, however, CXCL13-deficient animals showed normal CNS B cell recruitment, unaltered CNS virus replication and clearance, and intact peripheral anti-viral antibody responses. During experimental autoimmune encephalomyelitis (EAE), CNS levels of CXCL13 increased as symptoms emerged and equivalent numbers of B cells were identified among the CNS infiltrates of CXCL13-deficient mice compared to control animals. However, CXCL13-deficient mice did not sustain pathogenic anti-myelin T cell responses, consistent with their known propensity to develop more self-limited EAE. These data show that CXCL13 is dispensable for CNS B cell recruitment in both models. The disease course is unaffected by CXCL13 in a CNS infection paradigm that depends on a pathogen-specific B cell response, while it is heightened and prolonged by CXCL13 when myelin-specific CD4+ T cells drive CNS pathology. Thus, CXCL13 could be a therapeutic target in certain neuroinflammatory diseases, but not by blocking B cell recruitment to the CNS.
Keywords: B cell homing, Central nervous system, CXCL13, Experimental autoimmune encephalomyelitis, Alphavirus encephalitis
1. Introduction
Lymphoid chemokines, also known as homeostatic chemokines, are constitutively expressed in lymphoid organs and control the recruitment and compartmentalization of lymphocytes and antigen presenting cells within these specialized structures (Yoshie et al., 1997, Zlotnik et al., 1999). CCL19 and CCL21 bind to CCR7 and recruit T cells and dendritic cells (DCs) to T cell areas of secondary lymphoid tissues (Yoshie et al., 1997, Muller et al., 2003). CXCL12 binds to CXCR4 and attracts multiple immune cell types to lymph nodes and spleen, and along with CXCL13, drives the formation of germinal centers (Campbell et al., 2003, Allen et al., 2004). CXCL13 is made by stromal cells in B cell follicles, and recruits both B cells and follicular helper T cells to these compartments by acting through its cognate receptor, CXCR5 (Forster et al., 1996, Legler et al., 1998, Ansel et al., 2000, Moser et al., 2002, Muller et al., 2003). The expression of all lymphoid chemokines in both T and B cell areas of lymphoid organs depends on lymphotoxin (LT) α1β2 and tumor necrosis factor (TNF) signaling in stromal cells that are a main source of these chemoattractants (Mackay and Browning, 1998, Ngo et al., 1999, Fu and Chaplin, 1999).
Beyond their role in the development and maintenance of lymphoid tissues, the lymphoid chemokines have also been implicated in the perpetuation of tissue inflammation. During chronic inflammation of the synovium and salivary gland, ectopic structures resembling the T and B cell follicles of lymphoid organs have been observed (Hjelmstrom, 2001). Evidence supporting a role for lymphoid chemokines in this process of “lymphoid neogenesis” derives in part from studies showing that transgenic expression of CCL21 or CXCL13 in organs such as the pancreas or thyroid gland is sufficient to cause leukocyte recruitment and the formation of lymphoid structures at these sites (Fan et al., 2000, Luther et al., 2000, Luther et al., 2002, Chen et al., 2002, Martin et al., 2004). CXCL13 is expressed in gastric mucosa-associated lymphoid structures that develop in response to Helicobacter pylori infection (Mazzucchelli et al., 1999), and has also been found in B cell aggregates that develop in the inflamed meninges of mice with experimental autoimmune encephalomyelitis (EAE) and humans with progressive multiple sclerosis (MS) (Magliozzi et al., 2004, Magliozzi et al., 2007, Serafini et al., 2004, Aloisi et al., 2008). Such data have fueled interest in the role played by CXCL13 in local B cell recruitment during organ-specific infectious and autoimmune diseases (Lalor and Segal, 2010).
Intrathecal accumulation of B cells and immunoglobulins (Ig) is a prominent feature of many infectious and inflammatory disorders of the CNS, even when ectopic lymphoid follicles have not been convincingly identified. In humans, elevated cerebrospinal fluid (CSF) levels of CXCL13 are found not only in MS, but also in Lyme neuroborreliosis and primary central nervous system (CNS) lymphomas (PCNSL) where the tumors are almost always of B cell origin (Krumbholz et al., 2006, Ljostad and Mygland, 2008, Fischer et al., 2009, Rupprecht et al., 2009, Sellebjerg et al., 2009). In many of these settings, the magnitude of CXCL13 elevation correlates directly with the number of B cells present in the CSF sample (Krumbholz et al., 2006), while successful treatment is associated with parallel declines of both CSF CXCL13 concentrations and CSF B cell numbers (Ljostad and Mygland, 2008, Fischer et al., 2009, Sellebjerg et al., 2009). Still, the relative contribution of CXCL13 to recruiting CXCR5+ B cells into the CNS versus the coordinating their localization and interactions inside the CNS remains unresolved. One study in EAE showed that treating mice with a LTβR-Ig fusion protein that blocked LTα1β2 signaling could prevent the induction of CXCL13 in the meninges and stop the formation of organized B cell follicles at that site, but did not affect overall meningeal B cell migration (Columba-Cabezas et al., 2006). Continued uncertainty regarding the role of CXCL13 in B cell recruitment to the inflamed CNS prompted us to more thoroughly investigate this question in two well-established animal models of neuroinflammation. Despite its induction in target tissues, we find that CXCL13 is dispensable for B cell recruitment to the CNS during both acute viral encephalitis and EAE, even though it may still be a therapeutic target in CNS autoimmune disease driven by CD4+ T cells.
2. Materials and methods
2.1. Mice
Inbred C57BL/6 mice were obtained from Charles River or Harlan Laboratories. CXCL13-deficient mice generated on a C57BL/6 background were a generous gift from Dr. Jason Cyster (University of California, San Francisco) (Ansel et al., 2000, Ansel et al., 2002). Animals were housed under specific pathogen-free, barrier facility conditions on a 10/14-h light/dark cycle with food and water available ad libitum. All procedures were performed under isoflurane or avertin anesthesia and were conducted in strict accordance with protocols approved by the University Committee on Use and Care of Animals.
2.2. Peptides and viruses
The 35–55 amino acid peptide fragment of myelin oligodendrocyte glycoprotein (MOG) (MOG35–55), having an amino acid sequence of MEVGWYRSPFSRVVHLYRNGK, was synthesized by Bio-Synthesis Incorporated and purified by HPLC. Wild-type Sindbis virus (SV) strain AR339, biologically cloned stock SV1A, was grown and assayed for plaque formation in BHK-21 cells. Stock titers of 5 × 107 plaque-forming units (PFU)/ml were stored at −80 °C until use. All virus aliquots were used once and not subjected to repeated freeze–thaw cycles.
2.3. Induction of viral encephalitis
To induce SV encephalitis, mice were injected with 1000 PFU of SV suspended in 20 μl of phosphate-buffered saline (PBS) via a direct intracerebral route into the right cerebral hemisphere. PBS alone was used as a sham inoculation control. Neither the inoculation procedure nor the disease itself caused any overt neurological symptoms.
2.4. Induction of EAE via active immunization
Active EAE was induced in mice by subcutaneous immunization with 100 μg of MOG35–55 peptide emulsified in Complete Freund’s Adjuvant (CFA) containing 4 mg/ml heat-killed Mycobacterium tuberculosis H37Ra (Sigma–Aldrich) at four sites over the flanks. Sham immunized mice received CFA alone. Each mouse also received 300 ng of Bordetella pertussis toxin (List Biological Laboratories) injected intraperitoneally in 0.15 ml of PBS on day 0 and day 2 post-immunization. Mice were examined daily for signs of EAE and the severity of neurological impairment rated on a standard six-point scale (Bagaeva et al., 2006) as follows: 0, no discernible deficit; 1, limp tail; 2, impaired gait and/or ability to flip over from a supine position; 3, partial hind-limb paralysis; 4, total hind-limb paralysis; 5, moribund; 6, dead. Mice were euthanized once they reached a moribund state.
2.5. Preparation of primary microglial and astrocyte cultures
Primary microglia and astrocytes were isolated and cultured from the cortices of 2–3-day-old C57BL/6 mice as previously described (Esen and Kielian, 2005). In brief, when mixed glial cell cultures reached confluency after 7–10 days, flasks were shaken overnight at 200 rpm at 37 °C to detach microglia from the more firmly adherent astrocytes. Cells in suspension were collected and 2 × 105 cells plated into each well of 96-well plates. Cells retained in the culture flasks after three shakings were treated with 0.1 mM l-leucine methyl ester (l-LME) and passaged three times in the presence of l-LME to eliminate all remaining microglia. Cells (>95% astrocytes) were then trypsinized and 1 × 105 cells plated into each well of 96-well plates. The next day, microglia and astrocyte monolayers were washed and stimulated for 24 h either with SV or a known Toll-like receptor (TLR) ligand in a total volume of 200 μl as follows: 1 × 106 PFU SV (virus:cell ratio of 10:1), 100 ng/ml Escherichia coli lipopolysaccharide (LPS) (List Biological Laboratories) (a TLR4 stimulus), 25 mg/ml poly(I:C) (Invivogen) (a TLR3 stimulus), 1 mM loxoribine (Invivogen) (a TLR7 stimulus), or 3 mM unmethylated DNA oligonucleotides bearing CpG motifs (Invivogen) (a TLR9 stimulus). At the end of the 24-h treatment period, culture supernatants were collected and CXCL13 levels were measured by enzyme-linked immunosorbent assay (ELISA) as described below.
2.6. Measurement of chemokine concentrations in tissues and culture supernatants
Infected, immunized, or sham-inoculated mice were perfused extensively with PBS, and brains and spinal cords extracted, snap-frozen on dry ice, and stored at −80 °C until use. Thawed tissue was later minced and homogenized in 0.5 ml of PBS containing a protease inhibitor cocktail and an RNase inhibitor (Sigma–Aldrich). After homogenates were centrifuged to pellet remaining tissue debris, total protein content was measured in each extract and supernatants were diluted in PBS and assayed for CXCL9, CXCL10, CXCL12 and CXCL13 levels using commercially available ELISA kits (R&D Systems). Microglial and astrocyte culture supernatants were used undiluted in these assays without further manipulation. Results presented reflect the mean ± standard error of the mean (SEM) of pg chemokine/mg of protein extracted from tissue for a minimum of three animals at each time point for the tissue samples, or the mean ± SEM pg chemokine/ml culture supernatant from triplicate culture wells.
2.7. Isolation and flow cytometric analysis of CNS and other tissue mononuclear cells
Anesthetized mice underwent thorough intracardiac perfusion with PBS. Brains and/or spinal cords were collected, minced into small fragments, and pressed through a 70-μM mesh sieve into Hank’s balanced salt solution (HBSS) containing 10% fetal bovine serum (FBS) before digestion with collagenase (0.2 mg/ml; Worthington Biochemicals) and DNase (28 U/ml; Sigma–Aldrich) for 40 min at 37 °C. Mononuclear cells (MNC) were then isolated over a 30%/70% Percoll gradient (GE Healthcare Life Sciences) and washed with HBSS. Peripheral blood MNC were prepared from whole blood subjected to hypotonic lysis of red blood cells (RBC). Splenic MNC were collected by homogenizing whole spleen through a fine mesh screen and eliminating contaminating RBC by hypotonic lysis. All cells were resuspended in PBS containing 2% FBS and stained with fluorescently conjugated primary antibodies followed by flow cytometric analysis on a FACSCanto II flow cytometer (Becton–Dickinson). To detect B cells, cell suspensions were stained with antibodies against CD45 (eBiosciences) and CD19 (Becton–Dickinson). Peripheral B cell activation was quantified by enumerating CD19+ cells expressing CD86 (Becton–Dickinson) compared to staining with an isotype control (Becton–Dickinson). The total B cell content of each brain or spinal cord was calculated by multiplying the total number of gradient-isolated cells from each sample (counted on a hemacytometer) by the proportion of cells in the forward scatter/side scatter lymphocyte gate and then by the proportion of CD45+/CD19+ cells in each sample. A minimum of 10,000 gated events were analyzed for each experimental sample. Results presented reflect either the percentages or the absolute numbers of cells collected from a minimum of three animals at each experimental time point. For experiments involving cell sorting, CNS MNC were stained with a combination of CD45 and CD11b (eBiosciences), and separated into CD45dim/CD11b+ (microglial cells), CD45high/CD11b+ (infiltrating myeloid cells), and CD45high/CD11b− (infiltrating lymphoid cells) populations using a MoFlo XDP High-Speed Cell Sorter (Beckman-Coulter). Separated cells were stored in PrepProtect RNA stabilization solution (Miltenyi Biotec) at −20 °C until RNA isolation was performed.
2.8. Quantitative PCR analysis of chemokine expression by distinct CNS-derived mononuclear cell populations
Flow sorted cell populations were thawed, pelleted, and carefully removed from the PrepProtect solution. Total RNA was isolated from CD45dim/CD11b+ (microglial cells), CD45high/CD11b+ (infiltrating myeloid cells), and CD45high/CD11b− (infiltrating lymphoid cells) populations pooled from the brains of five SV-infected mice using QIAshredder Kit and the RNeasy Mini Kit according to the manufacturer’s instructions (QIAGEN). cDNA was then generated using the SuperScript® III First Strand Synthesis System for reverse transcriptase PCR (Invitrogen). Quantitative PCR (qPCR) was undertaken to measure CXCL13 mRNA transcripts using the MyiQ Single Color Real-Time PCR Detection System (Bio-Rad) and the CXCL13 primer/probe set in the TaqMan® Gene Expression Assay (Applied Biosystems). Standard curves for both CXCL13 and β-actin were prepared in order to quantify the amount of each transcript in each experimental sample. For comparison purposes, CXCL13 copy number was normalized to 106 copies of β-actin mRNA.
2.9. Immunostaining for B cells in CNS tissue sections
Anesthetized mice were sequentially perfused with PBS and then PBS containing 4% paraformaldehyde. Tissues were dissected, post-fixed overnight, cryopreserved in 30% sucrose, and flash frozen in optimal cutting temperature (OCT) compound for cryosectioning. Ten micrometers axial sections of spinal cord were first treated with 1% hydrogen peroxide in methanol to block all endogenous peroxidase then blocked in 2% normal goat serum (Vector Laboratories). Slides were washed, incubated with 1:100 dilutions of the B cell marker, CD19 (Becton–Dickinson), for 1 h at room temperature (RT), washed extensively again, and then treated with biotin-labeled goat anti-rat IgG (Vector Laboratories) at a 1:200 dilution for another hour at RT. This step was followed by sequential incubations with avidin-DH-biotin complex (Vector Laboratories) and then 0.5 mg/ml 3,3′-diaminobenzidine (Sigma–Aldrich) in PBS containing 0.01% hydrogen peroxide. All slides were counterstained with hematoxylin and mounted with coverslips for light microscopy. The number of immunoreactive B cells per cross section of spinal cord was counted in five sections from each animal, and the mean ± SEM of B cell counts from triplicate mice derived from each experimental group is shown. Selected slides were imaged using a Nikon Ti-U inverted microscope equipped with a DS-Fi-1 digital camera and supported by the NIS-Elements Basic Research acquisition and analysis software package (Nikon).
2.10. Tissue viral titrations and measurement of serum anti-viral neutralizing antibody titers
To measure the amount of infectious virus present in CNS tissues, animals were perfused extensively with PBS and brains were extracted, weighed, snap-frozen on dry ice, and stored at −80 °C until virus titrations assays were performed. At this time, 20% (w/v) homogenates of each sample were prepared in PBS, and serial 10-fold dilutions of each homogenate were assayed for plaque formation on monolayers of BHK-21 cells. Results presented are the mean ± SEM of the log10 of viral plaque-forming units per gram of tissue derived from a minimum of three animals at each time point. To calculate serum neutralizing antibody titer, blood was collected from infected mice by cardiac puncture. Serial 2-fold dilutions of serum were incubated with 50 PFU of SV. Serum-virus mixtures were then plated on BHK-21 cell monolayers as described above. Neutralization was defined as the highest dilution at which the serum sample reduced the number of plaques formed by a minimum of 50%. Each result reflects the mean ± SEM of the neutralization response from at least three animals at each time point.
2.11. Enumeration of MOG-specific T helper (Th)1 and Th17 cells
Splenic MNC were harvested from CXCL13−/− and CXCL13+/+ mice at peak disease and 2 weeks later. Cells from individual animals were cultured in replicate for 18 h in 96-well filtration plates (Millipore) at a densities ranging from 5 × 105 cells/well to 0.5 × 105 cells/well with or without the addition of 50 μg/ml MOG35–55 peptide. Cell densities of 2.5 × 105 cells/well and 1 × 105 cells/well provided optimal cytokine detection on day 13 and day 28, respectively. The following antibodies from eBiosciences were used to detect individual cell cytokine production by ELISpot assay: IL-17 (clone TC11-18H10), IL-17-biotin (clone TC11-8H4), IFN-γ (clone AN18), IFN-γ-biotin (clone R4-6A2), IL-2 (clone JES6-1A12), IL-2-biotin (clone JES6-5H4), GM-CSF (clone MP1-22E9), and GM-CSF-biotin (clone MP1-31G6). Streptavidin–alkaline phosphatase (Southern Biotech) and an alkaline phosphatase substrate kit (Vector Laboratories) were used to identify labeled cells. Spots were counted with a CTL ImmunoSpot Analyzer using ImmunoSpot software (Cellular Technology). Counts from cells cultured in media alone were subtracted from those stimulated with MOG35–55 to determine the proportion of antigen-specific T cells having either a Th1 (IFN-γ production) or a Th17 (IL-17 production) phenotype. Results presented reflect the numbers of cells collected from a minimum of three animals at each experimental time point.
2.12. Statistical comparisons
Student’s t-test was used when comparing a single group under two experimental conditions, while a two-way analysis of variance (ANOVA) test was applied when comparing the effects of different experimental findings between two groups over time. In both cases, differences at a p < 0.05 level were considered significant.
3. Results
3.1. Sustained induction of CXCL13 in the brains of mice with acute alphavirus encephalitis and in the spinal cords of mice with EAE
To understand the molecular signals that might serve to attract B cells into the brains of mice with SV encephalitis, ELISA assays were used to measure two candidate lymphoid chemokines, CXCL12 and CXCL13, as well as two non-lymphoid chemokines implicated in CNS B cell recruitment, CXCL9 and CXCL10 (Tschen et al., 2006), directly in tissue extracts. Although CXCL12 was constitutively found in naïve brain samples, it was not increased by infection (Fig. 1 A). In contrast, CXCL13 levels rose steadily and were sustained in the brains of SV-infected mice over time compared to sham inoculated controls (Fig. 1B). Interestingly, CXCL9 also did not increase with infection (Fig. 1C), while CXCL10 levels rose quickly and then declined as peak brain inflammation developed (Fig. 1D). Overall, these data suggest that CXCL13 could be important for B cell recruitment to the CNS of SV-infected animals based on its sustained expression over the course of acute infection.
Fig. 1.
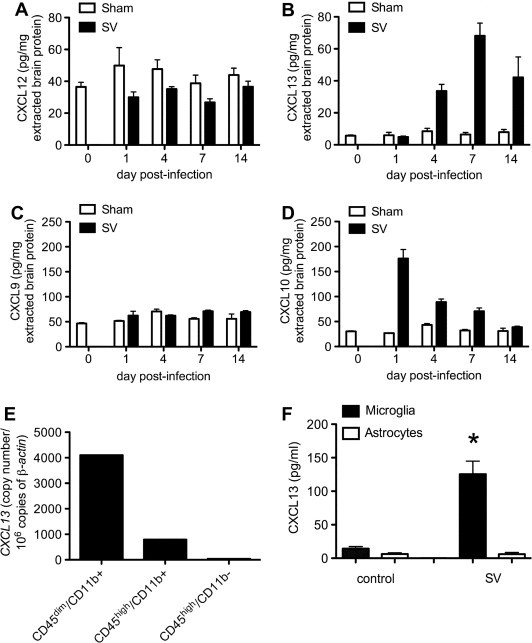
Induction of the lymphoid chemokine, CXCL13, is sustained in the brains of mice with SV encephalitis and is produced mainly by microglial cells in vivo. ELISA assays show that a related lymphoid chemokine, CXCL12, is not up-regulated in the brains of SV-infected compared to sham-inoculated mice (not significant (NS) by two-way ANOVA) (A). CXCL13 levels, however, increase and are sustained over the course of acute infection compared to controls (p < 0.0001 by two-way ANOVA) (B). A non-lymphoid chemokine previously implicated in CNS B cell recruitment, CXCL9, is not induced (NS by two-way ANOVA) (C), while another non-lymphoid chemokine previously implicated in this process, CXCL10, is quickly induced but not sustained in CNS tissue (p < 0.0001 by two-way ANOVA) (D). Quantitative PCR analysis of RNA extracted from FACS-sorted CNS cell populations shows that CXCL13 mRNA is found mostly in CD45dim/CD11b+ (microglial) cells, and to a lesser extent in CD45high/CD11b+ (infiltrating myeloid) cells following SV infection (E). Primary microglial cells produce CXCL13 following direct exposure to SV in vitro, while astrocytes do not respond in a similar manner (∗p < 0.05 by Student’s t-test) (F). All bars represent the mean ± SEM of concentrations measured from at least triplicate samples.
To determine if resident microglia and/or infiltrating mononuclear cell subsets were sources of CXCL13 during SV encephalitis, cells derived from the brains of infected animals were sorted into CD45dim/CD11b+ (microglia), CD45high/CD11b+ (infiltrating myeloid cells), and CD45high/CD11b− (infiltrating lymphoid cells) populations by flow cytometry. Quantitative PCR analysis of RNA isolated from each group of pooled cells revealed that CXCL13 mRNA was found primarily in microglia and to a lesser degree in infiltrating myeloid cells with limited expression in the lymphoid population (Fig. 1E). When primary cultures of microglia and astrocytes were directly exposed to SV in vitro, microglia readily produced CXCL13 under these conditions, while astrocytes did not (Fig. 1F). Conversely, synthetic TLR ligands provoked modest CXCL13 production by astrocytes and SV triggered the release of other inflammatory mediators by these cells (data not shown). Thus, microglia are the main source of CXCL13 in the brain during SV encephalitis, with infiltrating myeloid cells contributing modest amounts as well. Furthermore, this microglial production may be the result of direct exposure to the virus rather than being triggered by the actions of another mediator.
Spinal cord levels of CXCL12 did not change acutely following the induction of EAE (Fig. 2 A), even though baseline levels in naïve spinal cord were much higher than in brain (Fig. 1A). Concentrations actually fell below baseline during chronic disease, although CXCL12 levels also fell in samples derived from control animals at this time point for unclear reasons (Fig. 2A). In contrast, spinal cord levels of CXCL13 rose coincident with the onset of clinical signs and remained elevated over time (Fig. 2B). Prior studies demonstrated that CNS-infiltrating CD11c+ DCs are important CXCL13 producers during EAE, although microglia and other infiltrating myeloid cell populations likely contribute to local CXCL13 production as well (Bagaeva et al., 2006). Taken together, these data show that induction of the putative B cell chemoattractant, CXCL13, occurs in the CNS during two well-established models of neuroinflammation, and that both endogenous glial cells and infiltrating myeloid cells are sources of this mediator.
Fig. 2.

CXCL13 is induced in the spinal cords of mice with EAE. Levels of CXCL12 do not change over the acute stages of EAE compared to mice immunized with CFA alone (NS by two-way ANOVA) (A). Conversely, CXCL13 is induced in the spinal cord during acute EAE as paralysis develops and is sustained over time (p = 0.03 by two-way ANOVA) (B). Clinical course: d0 = naïve, d7 = pre-clinical, d13 = peak paralysis, d28 = chronic disease. All bars represent the mean ± SEM of concentrations measured from at least triplicate samples.
3.2. Peripheral B cells traffic to the brains of mice with acute alphavirus encephalitis and the spinal cords of mice with EAE
The intracerebral inoculation of SV causes acute encephalitis that results in MNC infiltration of the CSF, meninges, and brains of infected hosts (Moench and Griffin, 1984). B cells can be identified in this parenchymal infiltrate by immunostaining (Tyor et al., 1989), and anti-viral antibodies act within the CNS to clear virus from infected neurons in a non-cytolytic manner (Levine et al., 1991, Griffin et al., 1997). Virus-specific antibody-secreting cells arise in the spleens of SV-infected mice before appearing in the brain (Tyor and Griffin, 1993). To further characterize CNS B cell infiltration during SV encephalitis, flow cytometry was used to investigate the expression of activation markers on peripheral B cells and to enumerate B cells within CNS MNC isolates derived from infected animals. These assays showed that a subset of both splenic and peripheral blood B cells become activated in the first 7 days after SV infection as evidenced by their increased expression of the activation marker, CD86 (Fig. 3 A). This peripheral activation occurs coincident with escalating MNC infiltration of the brain (Fig. 3B). Phenotyping of the isolated brain MNC showed that B cells constitute a small but measurable fraction of the infiltrates (Fig. 3C and D). Prior studies of disease recovery have shown that while total B cell numbers decline in the CNS 3–4 weeks after SV infection (well beyond when infectious virus has been cleared from the brain), a small population is retained in the brain for many months thereafter and is highly enriched for those B cells producing anti-viral antibodies (Tyor et al., 1992, Griffin et al., 1997). The present data show that B cells traffic into the brain during acute SV encephalitis in parallel to rising tissue levels of CXCL13.
Fig. 3.
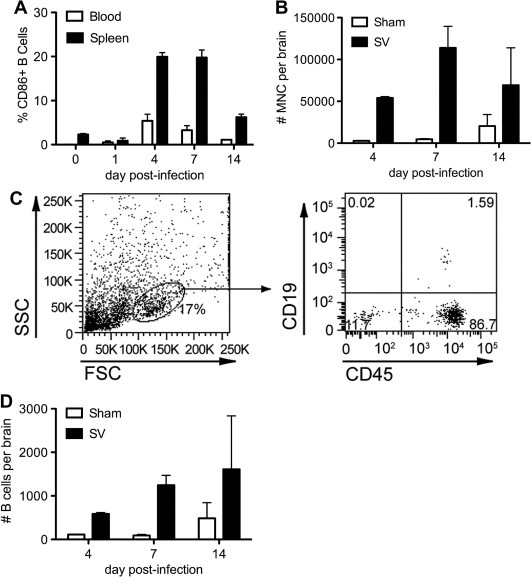
Peripheral B cells become activated and traffic to the CNS during acute SV encephalitis. Flow cytometry quantifies the proportion of CD19+ B cells in the spleen and peripheral blood expressing the known activation marker, CD86, over the course of acute infection (A). Total brain MNCs are simultaneously quantified from infected and sham-inoculated mice at three time points (B). Flow cytometry then determines the proportion of CD19+/CD45+ B cells (right panel) found among the cells present in the forward/side scatter lymphocyte gate (left panel) within the brains of SV-infected animals (C). The total number of B cells per brain is calculated as described in Section 2 (D). All bars represent the mean ± SEM of percentages or numbers measured from at least triplicate animals.
During the acute stages of MOG-induced EAE, B cells were difficult to detect among spinal cord infiltrates either before the onset of symptoms or just when animals were beginning to show the first evidence of abnormal neurological signs (data not shown). At peak paralysis, immunostaining procedures showed that a few B cells could be found infiltrating spinal white matter (Fig. 4 B), and flow cytometry demonstrated that B cells could be enumerated in spinal cord MNC samples pooled from multiple animals (Fig. 4D). Together, these data show that in a model where the local humoral immune response directly influences acute disease outcome (viral clearance), CNS-infiltrating B cells are found in moderate numbers. In a model where CD4+ T cells drive disease symptoms (paralysis), small numbers of B cells still traffic to the principal site of inflammation. In both cases, B cell influx into the CNS occurs in parallel to rising tissue levels of CXCL13.
Fig. 4.

CXCL13 deficiency has little effect on B cell infiltration into the brains of mice with SV encephalitis or the detection of B cells in the spinal cords of mice with acute EAE. Flow cytometry enumerates B cells in the brains of CXCL13+/+ and CXCL13−/− mice during SV encephalitis. Cell counts are normalized to a constant number of brain MNC cells to account for slight differences in the total amount of brain inflammation (A). Immunohistochemistry identifies occasional CD19+ B cells (arrow) in the spinal cords of CXCL13+/+ mice at the peak of EAE symptoms (B). Quantification of labeled cells in cross sections of lumbar spinal cord shows no difference in the numbers of B cells detected between CXCL13+/+ and CXCL13−/− mice (C). Flow cytometry conducted on pooled spinal cord samples demonstrates equivalent numbers of B cells among spinal cord MNC from these two hosts (D). All bars represent the mean ± SEM of numbers measured from at least triplicate animals.
3.3. Consequences of CXCL13 deficiency on CNS B cell infiltration and disease outcome in mice with acute alphavirus encephalitis and during EAE
Because CXCL13 levels increased in the CNS during the onset of inflammation in these two models, the effects of CXCL13 deficiency on B cell infiltration and disease outcome were investigated. During acute SV encephalitis, B cells proved to be at least as common in the brains of infected CXCL13−/− mice as they were in CXCL13+/+ control animals (Fig. 4A). Likewise, equivalent numbers of B cells were found in the spinal cords of both hosts at peak EAE when quantified in tissue sections and enumerated in pooled tissue MNC isolates by flow cytometry (Fig. 4C and D). Thus, the absence of CXCL13 appears to have no effect on the migration of B cells to the CNS in both models of acute neuroinflammation.
When the virological consequences of CXCL13 deficiency were examined in the SV encephalitis model, no differences in the kinetics of CNS virus replication or clearance were observed between CXCL13−/− and CXCL13+/+ mice (Fig. 5 ). During active immunization EAE, the effects of CXCL13 deficiency were more apparent. Prior studies showed that the timing of both disease onset and peak disease severity was similar between wild type and knockout hosts, although peak severity itself was slightly lower in CXCL13−/− animals (Bagaeva et al., 2006). More notably, however, CXCL13−/− mice demonstrated an accelerated and more complete clinical recovery following MOG immunization compared to control mice where neurological deficits often persisted for more than 30 days (Bagaeva et al., 2006). Thus, disease is unaffected by CXCL13 in a CNS infection paradigm that depends on a pathogen-specific B cell response, while its course is heightened and prolonged by CXCL13 when myelin-specific CD4+ T cells drive CNS pathology.
Fig. 5.
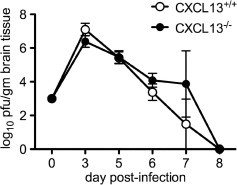
Replication and clearance of SV from the brains of CXCL13−/− mice is similar when compared to CXCL13+/+ controls. Brain tissue was collected from SV-infected animals and tissue viral titers were measured by plaque titration assays as described in Section 2. Both peak titer and the rate of viral clearance are unaltered by the loss of CXCL13 (curves are not significantly different from each other as measured by two-way ANOVA, although both groups changed significantly over time). All values represent the mean ± SEM of titers measured from at least triplicate samples.
3.4. The role of CXCL13 in the immunological response to the inciting antigen during acute alphavirus encephalitis and EAE
CXCL13−/− mice have disorganized B cell follicles in secondary lymphoid tissue and may be impaired in their capacity to mount certain natural antibody responses (Ansel et al., 2000, Ansel et al., 2002). Because anti-viral antibodies are crucial to non-cytolytic clearance of SV from infected neurons (Levine et al., 1991, Griffin et al., 1997), the serum neutralizing anti-viral antibody response was measured in both CXCL13+/+ and CXCL13−/− mice at various time points following infection. Consistent with their capacity to clear virus normally from the CNS (Fig. 5), CXCL13−/− mice mounted an early serum neutralizing antibody response equivalent to that measured in CXCL13+/+ hosts (Fig. 6 ). Only at a late disease stage, well after infectious virus had been fully cleared from the CNS (Fig. 5), did the systemic anti-viral antibody response wane slightly, but not significantly, in CXCL13−/− mice compared to CXCL13+/+ controls (Fig. 6). This slight drop was not sufficient to allow any recrudescence of infection to occur (data not shown). Thus, CXCL13 does not meaningfully influence the host’s ability to mount an effective anti-viral antibody response during SV encephalitis.
Fig. 6.
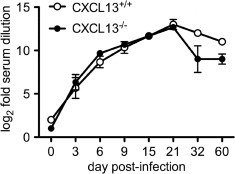
Generation of the serum neutralizing anti-viral antibody response in CXCL13−/− mice mirrors that of CXCL13+/+ controls until late in disease. Blood was collected from SV-infected animals and serum neutralization assays were performed as described in Section 2. While the response in both hosts was virtually identical over the first 21 days of infection, antibody titers dropped slightly in the CXCL13−/− mice thereafter (although curves were not significantly different from each other as measured by two-way ANOVA, even though both changed significantly over time). All values represent the mean ± SEM of titers measured from at least triplicate samples.
In EAE, myelin antigen-specific CD4+ T cells of a Th1 (IFN-γ producing) or a Th17 (IL-17 producing) phenotype are required to initiate disease. When MOG-specific Th1 and Th17 responses were quantified among peripheral lymphocytes from CXCL13+/+ and CXCL13−/− mice following the induction of EAE, both subsets of encephalitogenic CD4+ T cells were found in equivalent numbers at peak disease (Fig. 7 A). Cells making two other T cell-derived cytokines, IL-2 and GM-CSF, in response to MOG peptide were likewise found at equivalent rates in these two hosts (Fig. 7A). This indicates that CXCL13 does not influence T cell priming or differentiation in response to a myelin autoantigen. However, the frequencies of T cells making each of these four cytokines were significantly lower in the CXCL13−/− mice 2 weeks after peak disease was reached (Fig. 7B). These waning pathogenic Th1 and Th17 responses in CXCL13−/− mice correspond with the known propensity of these animals to undergo accelerated and more complete EAE remission compared to control mice (Bagaeva et al., 2006). Further evidence supporting a primary role for CXCL13 in maintaining rather than generating pathogenic T cell responses comes from adoptive transfer studies where wild type, Th1-polarized, MOG-primed T cells caused a milder and more self-limited disease when introduced into CXCL13−/− compared to CXCL13+/+ mice (Bagaeva et al., 2006). Thus, in contrast to the anti-SV antibody response that develops and is sufficiently maintained in the absence of CXCL13, the pathogenic CD4+ T cell response elicited during EAE requires CXCL13 to be sustained long term after its initial priming.
Fig. 7.

MOG-specific T cell cytokine production in CXCL13+/+ and CXCL13−/− mice during active immunization EAE. ELISpot assays were used to enumerate the number of splenic T cells producing each cytokine ex vivo following stimulation with MOG35–55. At peak disease, cells producing both IFN-γ (Th1) and IL-17 (Th17) were present in equivalent numbers in both hosts (A). Two weeks later, both Th1 and Th17 responses had waned in the CXCL13−/− mice compared to the CXCL13+/+ controls. ∗p < 0.05 by Student’s t-test for the indicated cytokines compared to CXCL13+/+ controls. All bars represent the mean ± SEM of numbers measured from at least triplicate samples.
4. Discussion
The findings in our study demonstrate that CXCL13, a lymphoid chemokine important for B cell recruitment into lymphoid follicles and commonly induced in the inflamed CNS, is surprisingly dispensable for the initial recruitment of B cells into the brains of mice with acute viral encephalitis and the spinal cords of mice with EAE (Fig. 4). Thus, while CXCL13 levels increase at the main sites of inflammation during both disorders (Fig. 1, Fig. 2B), its absence has no impact on either peripheral or CNS humoral responses during an infection model whose recovery depends primarily on this arm of the immune system (Fig. 4, Fig. 6). Conversely, CXCL13 acts to sustain pathogenic immunity (Fig. 7), as well as tissue inflammation and disease symptoms in a model where CD4+ T cells drive CNS pathology (Bagaeva et al., 2006). Taken together, these data suggest that CXCL13 plays a rather limited role in the development of neuroinflammation, but may instead be relatively more important in the maintenance of immune responses within the CNS.
Mechanisms of B cell trafficking through the normal and inflamed CNS are incompletely understood. While CD4+ and CD8+ T cells may be the main effectors of immune surveillance in the normal CNS (Ransohoff et al., 2003), B cells also contribute to this process. Studies in patients receiving B cell-depleting immunotherapies for the treatment of systemic autoimmune disorders or hematopoietic malignancies have shown them to be at increased risk of developing progressive multifocal leukoencephalopathy (PML), a lethal encephalitis caused by the polyomavirus, JC virus (Carson et al., 2009, Gea-Banacloche, 2010). In this setting, the systemic depletion of virus-specific B cells likely disrupts their ongoing recirculation through the brain where latent JC virus resides, thereby reducing the host’s capacity to keep the pathogen in check and augmenting the risk of viral recrudescence. The molecular determinants through which B cells provide normal immune surveillance of the CNS remain unknown, and may or may not involve a chemokine-directed recruitment process.
Beyond immune surveillance, B cells infiltrate various CNS compartments (meninges, subarachnoid space, brain and spinal cord parenchyma) during a wide variety of neuroinfectious and neuroinflammatory disorders. In human disease settings, convincing evidence of early B cell recruitment and clonal expansion within the intrathecal space is provided by the detection of oligoclonal populations of IgG molecules (oligoclonal bands) and high IgG:albumin ratios (IgG index) in CSF samples obtained via lumbar puncture. During infection, these humoral responses presumably target the invading pathogen. In disorders such as MS, their frequent identification has long been interpreted as evidence supporting an immune-mediated basis of the disease. A role for B cells in the pathogenesis of MS is highlighted by the clinical benefit of therapies that block adhesion receptors mediating lymphocyte passage across the blood–brain barrier (BBB) or that deplete systemic B cells (Polman et al., 2006, Hauser et al., 2008). These findings naturally raise questions as to the molecular signals that recruit B cells into the inflamed CNS, and our data would suggest that recruitment molecules other than CXCL13 are involved.
During acute alphavirus encephalitis, virus-specific antibodies act locally within the CNS to clear virus from infected neurons in a non-cytolytic manner (Levine et al., 1991, Griffin et al., 1997). The B cells producing these antibodies are first detected in the spleens of infected animals before appearing in the brain (Tyor and Griffin, 1993), although their need to physically traverse the BBB in order for antibodies to reach foci of infection is debatable. Indeed, prior studies have shown that the intravenous transfer of immune serum or monoclonal anti-viral antibodies is sufficient to promote viral clearance and recovery in SV-infected mice, provided that enough material is given (Stanley et al., 1986, Levine et al., 1991). In this disease setting, the influx of B cells into the CNS may be more important to establish a local population of anti-viral effectors that are retained at that site well after viral clearance has been achieved in order to prevent late viral recrudescence (Tyor et al., 1992, Griffin et al., 1997). By comparison to other viral encephalitis models, CXCL13 was not induced in the CNS during experimental coronavirus encephalomyelitis despite prominent accumulation of virus-specific antibody-secreting cells in the target tissue (Tschen et al., 2006). Here, CXCL9 and CXCL10, both acting on plasmablasts through CXCR3, were implicated in CNS B cell recruitment (Tschen et al., 2006). Our data would suggest that neither CXCL9 nor CXCL10 are likely responsible for B cell recruitment to the CNS of SV-infected animals, since one mediator (CXCL9) is simply not induced by infection and the other (CXCL10) only goes up transiently and then declines as B cell influx begins in earnest (Fig. 1C and D). Ongoing studies aim to investigate the role of other chemokines in CNS B cell recruitment in the SV encephalitis model.
The precise role of B cells in EAE pathogenesis depends on the model under investigation. Early studies in C57BL/6 mice showed that genetic deficiency of B cells had no effect on chronic disease triggered by the MOG35–55 peptide, although B cell-deficient animals were resistant to EAE induced by the whole MOG protein (Hjelmstrom et al., 1998, Lyons et al., 1999). Germline deletion of B cells also had little effect on a relapsing EAE model (Dittel et al., 2000). More recently, however, very different effects on the clinical course of MOG-induced disease in C57BL/6 mice were observed when acute B cell depletion was undertaken at different intervals relative to disease initiation (Matsushita et al., 2008). These findings led to the conclusion that B cells exert reciprocal roles in EAE pathogenesis; sometimes serving an immunoregulatory function while at other times actually helping to prime MOG-specific CD4+ T cells (Matsushita et al., 2008). In both circumstances, however, these actions of B cells could readily occur outside the CNS and not require their passage across the BBB. Indeed, only in the more chronic phases of both EAE and MS are actual B cell aggregates found in the CNS, and then only in the meninges surrounding the parenchyma (Magliozzi et al., 2004, Magliozzi et al., 2007, Serafini et al., 2004, Aloisi et al., 2008). CXCL13 is present in these B cell aggregates (Magliozzi et al., 2004, Magliozzi et al., 2007, Serafini et al., 2004, Aloisi et al., 2008), so its primary role may be to help form ectopic lymphoid follicles rather than to actually recruit B cells into the CNS. The therapeutic benefit of disrupting these follicles via CXCL13 neutralization is unclear; in the non-obese diabetic (NOD) mouse model of type-1 diabetes, such an action prevented ectopic follicle formation in the pancreas but had no effect on diabetes progression (Henry and Kendall, 2010). The pathogenic significance of these lymphoid structures in experimental and human demyelinating disease awaits further study.
An important finding described here (Fig. 7) and elsewhere (Bagaeva et al., 2006) is that CXCL13 acts to sustain the pathogenic CD4+ T cell responses that drive EAE. How this happens remains poorly understood, although CXCL13 acts to position lymphocytes and DCs within T and B cell zones in lymphoid organs (Forster et al., 1996, Legler et al., 1998, Ansel et al., 2000), so it may serve a similar function within the CNS. One study suggested that CXCL13 could be an important effector that brings CD11c+ DCs and autoreactive CXCR5+ T cells together, increasing the likelihood of the cognate interactions that drive EAE (Bagaeva et al., 2006). Since CD11c+ DCs are an important source of CXCL13 in this disorder, and since most of the CXCR5+ cells found in CNS infiltrates are CD3+ T cells, the absence of CXCL13 may prevent sufficient numbers of myelin-specific T cells from reactivating to sustain disease beyond its initial stages (Bagaeva et al., 2006). This makes CXCL13 a potential therapeutic target in autoimmune demyelination of the CNS, but for reasons other than altered B cell recruitment into the target tissue. Further studies are required to clarify its role in EAE and related disorders.
In summary, our study shows that CXCL13 does not influence B cell recruitment into the inflamed CNS in common animal models of infectious and inflammatory demyelinating disease. The disease course is unaffected by CXCL13 in a CNS infection paradigm that depends on a pathogen-specific B cell response for recovery, while it is heightened and prolonged by CXCL13 when myelin-specific CD4+ T cells drive CNS pathology. The data suggest that alternative molecular mechanisms driving CNS B cell recruitment need to be pursued if this process is to be better understood.
Conflict of interest statement
All authors declare that there are no conflicts of interest.
Acknowledgments
This work was supported by Grants from the National Institutes of Health (AI057505 to D.I. and NS047687 to B.S.) and the National Multiple Sclerosis Society (RG3866-A-3 to B.S.).
References
- Allen C.D., Ansel K.M., Low C., Lesley R., Tamamura H., Fujii N., Cyster J.G. Germinal center dark and light zone organization is mediated by CXCR4 and CXCR5. Nat. Immunol. 2004;5:943–952. doi: 10.1038/ni1100. [DOI] [PubMed] [Google Scholar]
- Aloisi F., Columba-Cabezas S., Franciotta D., Rosicarelli B., Magliozzi R., Reynolds R., Ambrosini E., Coccia E., Salvetti M., Serafini B. Lymphoid chemokines in chronic neuroinflammation. J. Neuroimmunol. 2008;198:106–112. doi: 10.1016/j.jneuroim.2008.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansel K.M., Ngo V.N., Hyman P.L., Luther S.A., Forster J.D., Sedgwick J.D., Browning J.L., Lipp M., Cyster J.G. A chemokine-driven positive feedback loop organizes lymphoid follicles. Nature. 2000;406:309–314. doi: 10.1038/35018581. [DOI] [PubMed] [Google Scholar]
- Ansel K.M., Harris R.B., Cyster J.G. CXCL13 is required for B1 cell homing, natural antibody production, and body cavity immunity. Immunity. 2002;16:67–76. doi: 10.1016/s1074-7613(01)00257-6. [DOI] [PubMed] [Google Scholar]
- Bagaeva L.V., Rao P., Powers J.M., Segal B.M. CXC chemokine ligand 13 plays a role in experimental autoimmune encephalomyelitis. J. Immunol. 2006;176:7676–7685. doi: 10.4049/jimmunol.176.12.7676. [DOI] [PubMed] [Google Scholar]
- Campbell D.J., Kim C.H., Butcher E.C. Chemokines in the systemic organization of immunity. Immunol. Rev. 2003;195:58–71. doi: 10.1034/j.1600-065x.2003.00067.x. [DOI] [PubMed] [Google Scholar]
- Carson K.R., Evens A.M., Richey E.A., Haberman T.M., Focosi D., Seymour J.F., Laubach J., Bawn S.D., Gordon L.I., Winter J.N., Furman R.R., Vose J.M., Zelenetz A.D., Mamtani R., Raisch D.W., Dorshimer G.W., Rosen S.T., Muro K., Gottardi-Littell N.R., Talley R.L., Sartor O., Green D., Major E.O., Bennett C.L. Progressive multifocal leukoencephalopathy after rituximab in HIV-negative patients: a report of 57 cases from the Research on Adverse Drug Events and Reports project. Blood. 2009;113:4834–4840. doi: 10.1182/blood-2008-10-186999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.C., Vassileva G., Kinsley D., Holzmann S., Manfra D., Wiekowski M.T., Romani N., Lira S.A. Ectopic expression of the murine chemokines CCL21a and CCL21b induces the formation of lymph node-like structures in pancreas, but not skin, of transgenic mice. J. Immunol. 2002;168:1001–1008. doi: 10.4049/jimmunol.168.3.1001. [DOI] [PubMed] [Google Scholar]
- Columba-Cabezas S., Griguoli M., Rosicarelli B., Magliozzi R., Ria F., Serafini B., Aloisi F. Suppression of established experimental autoimmune encephalomyelitis and formation of meningeal lymphoid follicles by lymphotoxin β receptor-Ig fusion protein. J. Neuroimmunol. 2006;179:76–86. doi: 10.1016/j.jneuroim.2006.06.015. [DOI] [PubMed] [Google Scholar]
- Dittel B.N., Urbania T.H., Janeway C.A., Jr. Relapsing and remitting experimental autoimmune encephalomyelitis in B cell deficient mice. J. Autoimmun. 2000;14:311–318. doi: 10.1006/jaut.2000.0371. [DOI] [PubMed] [Google Scholar]
- Esen N., Kielian T. Recognition of Staphylococcus aureus-derived peptidoglycan (PGN) but not intact bacteria is mediated by CD14 in microglia. J. Neuroimmunol. 2005;170:93–104. doi: 10.1016/j.jneuroim.2005.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan L., Reilly C.R., Luo Y., Dorf M.E., Lo D. Cutting edge: ectopic expression of the chemokine TCA4/SLC is sufficient to trigger lymphoid neogenesis. J. Immunol. 2000;164:3955–3959. doi: 10.4049/jimmunol.164.8.3955. [DOI] [PubMed] [Google Scholar]
- Fischer L., Korfel A., Pfeiffer S., Kiewe P., Volk H.D., Cakiroglu H., Widmann T., Thiel E. CXCL13 and CXCL12 in central nervous system lymphoma patients. Clin. Cancer Res. 2009;15:5968–5973. doi: 10.1158/1078-0432.CCR-09-0108. [DOI] [PubMed] [Google Scholar]
- Forster R., Matis A.E., Kremmer E., Wolf E., Brem G., Lipp M. A putative chemokine receptor, BLR1, directs B cell migration to defined lymphoid organs and specific anatomic compartments of the spleen. Cell. 1996;87:1037–1047. doi: 10.1016/s0092-8674(00)81798-5. [DOI] [PubMed] [Google Scholar]
- Fu Y.-X., Chaplin D.D. Development and maturation of secondary lymphoid tissues. Annu. Rev. Immunol. 1999;17:399–433. doi: 10.1146/annurev.immunol.17.1.399. [DOI] [PubMed] [Google Scholar]
- Gea-Banacloche J.C. Rituximab-associated infections. Semin. Hematol. 2010;47:187–198. doi: 10.1053/j.seminhematol.2010.01.002. [DOI] [PubMed] [Google Scholar]
- Griffin D., Levine B., Tyor W., Ubol S., Despres P. The role of antibody in recovery from alphavirus encephalitis. Immunol. Rev. 1997;159:155–161. doi: 10.1111/j.1600-065x.1997.tb01013.x. [DOI] [PubMed] [Google Scholar]
- Hauser S.L., Waubant E., Arnold D.L., Vollmer T., Fox R.J., Bar-Or A., Panzara M., Sarkar N., Agarwal S., Langer-Gould A., Smith C.H., HERMES Trial Group B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N. Engl. J. Med. 2008;358:676–688. doi: 10.1056/NEJMoa0706383. [DOI] [PubMed] [Google Scholar]
- Henry R.A., Kendall P.L. CXCL13 blockade disrupts B lymphocyte organization in tertiary lymphoid structures without altering B cell receptor bias or preventing diabetes in nonobese diabetic mice. J. Immunol. 2010;185:1460–1465. doi: 10.4049/jimmunol.0903710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjelmstrom P. Lymphoid neogenesis: de novo formation of lymphoid tissue in chronic inflammation through expression of homing chemokines. J. Leukoc. Biol. 2001;69:331–339. [PubMed] [Google Scholar]
- Hjelmstrom P., Juedes A.E., Fjell J., Ruddle N.H. B cell-deficient mice develop experimental allergic encephalomyelitis with demyelination after myelin oligodendrocyte glycoprotein sensitization. J. Immunol. 1998;161:4480–4483. [PubMed] [Google Scholar]
- Krumbholz M., Theil D., Cepok S., Hemmer B., Kivisakk P., Ransohoff R.M., Hofbauer M., Farina C., Derfuss T., Hartle C., Newcombe J., Hohlfeld R., Meinl E. Chemokines in multiple sclerosis: CXCL12 and CXCL13 up-regulation is differentially linked to CNS immune cell recruitment. Brain. 2006;129:200–211. doi: 10.1093/brain/awh680. [DOI] [PubMed] [Google Scholar]
- Lalor S.J., Segal B.M. Lymphoid chemokines in the CNS. J. Neuroimmunol. 2010;224:56–61. doi: 10.1016/j.jneuroim.2010.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legler D.F., Loetscher M., Roos R.S., Clark-Lewis I., Baggiolini M., Moser B. B-cell attracting chemokine 1, a human CXC chemokine expressed in lymphoid tissues, selectively attracts B lymphocytes via BLR1/CXCR5. J. Exp. Med. 1998;187:655–660. doi: 10.1084/jem.187.4.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B., Hardwick J.M., Trapp B.D., Crawford T.O., Bollinger R.C., Griffin D.E. Antibody-mediated clearance of alphavirus infection from neurons. Science. 1991;254:856–860. doi: 10.1126/science.1658936. [DOI] [PubMed] [Google Scholar]
- Ljostad U., Mygland A. CSF B-lymphocyte chemoattractant (CXCL13) in the early diagnosis of acute Lyme neuroborreliosis. J. Neurol. 2008;255:732–737. doi: 10.1007/s00415-008-0785-y. [DOI] [PubMed] [Google Scholar]
- Luther S.A., Lopez T., Bai W., Hanahan D., Cyster J.G. BLC expression in pancreatic islets causes B cell recruitment and lymphotoxin-dependent lymphoid neogenesis. Immunity. 2000;12:471–481. doi: 10.1016/s1074-7613(00)80199-5. [DOI] [PubMed] [Google Scholar]
- Luther S.A., Bidgol A., Hargreaves D.C., Schmidt A., Xu Y., Paniyadi J., Maloubian M., Cyster J.G. Differing activities of homeostatic chemokines CCL19, CCL21, and CXCL12 in lymphocyte and dendritic cell recruitment and lymphoid neogenesis. J. Immunol. 2002;169:424–433. doi: 10.4049/jimmunol.169.1.424. [DOI] [PubMed] [Google Scholar]
- Lyons J.A., San M., Happ M.P., Cross A.H. B cells are critical to induction of experimental allergic encephalomyelitis by protein but not by a short encephalitogenic peptide. Eur. J. Immunol. 1999;29:3432–3439. doi: 10.1002/(SICI)1521-4141(199911)29:11<3432::AID-IMMU3432>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Mackay F., Browning J.L. Turning off follicular dendritic cells. Nature. 1998;395:26–27. doi: 10.1038/25630. [DOI] [PubMed] [Google Scholar]
- Magliozzi R., Columba-Cabezas S., Serafini B., Aloisi F. Intracerebral expression of CXCL13 and BAFF is accompanied by formation of lymphoid follicle-like structures in the meninges of mice with relapsing experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2004;148:11–23. doi: 10.1016/j.jneuroim.2003.10.056. [DOI] [PubMed] [Google Scholar]
- Magliozzi R., Howell O., Vora A., Serafini B., Nicholas R., Popolo M., Reynolds R., Aloisi F. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain. 2007;130:1089–1104. doi: 10.1093/brain/awm038. [DOI] [PubMed] [Google Scholar]
- Martin A.P., Coronel E.C., Sano G., Chen S.C., Vassileva G., Canastro-Chibuque C., Sedgwick J.D., Frenette P.S., Lipp M., Furtado G.C., Lira S.A. A novel model for lymphocytic infiltration of the thyroid gland generated by transgenic expression of the CC chemokine CCL21. J. Immunol. 2004;173:4791–4798. doi: 10.4049/jimmunol.173.8.4791. [DOI] [PubMed] [Google Scholar]
- Matsushita T., Yanaba K., Bouaziz J.-D., Fujimoto M., Tedder T.F. Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J. Clin. Invest. 2008;118:3420–3430. doi: 10.1172/JCI36030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzucchelli L., Blaser A., Kappeler A., Scharli P., Laissue J.A., Baggiolini M., Uguccioni M. BCA-1 is highly expressed in Helicobacter pylori-induced mucosa-associated lymphoid tissue and gastric lymphoma. J. Clin. Invest. 1999;104:49–54. doi: 10.1172/JCI7830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moench T.R., Griffin D.E. Immunocytochemical identification and quantification of the mononuclear cells in the cerebrospinal fluid, meninges, and brain during acute viral meningoencephalitis. J. Exp. Med. 1984;159:77–88. doi: 10.1084/jem.159.1.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser B., Schaerli P., Laetscher P. CXCR5+ T cells: follicular homing takes center stage in T-helper-cell responses. Trends Immunol. 2002;23:250–254. doi: 10.1016/s1471-4906(02)02218-4. [DOI] [PubMed] [Google Scholar]
- Muller G., Hopken U.E., Lipp M. The impact of CCR7 and CXCR5 on lymphoid organ development and systemic immunity. Immunol. Rev. 2003;195:117–135. doi: 10.1034/j.1600-065x.2003.00073.x. [DOI] [PubMed] [Google Scholar]
- Ngo V.N., Korner H., Gunn M.D., Schmidt K.N., Riminton D.S., Cooper M.D., Browning J.L., Sedgwick J.D., Cyster J.G. Lymphotoxin-α/β and tumor necrosis factor are required for stromal cell expression of homing chemokines in B and T cell areas of the spleen. J. Exp. Med. 1999;189:403–412. doi: 10.1084/jem.189.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polman C.H., O’Connor P.W., Havrdova E., Hutchinson M., Kappos L., Miller D.H., Phillips J.T., Lublin F.D., Giovannoni G., Wajgt A., Toal M., Lynn F., Panzara M.A., Sandrock A.W., Investigators A.F.F.I.R.M. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N. Engl. J. Med. 2006;354:899–910. doi: 10.1056/NEJMoa044397. [DOI] [PubMed] [Google Scholar]
- Ransohoff R.M., Kivisakk P., Kidd G. Three or more routes for leukocyte migration into the central nervous system. Nat. Rev. Immunol. 2003;3:569–581. doi: 10.1038/nri1130. [DOI] [PubMed] [Google Scholar]
- Rupprecht T.A., Plate A., Adam M., Wick M., Kastenbauer S., Schmidt C., Klein M., Pfister H.W., Koedel U. The chemokine CXCL13 is a key regulator of B cell recruitment to the cerebrospinal fluid in acute Lyme neuroborreliosis. J. Neuroinflamm. 2009;6:42–51. doi: 10.1186/1742-2094-6-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellebjerg F., Bornsen L., Khademi M., Krakauer M., Olsson T., Frederiksen J.L., Sorensen P.S. Increased cerebrospinal fluid concentrations of the chemokine CXCL13 in active MS. Neurology. 2009;73:2003–2010. doi: 10.1212/WNL.0b013e3181c5b457. [DOI] [PubMed] [Google Scholar]
- Serafini B., Rosicarelli B., Magiozzi R., Stigliano E., Aloisi F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. 2004;14:164–174. doi: 10.1111/j.1750-3639.2004.tb00049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley J., Cooper S.J., Griffin D.E. Monoclonal antibody cure and prophylaxis of lethal Sindbis virus encephalitis in mice. J. Virol. 1986;58:107–115. doi: 10.1128/jvi.58.1.107-115.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschen S.-I., Stohlman S.A., Ramakrishna C., Hinton D.R., Atkinson R.D., Bergmann C.C. CNS viral infection diverts homing of antibody-secreting cells from lymphoid organs to the CNS. Eur. J. Immunol. 2006;36:603–612. doi: 10.1002/eji.200535123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyor W.R., Griffin D.E. Virus specificity and isotype expression of intraparenchymal antibody-secreting cells during Sindbis virus encephalitis in mice. J. Neuroimmunol. 1993;48:37–44. doi: 10.1016/0165-5728(93)90056-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyor W.R., Moench T.R., Griffin D.E. Characterization of the local and systemic B cell response of normal and athymic nude mice with Sindbis virus encephalitis. J. Neuroimmunol. 1989;24:207–215. doi: 10.1016/0165-5728(89)90118-5. [DOI] [PubMed] [Google Scholar]
- Tyor W.R., Wesselingh S., Levine B., Griffin D.E. Long term intraparenchymal Ig secretion after acute viral encephalitis in mice. J. Immunol. 1992;149:4016–4020. [PubMed] [Google Scholar]
- Yoshie O., Imai T., Nomiyama H. Novel lymphocyte-specific CC chemokines and their receptors. J. Leukoc. Biol. 1997;62:634–644. doi: 10.1002/jlb.62.5.634. [DOI] [PubMed] [Google Scholar]
- Zlotnik A., Morales J., Hedrick J.A. Recent advances in chemokines and chemokine receptors. Crit. Rev. Immunol. 1999;19:1–47. [PubMed] [Google Scholar]