

Abstract
The linear rheological properties of networks formed by adding bis-Pd(II) cross-linkers to poly(4-vinylpyridine) (PVP) solution are examined, and the scaling law relationships between the zero shear viscosity (η0) of the networks versus the concentration of PVP solution (CPVP), the concentration of cross-linkers (CX), and the number density of elastically active chains (vphantom) are experimentally determined. The scaling law relationships are compared to the theoretical expectations of the Sticky Rouse and Sticky Reptation models (Macromolecules 2001, 34, 1058-1068), and both qualitative and quantitative differences are observed.
Introduction
Solutions of associating polymers link two major classes of polymeric systems—solutions and networks—through reversible cross-linkers.1 The presence of reversible cross-linkers endows solutions of associative polymers with rich rheological properties and fuels their use in a wide range of applications, including rheology modifiers for paintings, cosmetics, pharmaceutics, and coatings.2,3 The general characteristics of such reversible networks, compared with that of polymers that do not have associating groups, include their longer relaxation time and higher plateau moduli.4 A fundamental goal, therefore, is to identify the relationship between the microscopic kinetics of the cross-linkers and the macroscopic rheological properties of the network.4–11
Relative to theoretical work,1, 4–8 experimental studies regarding the dynamics in associating polymers have been largely qualitative, because the lifetime (τ0,τ0=1/kd) of the associative groups was often not clearly characterized.12–15 Our group has previously demonstrated a useful method for probing the contributions of molecular reversibility to the rheological properties of polymer networks. 16–20 This method takes advantage of steric effects at the N-alkyl positions of N,C,N-pincer Pd(II) or Pt(II) complexes (Figure 1), through which the dissociation rate of the cross-linkers can be changed by orders of magnitude independently of the association constant.16,21 As the equilibrium association constant (Keq, Keq=ka/kd) and dissociation rate constant (kd) of these cross-linkers can be quantitatively characterized and the associative rate constant ka can be calculated by the above relationship,16, 21 this system provides a useful model to explore the relaxation dynamics of associative polymer networks.
Figure 1.
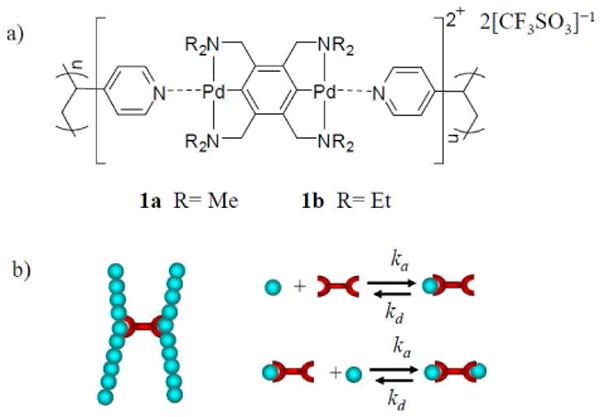
(a) Schematic picture of networks formed from poly(4-vinylpyridine) (PVP) chains and bis-Pd(II) cross-linkers (1a or 1b); (b) Schematic picture of the kinetic parameters of the supramolecular interaction underlying network formation. Each pyridine unit in the PVP chain is represented by a bead.
In this work, we consider the behavior of networks formed from both unentangled and entangled semidilute polymer solutions. We also consider networks with concentrations of cross-linkers that range from just above the gel point to several active cross-linkers per polymer chain. Much of this regime falls on the border of or just outside of the territory that has been explicitly examined theoretically in the context of the Sticky Rouse and/or Sticky Reptation models,1 but it comprises regions of structure space that are common in experimental systems and that have potential technological utility. It is therefore of interest to first establish empirical scaling law behaviors and, second, to compare them to the existing theoretical framework in order to determine its applicability to these and similar systems.
The characteristics of the supramolecular polymer network are shown in Scheme 1a.16–19 The bis-Pd cross-linkers are small molecules, and they bind through each of the two Pd atoms to any two of the pyridine units of the poly(4-vinylpyridine) (PVP) chain, forming a cross-link. There are four states of cross-linkers: free, dangling, intrachain bound, and interchain bound cross-linkers. Of these, only interchain cross-linkers are elastically active.18 In the equilibrium state, the fraction of free cross-linkers is very small, and can be neglected.18 If a cross-linker dissociates, it changes temporarily to the dangling state, from which it can bind again to an available pyridine residue elsewhere. The characteristics of our supramolecular polymer network (Scheme 1a) are superficially similar to the characteristics of the Sticky Rouse or Sticky Reptation models of associative polymer networks commonly discussed in the literature.1,4 The characteristics of the Sticky Rouse or Sticky Reptation models are shown in Scheme 1b.1, 4 There are S (S≫1) stickers covalently grafted to linear polymer chains. Two stickers associate with each other to form a cross-linking point. There are three states of cross-linkers: dangling, intrachain bound and interchain bound cross-linkers. As in the experimental system, only interchain cross-linkers are elastically active, 1, 4 and cross-linker dissociation allows the diffusion of its parent chain followed by re-association with another sticker (or, in certain cases, to its previous partner). 1,4 There are, however, significant differences between our supramolecular polymer network and the Sticky Rouse or Sticky Reptation models. In our supramolecular polymer network, the cross-linkers are physically rather than covalently attached to the PVP chain (which we term here a “diffusive polymer network”), so it is possible that the cross-linkers can migrate independently of the movement of polymer chains, and bind again with a new pyridine partner, whereas in the Sticky Rouse or Sticky Reptation models the movement of cross-links is always restricted by the dynamics of the polymer chain. 1
Scheme 1.

(a) Schematic of the supramolecular polymer network (1·PVP) in which polymer chains are cross-linked by bisfunctional recognition units. Each pyridine unit in the polymer chain is represented by a bead. Four different states of association groups are pictured; (b) Reversible network with pairwise associating groups in Sticky Rouse model.1 One side of each associating group is covalently bound to a polymer chain. Three different states of association groups are pictured.
In this work, the scaling law relationships between the zero shear viscosity (η0) of the diffusive polymer networks were experimentally determined as a function of the concentration of PVP solution (CPVP), the concentration of cross-linkers (CX), and the number density of elastically active chains (vphantom). The results are compared with the Sticky Rouse or Sticky Reptation models and differences in behavior are observed.1 We anticipate that the experimental results described here will motivate future theoretical work to explore these and other scaling laws for these networks.
Experimental Section
Materials
The bis-Pd(II) cross-linkers [2,3,5,6-tetrakis{(dimethylamino)methy}phenylene-1,4-bis(palladiumtrifluoromethane-sulfonate)] (1a) and [2,3,5,6-tetrakis{(diethylamino)methy}phenylene-1,4-bis(palladiumtrifluoromethane-sulfonate)] (1b) were synthesized as reported elsewhere.21 The binding thermodynamics and exchange kinetics of the metal–pyridine interactions have been characterized previously.16,21 The equilibrium association constants (Keq) and dissociation rate constants (kd) for 1a·pyridine in DMSO at 25 °C are 29 M−1 and 1450 s−1, respectively. Keq and kd for 1b·pyridine in DMSO at 25 °C are 33 M−1 and 17 s−1, respectively.16,21 Dimethyl sulfoxide (DMSO), and poly(4-vinylpyridine) (PVP) with Mw of 60,000 Da as reported from producer, were used as received from Aldrich. The Mn and Mw of above PVP from Aldrich have been reported previously to be 22,000 and 64,300 g/mol (polydispersity index (PDI) = 2.92), respectively, in the literature.22 We carried out an independent analysis of the specific sample used in our experiments (see Figure S1 in SI) and obtained values of Mn of 33,000 and Mw of 44,500 g/mol (PDI = 1.35). Another PVP with Mn of 36,300 and Mw of 42,500 g/mol (PDI = 1.17) (see Figure S2 in SI) was used as received from Polymer Source, Inc. (Canada). Unless otherwise noted, the results in this paper are from the samples made with the PVP from Aldrich.
Sample Preparation
Samples were prepared by mixing PVP solutions with cross-linker solutions, as previously reported.18 The concentration of cross-linkers in the samples varied from 1% to 5% (reported as the ratio of Pd atoms in the cross-linkers to N atoms in PVP), all of which are near or above the critical concentration to form a gel (shown previously to be approximately 0.8 % for ∼ 0.10 g/mL PVP/DMSO solutions).23 The mass concentration of PVP in the samples varies from ∼0.08 (semidilute unentangled regime) to ∼0.27 g/mL (semidilute entangled regime). Details regarding the concentration regimes of PVP (from Aldrich) solution have been reported previously.20 The critical concentration of entanglement of PVP/DMSO solution is 0.155 g/mL at 25 °C. 20 The average number of physical entanglements per polymer chain is calculated to be from 1 to 3 for PVP concentrations from ca. 0.16 g/mL to 0.27 g/mL.20,24 In Figure S3 in the Supporting Information, the concentration regimes of the PVP obtained from Polymer Source is characterized, and the critical concentration for entanglement of these PVP solution is 0.152 g/mL at 25 °C.
Rheological Measurements
Rheological data were obtained using an AR-G2 rheometer (TA Instruments) with cone–plate geometry (diameter of 20 mm, cone angle of 2°, truncation height of 49 μm). Strain sweep experiments were performed at a frequency of 10 rad/s to determine the region of linear response. Oscillatory frequency sweeps from 0.1 to 500 rad/s were carried out with appropriate strain in the linear region. Steady shear measurements were performed over a range of shear rates between ∼10−3 and ∼103 s−1. Experiments were carried out at 25 °C.
Results and Discussion
Conditions to Form a Supramolecular Polymer Network
First, the conditions under which the samples form a network are determined. Here, a supramolecular polymer network is defined as corresponding to conditions under which the relaxation of the network is dominated by the dissociation kinetics of cross-linkers. As in our previous work, this condition is characterized through linear oscillatory frequency sweep rheology of samples with identical concentration of 1a or 1b in the same concentration of PVP solution. If the frequency sweep data can be superposed onto a single master curve by the scaled frequency (ω/kd),16–18,20 i.e., the relaxation time (τ) of the 1·PVP network is proportional to the lifetime (τ0, τ0=1/kd) of the cross-linker, 16–18,20 we conclude that the relaxation of the network requires cross-link dissociation. If the dynamic mechanical properties of the samples with identical concentration of 1a or 1b in the same concentration of PVP solution cannot be superposed onto a single master curve by the scaled frequency (ω/kd), relaxation is not restricted by the dissociation kinetics of the cross-linkers,18,20 indicating the presence of other relaxation dynamics such as the diffusion of un-networked polymer chains. The superposition of linear oscillatory frequency sweep results of samples with identical concentration of 1a or 1b in the same concentration of PVP solution is therefore used as the primary criterion for network structure.
In Figure 2, superposition of storage moduli (G′) and loss moduli (G″) versus scaled frequency (ω/kd) for ∼0.11 g/mL PVP with different concentration of 1a or 1b is shown. We note that the data for ∼0.11 g/mL PVP with 1% 1a or 1% 1b cannot be superposed, consistent with previous observations regarding cross-linker concentrations that are near the gel point.18,20 For ∼0.11 g/mL PVP with concentration of cross-linker from 2% to 5%, the G′ and G″ for 1a·PVP and 1b·PVP can be superposed by scaled frequency (ω/kd),18,20 and thus, we conclude that the relaxation of these networks are dominated by the kinetics of cross-linkers. In Figure S4-S9 in the Supporting Information, the superposition of storage moduli (G′) and loss moduli (G″) versus scaled frequency (ω/kd) for different concentrations of PVP (from ∼0.08 g/mL to ∼0.27 g/mL) with 1a or 1b is shown.
Figure 2.
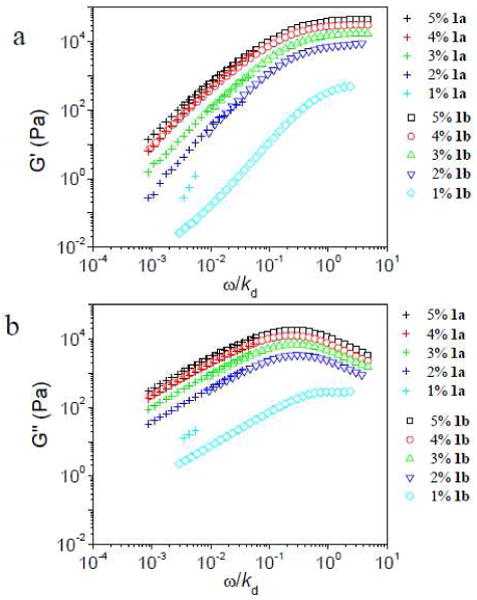
Storage (G′) and loss (G″) moduli versus scaled frequency (ω/kd) for ∼0.11 g/mL PVP with different concentration of 1a or 1b. T = 25 °C. For 1a, kd = 1450 s−1, and for 1b, kd = 17 s−1.
Whether or not the storage (G′) and loss (G″) moduli are superposed through the scaled frequency (ω/kd) for all examined samples is summarized in Figure 3. The influence of cross-linker concentration and PVP concentration on the superposition behavior is discussed in terms of the mean number of elastically active subchains per polymer chain (feas) below.
Figure 3.
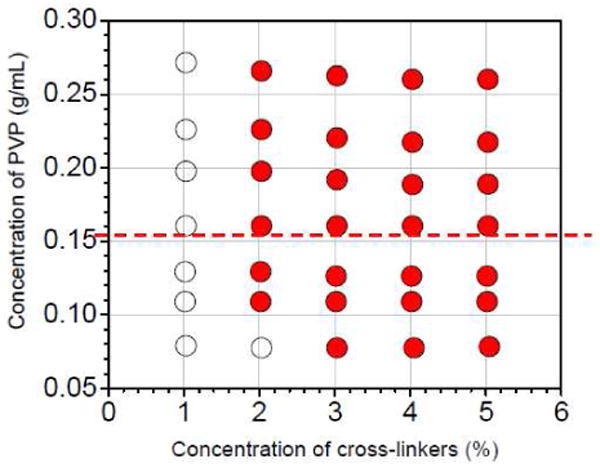
Superposition behavior of storage moduli (G′) and loss moduli (G″) versus scaled frequency (ω/kd) for samples with 1a or 1b as a function of the concentration of cross-linkers and the concentration of PVP in DMSO. Unfilled circles represent the conditions under which G′ and G″ of 1a·PVP and 1b·PVP are not superposed by scaled frequency (ω/kd). Filled circles represent conditions under which G′ and G″ of samples with 1a or 1b are superposed by scaled frequency (ω/kd). The dashed line indicates the critical concentration of entanglement (0.155 g/mL).
Nature of the Network
To further characterize the formation of the network, we considered the extent of cross-linking. For simplicity, the affine network model was used previously to calculate the apparent number density of elastically active chains in these supramolecular polymer networks.18,20 From the frequency sweep results of samples with cross-linker 1b (Figure 2 and Figure S4–S9 in SI), the plateau moduli (G0) are obtained from the constant value of G′ at high frequency, and the apparent number density of elastically active chains (vaffine) are then calculated from the affine network model:24
| (1) |
where kb is Boltzmann's constant, and T is the temperature. Unlike permanent cross-linkers, which remain intact in chemical cross-linked networks, the reversible cross-linkers in these supramolecular polymer networks may spontaneously break away and then reattach to allow the bound segment, and hence the polymer, to move.5 Thus, a phantom network model is more appropriate for associative polymer networks,5, 24 and the number density of elastically active chains (vphantom) is given by:24
| (2) |
In equation 2, f is the functionality of the cross-links. As shown in Figure 1b, four polymer strands join at one cross-linker, so a functionality of four (f=4) is used here, and the calculated vphantom is twice the value of vaffine for a given G0. Because the cross-linker 1 is both small and rigid, we do not treat it as an elastically active segment here.
In the semidilute unentangled regime, G0 is determined by the interchain bond cross-linkers, whereas in the semidilute entangled regime, G0 may be determined by both the interchain bond cross-linkers and entanglements between polymer chains.20 The lifetime of the physical entanglements between polymer chains is denoted τe — the Rouse relaxation time of a chain of length equal to one tube segment.24 The change in τe across concentrations from 0.155 to 0.27 g/mL is small, with an average value τ̄e of ∼ 2.6 × 10−5 s, as characterized previously.20 The lifetime of physical entanglements between polymer chains (τ̄e) is therefore much smaller than the lifetime of cross-linker 1b used in this work (τ0 ≈ 6 × 10−2 s). As seen in Figure 2, the frequency at which the plateau value of G′ is characterized is faster than the rate of dissociation of cross-linker 1b (ω/kd > 1) but is much slower than the rate of physical disentanglement between polymer chains.
The concentration and/or lifetime of cross-linkers 1b, however, have an influence on the lifetime of physical entanglements between polymer chains (τ̄e), resulting in a renormalized lifetime of physical entanglements between polymer chains (τ̄e *).1 When τ̄e * is close to the lifetime of cross-linker 1b (τ̄e * ≈ τ0), physical entanglements between polymer chains should also contribute to G0.17 We note here that τ̄e * is considered in the context of the linear rheological regime in this paper, in comparison to the nonlinear shear thickening/shear thinning behavior we have reported previously.20 That physical entanglements contribute to G0 in the semidilute entangled regime for these polymers has been inferred from the observation that the number of elastically active chain segments (calculated from the plateau modulus) exceeds that expected from the number of cross-linkers added to solution.20 The superposition of viscoelastic data and the measured relaxation rates of the 1a and 1b networks (Figure S6-S9 in SI) affirm that any physical entanglements of this type are effectively limited by cross-link dissociation, i.e. τ̄e *≈ τ0. We therefore treat such elastically active entanglements as cross-linkers with lifetime τ0. As shown in the following sections, the validity of this treatment is further supported by scaling law dependencies that remain unchanged above and below the critical concentration for entanglement.
Mean Number of Elastically Active Subchains per Polymer Chain
As described by Flory, the conditions for network formation (gel point) correspond to one interstrand cross-linker per polymer chain (i.e., two “shared” cross-linkers).25 At the gel point, therefore, there is on average one elastically active subchain per polymer chain. We therefore define the mean number of elastically active subchains per polymer chain (feas) according to equation 3:
| (3) |
where vphantom is taken from equation 2, np is the number of polymer chains per 1 mL volume, CPVP is the concentration of PVP (g/mL), NA is Avogadro's number, and Mn is the number average molecular weight of PVP.
The value of feas provides a second criterion for the formation of a network structure,25 and one that is independent of the previously discussed superposition of dynamic moduli. As seen in Figure 4, feas > 2 for those networks for which the dynamic moduli are superposed by scaled frequency (ω/kd) in Figure 3, whereas feas < 1.5 for those networks for which the superposition fails. This analysis therefore supports our earlier characterization, and we use this subset of 1·PVP networks (feas > 2; viscoelastic data superposed by cross-linker dissociation rates) to examine the scaling law relationships related to viscosity.
Figure 4.
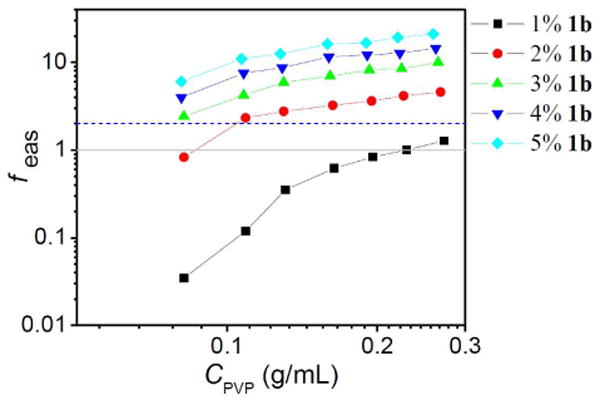
Mean number of elastically active subchains per polymer chain (feas) in 1b·PVP versus the concentration of PVP solution (CPVP). Critical concentration of entanglement is 0.155 g/mL. Mn(PVP) = 33,000 g/mol. The dotted line corresponds to feas = 2, used as a secondary criterion for network formation.25
We note that the networks are necessarily heterogeneous, and, in all cases, there must be some steady-state fraction of small, discrete polymers and polymer aggregates that are dissociated from the network.25 The presence of such “non-networked” fragments should not affect the main features of the analyses that follow, because they do not contribute substantially to the bulk viscoelastic properties of the samples and because the cross-link density is inferred directly from bulk measurements that reflect that network structure. Additionally, we point out that the heterogeneity means that the value of feas as calculated above underestimates the true average number of active subchains per polymer chain within the active network structure, because it is the ratio of the number of active segments (all of which are in the active network) to the total number of polymer chains (some of which are in the active network, and some of which are not).
Scaling Laws in Sticky Rouse and Sticky Reptation model
The scaling law relationships between zero shear viscosity (η0) of the networks versus the concentration of polymer in solution (ϕ) and the number of cross-linkers per chain (S) have been explored previously by Rubinstein and Semenov in the context of the Sticky Rouse and Sticky Reptation models.1 The detailed scaling law relationships (η0 ∼ Smϕn) in the Sticky Rouse and Sticky Reptation models are summarized in Table S1 in Supporting Information. For convenience, those scaling law relationships (η0 ∼ Smϕn) that are compared with our experimental results are summarized in Table 1; these relationships assume that the degree of polymerization of the polymer (N) is fixed. In Table 1, ϕs is the overlap concentration of the strands between stickers (not the overlap concentration of the polymer solution); ϕe is the critical entanglement concentration of polymer solution; ϕle is the entangled concentration of the strands between interchain bonds.1 Two further regimes are considered, one in which the lifetime (τ0) of cross-linkers depends on the probability of finding a new open sticker with which to associate (renormalized bond lifetime) and one in which it does not (unrenormalized bond lifetime).1
Table 1.
Relevant scaling laws (η0 ∼ Smϕn) in Sticky Rouse and Sticky Reptation models in the case of unrenormalized and renormalized bond lifetime, respectively.1
| Models | Concentration of polymer (ϕ) | Solvent property | Scaling law (η0 ∼Smϕn) | |
|---|---|---|---|---|
| unrenormalized bond lifetime | renormalized bond lifetime | |||
| Sticky Rouse | ϕ<ϕe and ϕ<ϕs | θ solvent | η0 ∼ Sϕ3 | η0 ∼ S1.5ϕ3.5 |
|
| ||||
| Sticky Reptation | ϕs<ϕ<ϕle | θ solvent | η0 ∼ S2ϕ3.67 | η0 ∼ S3ϕ3.17 |
Experimental Scaling Laws in 1·PVP Supramolecular Polymer Networks
Because the plateau moduli (G0) of the 1a·PVP networks cannot be obtained directly,18,20 the 1b·PVP networks are used to explore the scaling law relationships. Many of these data have been reported previously and examined in other contexts. For convenience, the value of the plateau modulus (G0) and zero shear viscosity (η0) from the results in ref. 20, the maximum theoretical number of elastically active point per unit volume (v0), as well as the number density of elastically active chains (vphantom) of the 1b·PVP networks are summarized in Tables S2–S6 in the Supporting Information.
As shown in previous work, at equilibrium the fraction of free cross-linkers (Scheme 1a) is very small and can be neglected.18 The concentration of cross-linkers (CX, molar ratio between Pd atom and N atom) is related to the degree of polymerization of the polymer (N) and the number of cross-linkers per chain (S) by
| (4) |
In this work, the degree of polymerization of the polymer (N) is a constant. So, assuming that binding of the cross-linkers is independent of the extent of cross-linking, CX is proportional to S at fixed CPVP, and we compare the scaling law relationship between viscosity and CX derived here (η0 ∼ Cxp) to that between viscosity and S (η0 ∼ Sm) in the Sticky Rouse and Sticky Reptation model.1 The scaling law relationship between viscosity and concentration of PVP solution CPVP (η0 ∼ CPVPu) are also compared to that between viscosity and concentration of polymer solution ϕ(η0 ∼ ϕn) in the Sticky Rouse and Sticky Reptation model.1
We first consider the scaling law describing the relationship between the zero shear viscosity (η0) and the number density of elastically active chains (vphantom) in the Sticky Rouse and Sticky Reptation models. Because the molar ratio of pyridine to cross-linker Pd is very high (≥ 20:1 for these samples), there is a surplus of new binding sites for each cross-linker upon dissociation, and so we explore the scaling relationships in the context of the unrenormalized bond lifetime models.1
In the Sticky Rouse model for the unentangled associative polymer network, the longest time of Rouse relaxation of polymer chain (τRouse, also called the sticky Rouse time) is equal to the lifetime (τo) of a reversible bond times the square of the number of elastically active cross-linkers per polymer chain1
| (5) |
where f* is the mean number of elastically active cross-linkers per chain. We note that while the concentration of PVP does not affect τRouse directly (since the Sticky Rouse time is defined for a chain), f* depends on the concentration of PVP.24
In the Sticky Rouse model, the modulus (G1) at the Rouse time (τRouse) is kBT per chain (kBT times the number density of chains)1
| (6) |
where np is the number of polymer chains per unit volume. The zero shear viscosity (η0) of the unentangled solution of associating polymers is proportional to the product of the modulus (G1) and the Sticky Rouse time (τRouse)1
| (7) |
At high concentrations of cross-linkers, f* is proportional to feas, and hence to vphantom (equation 3). Under these conditions, the zero shear viscosity (η0) of the network therefore follows
| (8) |
From equation 8, if the concentration of PVP solution (CPVP) is fixed, and only the concentration of cross-linker is varied, one obtains the Sticky Rouse expectation of . The same scaling law can be derived from the Sticky Reptation model for conditions in which the concentration of PVP solution (CPVP) is fixed and only the concentration of cross-linker is varied. 1
A slightly different expectation is derived from Marrucci et al's theoretical work for unentangled telechelic associative polymer networks, in which the zero shear viscosity (η0) is found to vary with the number density of elastically active chains (v) as η0∼v5/3.8 Superficially, the structure of our supramolecular polymer network (Scheme 1a) is quite different from that of a telechelic associative polymer network,8 and resembles more closely the Sticky Rouse model. We next compare the experimental scaling law relationship between zero shear viscosity (η0) and number density of elastically active chains vphantom (η0 ∼ vphantomr) to the expectations of these two models.
In Figure 5, the scaling law relationships between the zero shear viscosity (η0) of the networks as a function of the concentration of PVP solution (CPVP), the concentration of cross-linkers (CX), and the apparent number density of elastically active chains (vphantom) are shown. In Figure 5a are shown data for series of samples in which the concentration of PVP is held constant at seven different concentrations (from 0.08 to 0.27 g/mL) and CX is varied. The power law exponents p of η0 ∼ Cxp are relatively independent of polymer concentration. From low to high concentrations, p is 2.76, 2.59, 2.51, 2.46, 2.29, 2.62, and 2.53, with correlation coefficients above 0.993. The average value of p (pavg) is 2.5. In Figure 5b, data are shown as a function of CPVP for series of samples in which the relative concentration of cross-linkers is held constant at values of from 2% to 5%. The resulting power law exponents u of η0 ∼ CPVPu are 2.71, 2.85, 2.72, and 2.80, respectively, with correlation coefficients above 0.992. The average value of u (uavg) is 2.8. Most significantly, in Figure 5c, for series of samples in which the concentration of PVP solution is held constant from 0.08 to 0.27 g/mL, the power law exponents r of η0 ∼ vphantomr are 1.56, 1.52, 1.55, 1.42, 1.39, 1.62, and 1.56, respectively, with correlation coefficients above 0.993. The average value of r (ravg) is 1.5. Good scaling law relationships exist for these networks for the zero shear viscosity (η0) of the networks versus the concentration of PVP solution (CPVP), the concentration of cross-linkers (CX), and the number density of elastically active chains (vphantom).
Figure 5.

Zero shear viscosity (η0) of the 1b·PVP network in which the concentration of PVP is fixed and the concentration of cross-linkers (Cx) is varied (a); in which the concentration of cross-linkers is fixed and the concentration of PVP (CPVP) is varied (b); and as a function of the apparent number density of elastically active chains (vphantom) for all concentrations of polymer and cross-linker. For samples with concentration of PVP solution from 0.08 to 0.27 g/mL (a), the power law dependence p of η0 ∼ Cxp is 2.76, 2.59, 2.51, 2.46, 2.29, 2.62, and 2.53, respectively (pavg = 2.5), with correlation coefficients of linear fitting above 0.993. For samples with concentration of cross-linkers from 2% to 5% (b), the power law dependence u of η0 ∼ CPVPu is 2.71, 2.85, 2.72, and 2.80 for cross-linker concentrations of 2% to 5%, respectively (uavg = 2.8), with correlation coefficients of linear fitting above 0.992. (c) For samples with concentration of PVP solution from 0.08 to 0.27 g/mL, the power law exponent r of η0 ∼vphantomr is 1.56, 1.52, 1.55, 1.42, 1.39, 1.62, and 1.56, respectively (ravg = 1.5), with correlation coefficients of linear fitting above 0.993. Here, vphantom is reported as the number of elastically active chains per 1 mL volume.
Similar experimental work has been reported previously. Vermonden et al. studied the linear rheology of water-soluble reversible neodymium(III) coordination polymers, and scaling law relationships between zero-shear viscosity and concentration of polymer were observed.26 For their linear supramolecular polymer chains, the scaling law is in good agreement with the predictions of Cates' model that describes the dynamics of linear equilibrium polymers.26, 27 For the cross-linked supramolecular polymer network in Vermonden et al.'s work, however, the results are not consistent with the Cates model, although a good scaling law relationship between zero-shear viscosity and concentration of polymer was still observed.26, 27 Candau's group researched the rheological properties of associative polymer networks formed by hydrophobically modified poly(acrylamide)s.15, 28 A scaling law relationship between zero-shear viscosity and polymer concentration was observed and explained by the Sticky Reptation model.1, 15, 28
Influence of PVP Polydispersity and/or Molecular Weight
To explore the influence of polydispersity and/or molecular weight of PVP on the dynamics of the supramolecular polymer network, samples made from PVP with Mn of 36,300 and Mw of 42,500 g/mol (“PVP1.17” to reflect the polydispersity index (PDI) of 1.17) are compared with the prior samples, which were made from PVP with Mn of 33,000 and Mw of 44,500 g/mol (PDI = 1.35, see Figures S1-S2 in SI).22
In Figure 6, the concentration of PVP/DMSO solution is fixed at 0.11 g/mL and the concentration of 1b is varied from 2% to 5%. The power (p) of η0 ∼ Cxp is 2.66 for PVP1.17, in comparison to p = 2.59 for the original PVP. The power (r) of η0 ∼ vphantomr is 1.44 for PVP1.17 vs. 1.52 for the slightly more polydisperse PVP. Within this range, therefore, slight changes in molecular weight and/or polydispersity do not have a dramatic influence on the observed scaling law relationships.
Figure 6.
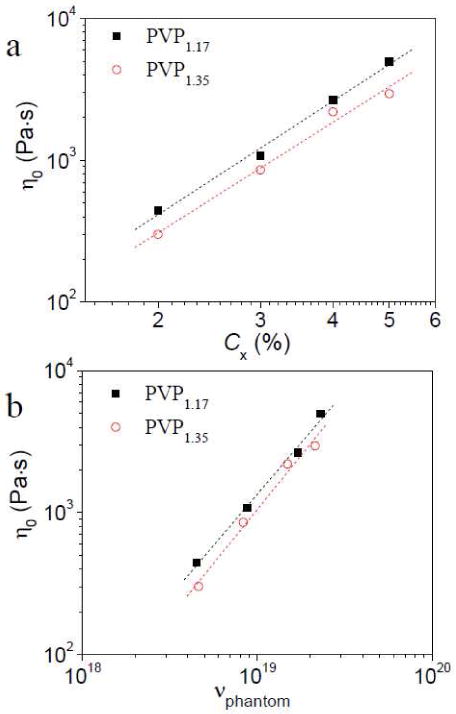
(a) Zero shear viscosity (η0) of the 1b·PVP network versus the concentration of cross-linkers (Cx) (a), and the apparent number density of elastically active chains (vphantom) (b) for ∼ 0.11 g/mL PVP solution with different concentration of cross-linkers (2% to 5%). Two different PVP from Polymer Source (Mn of 36,300 and Mw of 42,500 g/mol, PDI=1.17, abbreviated as “PVP1.17”) and Aldrich (Mn of 33,000 and Mw of 44,500 g/mol, PDI = 1.35, abbreviated as “PVP1.35”) are used, respectively. (a) The power (p) of η0 ∼ Cxp are 2.66 and 2.59 for PVP1.17 and PVP1.35, respectively. Correlation coefficients of linear fitting are 0.997 and 0.993, respectively. (b) The power (r) of η0 ∼vphantomr are 1.44 and 1.52 for PVP1.17 and PVP1.35, respectively. Correlation coefficients of linear fitting are 0.997 and 0.993, respectively. Here, vphantom is reported as the number of elastically active chains per 1 mL volume.
Discussion
We begin by pointing out some qualitative observations. First, there is no substantial difference in scaling law behavior as a function of polymer concentration, including comparisons of samples that are above and below the critical concentration for entanglement (0.155 g/mL). Second, the data in Figure 5c collapse nicely onto a single line. Because these data are presented in terms of the number of active cross-linkers for a range of polymer concentrations, they indicate that it is the number of active cross-linkers (vphantom), and not the number of active cross-linkers per polymer chain (f*), that is a primary determinant of network viscosity.
To further compare the data in Figure 5 with the Sticky Rouse and Sticky Reptation models in Table 1, the values of ϕs, ϕe and ϕle must be determined. In Sticky Rouse and Sticky Reptation models, the strand overlap concentration for the strands between stickers ϕs can be calculated by1
| (9) |
where ve is the excluded volume scaling exponent. As reported previously, ve = 0.53 for PVP in DMSO at 25 °C,20 and we therefore treat DMSO as a theta solvent (ve = 0.5). And ϕs can be estimated by the value of Cx0.5 by combining equations 4 and 9. For Cx = 1%, 2%, 3%, 4% and 5%, ϕs is 0.1, 0.141, 0.173, 0.2 and 0.224, respectively, and the relevant PVP concentrations are 0.110, 0.156, 0.190, 0.220 and 0.246 g/mL, respectively. As characterized previously, the critical entanglement concentration ϕe is 0.141 (or ∼ 0.155 g/mL). Also, the mean number of elastically active cross-linkers per chain (f*) in the semidilute entangled regime is larger than the average number of physical entanglements per polymer chain (between 1 and 3 20), so the strands between interchain bonds are not entangled in our experimental range, i.e. ϕ is smaller than ϕle. 1
Samples with concentration of PVP solution from 0.08 to 0.13 g/mL therefore meet the conditions of ϕ <ϕe and ϕ < ϕs. For a theta solvent, the Sticky Rouse expectation is η0 ∼ Sϕ3 and η0 ∼ S1.5ϕ3.5,1 respectively, for unrenormalized and renormalized bond lifetimes (Table 1). In Figure 5a, for samples with concentration of PVP solution from 0.08 to 0.13 g/mL, the exponent p of η0 ∼ Cxp is 2.76, 2.59 and 2.51, respectively, which is greater than the Sticky Rouse expectation1 of η0 ∼ S or η0 ∼ S1.5, as Cx is related to S. In Figure S10 in Supporting Information, for samples with concentration of cross-linkers from 2% to 5% in the semidilute unengangled PVP solution (from 0.08 to 0.13 g/mL), the power (u) of η0 ∼ CPVPu are 3.49, 3.70, 3.08, and 2.99, respectively. For samples with 4% and 5% cross-linker, the power (u) of η0 ∼ CPVPu are close to η0 ∼ ϕ3 expected for unrenormalized bond lifetimes in the Sticky Rouse model,1 whereas the 2% and 3% cross-linker samples are somewhat larger. In Figure 5c, for samples with concentration of PVP solution from 0.08 to 0.13 g/mL, the power (r) of η0 ∼vphantomr are 1.56, 1.52, and 1.55 respectively. The power (r) is smaller than η0 ∼vphantom2 in Sticky Rouse model.
Semidilute entangled samples in 0.26 g/mL PVP solution meet the condition ϕs < ϕ < ϕle of the Sticky Reptation model.1 For a theta solvent, the Sticky Reptation expectation is η0 ∼ S2ϕ3.67 and η0 ∼ S3ϕ3.17, respectively, for unrenormalized and renormalized bond lifetimes (Table 1). As shown in Figure 5a, for 0.26 g/mL PVP, the power (p) of η0 ∼ Cxp is 2.53, slightly greater than the expected value p = 2. Samples with PVP concentrations of 0.16 or 0.22 g/mL do not fall in a single concentration range in Table 1 and are not discussed here. Samples with 2% 1b in semidilute entangled PVP solution (from 0.163 to 0.268 g/mL) and samples with 3% 1b in the semidilute entangled PVP solution (from 0.193 to 0.265 g/mL) meet the condition as ϕs < ϕ < ϕle in Sticky Reptation model (Figure S11 in SI). The values of u for η0 ∼ CPVPu are 2.31 and 2.48, respectively, as compared to η0 ∼ ϕ3.67 in the Sticky Reptation model.1
Interestingly, although the structure of our supramolecular polymer network (Scheme 1a) is quite different from that of a telechelic associative polymer network in Marruci et al's work and the structure of elongated wormlike micelles in Turner et al.'s theoretical work,8,29 the scaling law η0 ∼vphantom1.5±0.1 in Figure 5c is close to η0 ∼ v5/3 in Marrucci et al's theoretical work and the scaling law η0 ∼ CPVP2.8±0.1 in Figure 5b is close to η0 ∼ϕ2.9 (“breathing regime” with end-interchange mechanism) or η0 ∼ϕ2.6 (“breathing regime” with bond-interchange mechanism) in Turner et al.'s work, respectively.8, 29 The agreement with these models suggests that Rouse-like or reptative dynamics may be secondary to the kinetics of cross-linker dissociation in these physical networks.
This point is supported further by considering the scaling exponents p and u for Cx and CPVP, respectively. The specific values of the exponents are perhaps less telling than is their (lack of) variation as a function of polymer concentration. In contrast to Sticky Rouse and/or Sticky Reptation type behavior, which differs above and below ϕs,1 these networks exhibit no substantial change in the scaling exponent as a function of concentration of polymer solution. The networks therefore appear to be in a regime where the limiting relaxation dynamics are not of the Sticky Rouse/Reptation type. The physics underlying the observed dynamics is not clear, although we speculate that the “diffusive network” character of the present system may play a role, because the spatial diffusion of entanglement points (cross-links) can occur without diffusion of the PVP chains. It is also possible that lower levels of cross-linking, relative to those typically assumed in Sticky Rouse/Reptation systems, are responsible for some or all of the behavior, although the apparent robustness of the scaling laws as a function of cross-linker content is perhaps a sign that this is not the sole contributor. We admit that these suppositions are speculative, but, mechanistic uncertainty notwithstanding, we believe that the empirical relationships reported here provide a foundation for further experimental and theoretical investigations of these and similar systems.
Conclusions
For the metallo-supramolecular polymer networks formed by bis-Pd(II) cross-linkers and solutions of poly(4-vinylpyridine) in dimethyl sulfoxide, the scaling law relationship between the zero shear viscosity (η0) of networks versus the concentration of PVP solution (CPVP), the concentration of cross-linkers (CX), and the number density of elastically active chains (vphantom) has been experimentally determined and compared with the expectation from the Sticky Rouse and Sticky Reptation models. It is found that the experimentally determined scaling law relationships in our work cannot be fully described by the Sticky Rouse and Sticky Reptation models. The different characteristics of the Sticky Rouse model and our metallo-supramolecular polymer networks include the presence vs. absence of a covalent attachment of the cross-linking groups to the polymer chains, which we propose contributes to the observed power law dependencies. The experiments provide benchmarks for future theoretical investigations of the rheological properties of these and similar supramolecular polymer networks.
Supplementary Material
Acknowledgments
S. C. thanks the NSF (CHE-0646670) and NIH (EB-001037) for financial support of this work. D. X. and S. C. are grateful for insightful comments from Prof. M. Rubinstein and the reviewers.
Footnotes
Supporting Information Available: Characterization of molecular weight of PVP; the concentration regime of PVP/DMSO solution for PVP from Polymer Source; superposition of storage moduli (G′) and loss moduli (G″) versus scaled frequency (ω/kd) for different concentration of PVP with 1a or 1b; scaling law relationships in Sticky Rouse and Sticky Reptation models; summary of original data for 1b·PVP with different concentrations of PVP and cross-linkers; scaling law relationships between zero shear viscosity (η0) and concentration of PVP (CPVP). This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Rubinstein M, Semenov AN. Macromolecules. 2001;34:1058–1068. [Google Scholar]
- 2.Berret JF, Calvet D, Collet A, Viguier M. Curr Opin Colloid Interface Sci. 2003;8:296–306. [Google Scholar]
- 3.Pellens L, Corrales RG, Mewis J. J Rheol. 2004;48:379–393. [Google Scholar]
- 4.Leibler L, Rubinstein M, Colby RH. Macromolecules. 1991;24:4701–4707. [Google Scholar]
- 5.Baxandall LG. Macromolecules. 1989;22:1982–1988. [Google Scholar]
- 6.Wang SQ. Macromolecules. 1992;25:7003–7010. [Google Scholar]
- 7.Tanaka F, Edwards SF. Macromolecules. 1992;25:1516–1523. [Google Scholar]
- 8.Marrucci G, Bhargava S, Cooper SL. Macromolecules. 1993;26:6483–6488. [Google Scholar]
- 9.Green MS, Tobolsky AV. J Chem Phys. 1946;14:80–92. [Google Scholar]
- 10.Lodge AS. Trans Faraday Soc. 1956;52:120–130. [Google Scholar]
- 11.Yamamoto M. J Phys Soc Jpn. 1956;11:413–421. [Google Scholar]
- 12.Feldman KE, Kade MJ, Meijer EW, Hawker CJ, Kramer EJ. Macromolecules. 2009;42:9072–9081. [Google Scholar]
- 13.Buhler E, Candau SJ, Kolomiets E, Lehn JM. Phys Rev E. 2007;76:061804. doi: 10.1103/PhysRevE.76.061804. [DOI] [PubMed] [Google Scholar]
- 14.Ng WK, Tam KC, Jenkins RD. J Rheol. 2000;44:137–147. [Google Scholar]
- 15.Candau F, Regalado EJ, Selb J. Macromolecules. 1998;31:5550–5552. [Google Scholar]
- 16.Yount WC, Loveless DM, Craig SL. J Am Chem Soc. 2005;127:14488–14496. doi: 10.1021/ja054298a. [DOI] [PubMed] [Google Scholar]
- 17.Loveless DM, Jeon SL, Craig SL. Macromolecules. 2005;38:10171–10177. [Google Scholar]
- 18.Xu D, Hawk J, Loveless DM, Jeon SL, Craig SL. Macromolecules. 2010;43:3556–3565. doi: 10.1021/ma100093b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu D, Craig SL. J Phys Chem Lett. 2010;1:1683–1686. doi: 10.1021/jz1004818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu D, Craig SL, Liu CY. Macromolecules. 2011;44:2343–2353. doi: 10.1021/ma2000916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jeon SL, Loveless DM, Yount WC, Craig SL. Inorg Chem. 2006;45:11060–11068. doi: 10.1021/ic061188b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen D, Handa H, Wan L, Mao G. Macromol Rapid Commun. 2007;28:1619–1623. [Google Scholar]
- 23.Loveless DM, Jeon SL, Craig SL. J Mater Chem. 2007;17:56–61. [Google Scholar]
- 24.Rubinstein M, Colby RH. Polymer Physics. Oxford University Press; 2003. [Google Scholar]
- 25.Flory PJ. Principles of Polymer Chemistry. Cornell University Press; 1953. [Google Scholar]
- 26.Vermonden T, van Steenbergen MJ, Besseling NAM, Antonius TM, Marcelis ATM, Hennink WE, Sudhölter EJR, Cohen Stuart MA. J Am Chem Soc. 2004;126:15802–15808. doi: 10.1021/ja0458928. [DOI] [PubMed] [Google Scholar]
- 27.Cates ME. Macromolecules. 1987;20:2289–2296. [Google Scholar]
- 28.Regalado EJ, Selb J, Candau F. Macromolecules. 1999;32:8580–8588. [Google Scholar]
- 29.Turner MS, Marques C, Cates ME. Langmuir. 1993;9:695–701. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.