

Summary
Tumor necrosis factor (TNF), a key effector in controlling tuberculosis, is thought to exert protection by directing formation of granulomas, organized aggregates of macrophages and other immune cells. Loss of TNF signaling causes progression of tuberculosis in humans and the increased mortality of Mycobacterium tuberculosis-infected mice is associated with disorganized and necrotic granulomas, though the precise roles of TNF signaling in the stages of pathogenesis preceding this endpoint remain undefined. We monitor transparent Mycobacterium marinum-infected zebrafish live to conduct a stepwise dissection of the effects of TNF signaling in mycobacterial pathogenesis. We find that loss of TNF signaling causes increased mortality even when only innate immunity is operant. In the absence of TNF intracellular bacterial growth and granuloma formation are accelerated followed by necrotic death of overladen macrophages and granuloma breakdown. Thus TNF is not required for tuberculous granuloma formation, but maintains granuloma integrity indirectly by restricting mycobacterial growth within macrophages and preventing their necrosis.
Introduction
Pathogenic mycobacteria are highly adapted intracellular pathogens that can survive for indefinite periods of time within their hosts. Infection results in the recruitment of host macrophages to the bacteria, their phagocytosis, and the transit of infected macrophages into deeper tissues (Cosma et al., 2003; Dannenberg, 1993). There the infected macrophages recruit additional macrophages and other immune cells to form tightly aggregated immune structures called granulomas, pathological hallmarks of tuberculosis (Cosma et al., 2003; Dannenberg, 1993). In certain host species, areas within tuberculous granulomas undergo necrosis, referred to as caseation, so that mycobacteria may occupy both an intracellular and extracellular niche during the course of tuberculosis (Cosma et al., 2003).
TNF was one of the first effector molecules found to be essential in the host protective response against tuberculosis (Flynn et al., 1995). Mice deficient either in TNF or TNF Receptor 1 have increased susceptibility to challenge with pathogenic mycobacteria (Bean et al., 1999; Flynn et al., 1995; Kaneko et al., 1999). The importance of TNF signaling in protection against human tuberculosis has become especially clear due to the increasing use of TNF-neutralizing drugs in treating a variety of immune and inflammatory conditions such as rheumatoid arthritis (Criscione and St Clair, 2002; Keane, 2005). Patients receiving TNF-neutralizing therapies therapy have an increased rate of reactivation of latent tuberculosis (Gardam et al., 2003). Similarly, TNF is required to prevent active disease in mouse models of latent tuberculosis (Botha and Ryffel, 2003; Chakravarty et al., 2008; Mohan et al., 2001), suggesting it is required for the control of active as well as latent tuberculosis.
TNF can be produced by a variety of immune cells including macrophages, neutrophils, and T-cells (Vassalli, 1992), and has pleiotropic effects in inflammation that affect cell activation and migration, apoptosis, and other biological processes (Locksley et al., 2001). Despite extensive investigation, the effects of the TNF pathway that play consequential roles in tuberculosis pathogenesis remain undefined. In vitro studies using cultured macrophages have implicated TNF in distinct and contradictory roles in mycobacterial pathogenesis, including induction of macrophage apoptosis that in turn leads to mycobacterial death (Fratazzi et al., 1999), activation of macrophage antimicrobial effectors to reduce mycobacterial numbers (Appelberg et al., 1994; Flesch and Kaufmann, 1990), or surprisingly, even enhancing mycobacterial growth (Engele et al., 2002). However, the in vivo studies that followed have led to the prevailing view that TNF is responsible for granuloma formation and maintenance, based on the finding of disorganized granulomas following infection of TNF-deficient mice (Algood et al., 2005; Bean et al., 1999; Chakravarty et al., 2008; Flynn and Chan, 2001; Kindler et al., 1989; Roach et al., 2002; Stenger, 2005). This hypothesis is consistent with its well-known role in orchestrating macrophage trafficking and leukocyte movement during inflammation (Kindler et al., 1989). The correlation of the protective role of TNF with the formation of granulomas, considered to be host protective structures, is also consistent with the concept that tuberculous granulomas benefit the host by containing and restricting mycobacteria (Flynn and Chan, 2001; Lawn et al., 2002; Ulrichs and Kaufmann, 2006). This model is supported by the concomitance of poorly formed granulomas and hypersusceptibility to infection that occur under various immunocompromising conditions in humans and mice (Flynn and Chan, 2001; Kaufmann, 2000; Lawn et al., 2002). More recently, studies of M. bovis, BCG (BCG) infection in TNF and T-cell deficient mice suggested that at least in the context of this attenuated strain, TNF exerts its effects primarily by dampening overactive T cell responses and reducing granuloma and lung tissue destruction, rather than acting during the innate immune phase of the response to mycobacterial infection (Zganiacz et al., 2004). Consistent with this anti-inflammatory role ascribed to TNF, other studies have suggested that TNF deficiency mediates pathology and granuloma destruction in tuberculosis that is independent of the increased bacterial numbers that are also found (Ehlers et al., 1999; Smith et al., 2002).
The accessibility and optical transparency of the developing zebrafish allows for a variety of unique experimental approaches to study pathogenesis, including vital microscopy, whole mount in situ hybridization, and modified antisense oligonucleotides (morpholinos) for functional gene knockdowns (Trede et al., 2004). Zebrafish develop tuberculosis-like disease when infected with Mycobacterium marinum, a natural pathogen of ectotherms and the closest genetic relative of the Mycobacterium tuberculosis complex (http://www.sanger.ac.uk/Projects/M_marinum/) (Stinear et al., 2008). The virulence factors tested to date suggest that M. marinum and M. tuberculosis share virulence mechanisms and effectors (Cosma et al., 2006; Stamm et al., 2003; Tobin and Ramakrishnan, 2008; Volkman et al., 2004). Infection of adult zebrafish produces disease with hallmarks of active human tuberculosis, including caseation necrosis (Swaim et al., 2006). Adult zebrafish have a complex adaptive immune system akin to that of mammals (Traver et al., 2003), and infection with M. marinum is moderated by adaptive immunity similar to the case with mammalian tuberculosis (Swaim et al., 2006). Advantageously, zebrafish can be infected and monitored during early development when adaptive immunity has not yet developed (Trede et al., 2004), allowing for the dissection of the contribution of innate and adaptive immunity to infection (Clay et al., 2007; Davis et al., 2002). M. marinum-infected zebrafish embryos develop disease with essential features of adult tuberculosis, including granuloma formation, showing that the early steps of mycobacterial pathogenesis result from interactions of mycobacteria with the innate immune system (Davis et al., 2002). This model allows for the sequential visualization of infection in single animals (Clay et al., 2007; Cosma et al., 2006; Davis et al., 2002; Volkman et al., 2004).
Using the zebrafish-M. marinum model, we previously demonstrated that TNF is induced in macrophages very early in infection and that innate macrophages can restrict mycobacterial growth without input from the adaptive immune system (Clay et al., 2007). Here, we use morpholinos to ablate TNF signaling to reveal a significant protective effect of TNF in mycobacterial infection that begins early and in the sole context of innate immunity. We find that macrophage trafficking and granuloma formation are not dependent on TNF signaling. Rather the absence of TNF signaling leads to an increase in intracellular bacterial burdens accompanied by accelerated granuloma formation and the necrotic death of infected macrophages within well-formed granulomas. Granuloma necrosis then renders the bacteria extracellular where their unrestricted growth continues to increase bacterial burdens.
Results
Morpholino knockdown of TNF Receptor 1 results in increased susceptibility to M. marinum in the context of innate immunity alone
The zebrafish genome encodes two putative TNF ligands (Zfin Database identification numbers ZDB-GENE-050317-1 and ZDB-GENE-050601-2). Due to higher sequence homology and the upregulation of the latter of these during infection with M. marinum and other pathogens (Clay et al., 2007; Pressley et al., 2005; Rojo et al., 2007), this gene has been identified as the zebrafish TNF homologue. While fluorescent in situ hybridization reveals TNF expression in macrophages (Clay et al., 2007), infected Pu.1 morphant embryos lacking macrophages were found to express TNF by endpoint reverse transcriptase PCR, suggesting that other cell types also produce TNF in response to M. marinum infection (data not shown).
Two splice blocking morpholinos (Draper et al., 2001) were designed against the TNF receptor 1 (TR1) homologue (ZDB-GENE-040426-2252) in order to completely abolish TNF signaling, including any that may occur via the alternate TNF gene in the zebrafish genome (Supplementary Figure 1). Injection of these morpholinos into fertilized eggs resulted in complete abrogation of native TR1 mRNA for five to six days post fertilization (dpf) with partial blocking thereafter for at least 10 dpf (Supplementary figure 1 and data not shown). These TR1 morphant embryos survived similarly to control embryos even when raised under conventional conditions in the presence of their normal commensal flora (Figure 1A) and appeared morphologically normal throughout the observation period. The morphants cleared intravenously injected nonpathogenic Escherichia coli at rates similar to controls in a dose dependent fashion. Both control and TR1 morphant embryos cleared one to ten CFU of E. coli within one day, ~50 CFU within two days, and ~100 CFU within three days of infection with no mortality of the embryos.
Figure 1.

TR1 morphant embryos are more susceptible to mycobacterial infection. (A) Control (Con) and TR1 morphant embryos were either mock injected (n=30 each) or infected with 108 +/- 11 colony forming units (CFU) of M. marinum on 1 dpf (n=50 each). Data are plotted as percentage of surviving embryos on each day. TR1 morphant embryos are significantly more susceptible to infection with M. marinum than controls (Hazard ratio 5.7, p < 0.0001, Kaplan Meier method with log-rank (Mantel-Cox) test). Survival was not statistically different between mock-injected TR1 and control embryos. (B) Mean bacterial loads per embryo for control (Con), TR1, and Pu.1 morphant embryos at 2, 4, and 6 days post injection (dpi) with 70 ± 13 CFU. p < 0.0001 by 1-way ANOVA. Data for Pu.1 embryos are only available for 2 and 4 dpi due to mortality. Error bars represent standard deviation from the mean. All morphant bacterial loads are significantly different from each other (p<0.05) at each individual time points (Control versus TR1, Control versus Pu.1, and TR1 versus Pu.1) by Student’s unpaired t-test. (C) Representative pictures of control, TR1, and Pu.1 morphant embryos at 4 dpi with fluorescence representing bacterial load. Scale bar, 500 μm.
Upon intravenous M. marinum infection, TR1 morphants succumbed to infection significantly faster than their control counterparts, reminiscent of the increased mortality of TNF-deficient mice infected with M. tuberculosis (Figure 1A) (Flynn et al., 1995). The TR1 morphants had a hazard ratio for death of 5.7 over the 12-day observation period and a median time to death of nine days vs. 12 days. Increased mortality of the embryos was also associated with up to ten-fold higher bacterial burdens in the TR1 morphants as compared to control embryos in the first six days (Figure 1B and C), comparable to the increased bacterial numbers seen in TNF-deficient mice early in infection (Flynn et al., 1995; Stenger, 2005). These results show that TNF signaling is important for the modulation of mycobacterial infection from its early stages and does not require adaptive immunity for protective effects in vivo.
TNF signaling is not required for macrophage migration in response to infection
Having confirmed that TNF deficiency in zebrafish embryos leads to the endpoint phenotypes of increased mortality and bacterial growth seen in TNF deficiency in adult mammals, we set out to determine the specific steps of mycobacterial pathogenesis at which TNF acts by taking advantage of the transparency of the zebrafish. As TNF signaling has been implicated in macrophage activation and migration (Vassalli, 1992), we first sought to determine whether or not mycobacteria could recruit phagocytes across epithelial barriers in the absence of TNF signaling by assessing macrophage recruitment to bacteria introduced into the hindbrain ventricle (Davis et al., 2002; Herbomel et al., 1999). The hindbrain ventricle is a neuroepithelial lined cavity that is structurally separated from the circulatory system and the yolk mesenchyme where macrophages arise during early development; this cavity that is normally devoid of macrophages in the first 24 hours post fertilization (Davis et al., 2002; Herbomel et al., 2001). We had previously demonstrated that phagocytes, readily identified by their morphology using Differential Interference Contrast (DIC) microscopy, are specifically recruited to this cavity upon injection of mycobacteria, but not like-sized latex particles (Clay et al., 2007; Davis et al., 2002). Similar numbers of phagocytes were recruited to the hindbrain ventricle after injection of M. marinum into TR1 morphant and control embryos (6 ± 1 macrophages in control and 6 ± 3 macrophages in TR1 morphant embryos) (Supplementary Figure 2). Macrophages phagocytosed the bacteria and migrated back into tissues normally in the TR1 morphants so that within 24 hours of hindbrain ventricle or caudal vein injection, the majority of bacteria had reached deeper tissues within macrophages (Figure 1C and data not shown). These results suggest that TNF signaling is not required for phagocyte trafficking across epithelial and endothelial barriers; macrophages are competent to respond and traffic to peripheral sites of infecting mycobacteria in the absence of TNF signaling.
Granuloma formation is accelerated in the absence of TNF signaling
The developing zebrafish is an ideal host in which to monitor early granuloma formation due to the ability to follow infected macrophages in individual animals over time (Davis et al., 2002; Volkman et al., 2004). Infection of developing zebrafish leads to the formation of macrophage aggregates easily identifiable by DIC microscopy that have characteristic pathological hallmarks of adult tuberculous granulomas, including epithelioid transformation of macrophages (Davis et al., 2002). In addition, several mycobacterial genes that are induced in granulomas of adult animals (granuloma activated genes; gags) are also induced rapidly and selectively upon macrophage aggregation into granulomas in the developing zebrafish (Davis et al., 2002). Hereafter, we will refer to the early granulomas in the developing zebrafish simply as granulomas.
In contrast with our expectation that TNF directs macrophage trafficking into granulomas, DIC microscopy revealed an abundance of granulomas in M. marinum infected TR1 morphant embryos (Figure 1C and 2A). To investigate this surprising result further we assayed granuloma formation by several methods. We first used the fluorescence of M. marinum-expressing transcriptional fusions of GFP to gag7 (RecC) and gag3.13 (homologue of M. tuberculosis Rv0133, a putative N-acetyltransferase) as sensitive indicators of granuloma formation (Davis et al., 2002) in the presence and absence of TNF signaling. Control and TR1 morphant embryos were infected with gag7::gfp and gag3.13::gfp M. marinum and the percentage of embryos with fluorescent bacteria was assessed at four days. A higher proportion of TR1 morphant embryos had formed gag-positive granulomas in comparison to control embryos at four days post infection (dpi) (Figure 2A and B) corroborating our impression from our initial qualitative DIC microscopy.
Figure 2.

Granulomas form faster in the absence of TNF signaling. (A) DIC (left) and fluorescence (right) images indicate activation of granuloma activated genes (gag3.13) in granulomas in both control and morphant embryos. Scale bars, 25 μm. (B) Three pools of ~30 embryos were injected with 56 ± 18 CFU of gag7 and four pools of ~30 embryos were injected with 64 ± 17 CFU of gag3.13. All groups were scored at four dpi for induction of gags as indicated by fluorescent activity. The average percentage of embryos with gag induction is plotted ± standard deviation of the mean. Averages for both gag7 and gag3.13 are significantly different between controls and morphants (p<0.05, Student’s unpaired t-test). (C-E) 23 Control and 14 TR1 morphant embryos were injected with 89 ± 4 CFU and followed sequentially for 5 dpi and monitored for granuloma formation and size. All dark bars represent control morphant data; light bars represent TR1 morphant data. Data is plotted as average ± standard error of the mean. (C) The percentage of embryos with at least one granuloma over time. TR1 morphant embryos have significantly more granulomas at 3 dpi (p<0.01) as analyzed by Fisher’s exact test of a contingency table. (D) The average number of granulomas identified by DIC and fluorescent microscopy over time. (E) TR1 morphant granulomas are significantly larger than control granulomas (p<0.05 at 3 dpi, p<0.0001 at 4 and 5 dpi by Student’s unpaired t-test, n=14 Control and 14 TR1 granulomas at 3 dpi, 75 control and 53 TR1 granulomas at 4 dpi, and 113 control and 72 granulomas at 5 dpi). Y axis represents granuloma diameter in μm. (F-G) Pools of 20 embryos were injected with 27 ± 6 wild-type (WT) and 51 ± 13 ΔRD1 M. marinum and assayed by microscopy at 4dpi for granuloma formation. (F) Representative images of control and TR1 morphant embryos. Arrowheads indicate granulomas. Scale bar, 200μm. (G) The average percentage of embryos with granulomas is plotted plus or minus standard deviation of the mean over four biological replicates. The percentage of embryos with granulomas is significantly higher when injected with WT (p<0.01 for Con; WT versus Con; ΔRD1 and p<0.001 for TR1; WT versus TR1; ΔRD1 by Student’s unpaired t-test).
In order to better characterize the increase in granuloma formation seen in the absence of TNF signaling, we monitored granuloma formation and size over time by DIC and fluorescence microscopy in control and TR1 morphant embryos. TR1 morphant embryos formed granulomas faster than controls (Figure 2C). At all time points, TR1 morphants had more granulomas than control embryos, although this difference was not statistically significant (Figure 2D). Moreover, the average size of granulomas was significantly larger in TR1 morphant embryos at all time points, suggesting a persistent acceleration in granuloma growth beyond the initial aggregation event (Figure 2E).
In the face of the widely held view that TNF signaling mediates granuloma formation (Algood et al., 2005; Flynn and Chan, 2001; Kindler et al., 1989; Lin et al., 2007; Roach et al., 2002), we wanted to investigate further our finding that granuloma formation is not impaired but rather enhanced in the absence of TNF. While the simplest explanation of this finding is that TNF does not mediate granuloma formation, we considered the alternative possibility that TNF signaling does mediate granuloma formation but that this phenotype is obscured by a distinct mechanism of granuloma formation that becomes operant in its absence. To investigate this question, we used the M. marinum mutant deficient in the ESX-1/RD1 secretion locus that produces infection with delayed and defective granulomas (Volkman et al., 2004). We compared granuloma formation by DIC and fluorescent microscopy assaying for discrete foci of tightly packed infected macrophage aggregates in control and TR1 morphant embryos infected with wild-type (WT) and ΔRD1 bacteria at four dpi. The aggregation defect of ΔRD1 was maintained in the presence and absence of TNF signaling (Figure 2F and G). These data showed that granuloma formation proceeds via a mycobacterial RD1-dependent fashion regardless of whether TNF signaling is present. Therefore, the abrogation of TNF signaling does not uncover a distinct pathway of accelerated granuloma formation. Rather these findings suggest that granulomas form in the course of normal infection by a pathway that is independent of TNF signaling. In summary, these data show that rather than preventing macrophage aggregation into granulomas, the loss of TNF signaling leads to an accelerated kinetics of granuloma formation and expansion. Notably, despite the absence of granuloma formation, we found some overall increase in bacterial burdens in the TR1 morphants during ΔRD1 infection (data not shown), suggesting that TNF signaling exerts its protective effect earlier than and independently of granuloma formation. This finding is also consistent with previous studies showing that TNF signaling exerts its protective effect in mice infected with M. bovis BCG that lacks RD1 (Kindler et al., 1989; Zganiacz et al., 2004).
TNF signaling exerts protection in vivo via its macrophage mycobactericidal activity
We have previously demonstrated that macrophages restrict mycobacterial growth from very early on during infection accompanied by TNF induction in infected macrophages (Clay et al., 2007). The increased total bacterial burdens found in TR1 morphant embryos from very early in infection even prior to granuloma formation further suggest that TNF signaling exerts a protective effect in vivo by mediating macrophage microbicidal activities (Figure 1B). This early increase in bacterial numbers in individual macrophages could then lead to accelerated granuloma formation in TR1 morphant embryos. This model is supported by our finding that higher infecting inocula lead to more rapid granuloma formation (data not shown).
We sought to determine if TNF signaling mediated macrophage microbicidal activities by enumerate the numbers of bacteria in individual macrophages during infection (Cosma et al., 2006). However, we were unable to make this determination using wild-type bacteria as even low dose inocula induced accelerated granuloma formation in the TR1 morphant (Figure 3C), with selective recruitment of highly infected macrophages into granulomas where they could not be scored, leaving insufficient numbers of highly infected individual infected macrophages available for analysis. To circumvent the rapid kinetics of granuloma formation with wild-type bacteria in the TR1 morphant, we took advantage of the attenuated M. marinum Erp mutant that is defective for growth in individual macrophages. The intramacrophage growth defect of the Erp mutant is responsible for its attenuation phenotype, which is rescued in Pu.1 morphant embryos lacking macrophages (Clay et al., 2007). Secondary to its macrophage growth defect, the Erp mutant also has reduced granuloma formation (data not shown). Therefore we reasoned that assessing the phenotype of the Erp mutant in TR1 morphant embryos would isolate TNF signaling-mediated macrophage mycobactericidal effects from its other possible protective effects on infection. First, we found overall increased growth of the Erp mutant bacteria in the TR1 morphants as evidenced by increased bacterial burdens by fluorescence microscopy (Figure 3A). Increased bacterial burdens were associated with increased mortality of the TR1 morphant embryos as compared to controls (mean time to death 9.6 vs. 10.8 days, p<0.01 by Student's unpaired t-test). Next we scored the bacterial burdens of individual infected macrophages in the embryos as previously described (Cosma et al., 2006). As predicted, the slower growth of the Erp mutant allowed for individual macrophage burdens to be assessed at a time point before all the macrophages had been recruited into granulomas, thereby allowing for a higher number of infected macrophages to be scored for bacterial burdens, which is not possible after macrophages are tightly aggregated. TR1 morphant embryos infected with Erp mutant bacteria had a significantly higher percentage of macrophages with high intracellular bacterial burdens (scored as more than ten bacteria per macrophage versus low intracellular bacterial burdens as less than ten bacteria per macrophage) compared to control embryos (Figure 3B-D). These results suggest that TNF signaling exerts a protective influence by restricting intracellular mycobacterial growth in vivo. Based on our finding that wild-type bacterial burdens in the TR1 morphants are significantly higher than in controls, but not as high as those in the Pu.1 morphants (Figure 1B and C), we conclude that TNF signaling is upstream of part, but not all, of macrophage effector mechanisms for combating mycobacterial growth in individual macrophages by enabling their microbicidal (or bacteriostatic) activity.
Figure 3.

Growth of Erp mutant bacteria is partially restored in TR1 morphant embryos. (A) Representative fluorescence images for control (Con) and TR1 morphant fish infected with 55 ± 7 CFU of Erp mutant (Erp) M. marinum at 4 dpi. Scale bar, 500 μm. (B-D) 50 individual macrophages were scored for multiplicity of infection (MOI) as either containing 10 or less bacteria (B) or more than 10 bacteria (C) at 3 dpi during infection with 55 ± 7 CFU of Erp mutant bacteria. Scale bar, 25 μm. (D) TR1 morphant embryos had a significantly higher percentage of macrophages with more than ten bacteria as compared to controls (p<0.05, Student’s unpaired t-test).
The effector molecule mostly widely ascribed to combating mycobacterial growth in macrophages is the reactive intermediate nitric oxide, made by the inducible nitric oxide synthase (iNOS)(Kwon, 1997). In order to determine whether or not iNOS is induced during infection of zebrafish embryos with M. marinum, we used fluorescent antibody detection of iNOS protein and co-localized infected cells with iNOS antibody staining. Both control and TR1 morphant embryos displayed iNOS staining that co-localized with a subset of infected macrophages (Supplementary Figure 3). We were unable to determine any differences in staining between control and TR1 morphant embryos, suggesting that TNF signaling is not required for iNOS expression in our system, consistent with previous reports in other systems (Bean et al., 1999; Bekker et al., 2000; Ehlers et al., 1999).
Nonapoptotic death of granuloma macrophages is increased in the absence of TNF
Our sequential imaging of live infected fish showed that while granulomas formed rapidly and appeared normal at first in the TR1 morphant embryos (Figure 2A), a combination of fluorescence and DIC microscopy revealed that they began to appear larger and more acellular than control granulomas soon after they had formed. To confirm this observation, we developed a quantitative assays to assess the cellularity of granulomas in control versus TR1 morphants using colorimetric whole mount in situ hybridization (WISH) analysis labeling the fms gene, a macrophage-specific marker encoding the macrophage colony stimulating factor receptor (M-CSFR) (Clay, 2005; Parichy et al., 2000). Only live macrophages will express fms mRNA and therefore hybridize to the probe (Figure 4A-B). WISH analysis was performed on control and TR1 morphant six dpi embryos. Granulomas were scored visually using DIC microscopy to identify bacterial masses in a blinded assay as being mostly cellular (greater than one third of the granuloma mass hybridizing to the fms probe) (Figure 4A) or acellular (one third or less of the granuloma mass hybridizing to the fms probe) (Figure 4B). Scoring of all of the individual granulomas from the two groups showed that twice as many of the TR1 morphant granulomas were acellular as compared to granulomas from control embryos (Figure 4C). While fms staining did not appear different between uninfected TR1 and control morphant embryos, there did appear to be an overall reduction in fms-positive cells in infected TR1 morphants (data not shown), suggesting that the accelerated macrophage death resulting from the loss of TNF signaling leads to their overall depletion.
Figure 4.

TR1 morphant embryos have significantly more acellular granulomas than controls. (A, B) Differential interference contrast overlay with fluorescence (left) of infected embryos then labeled using in situ hybridization for the macrophage marker fms (right). Green indicates M. marinum in fluorescent overlays. Purple staining of hybridized embryos indicates expression of the macrophage marker fms. (A) An example of a cellular granuloma in a control embryo before and after in situ hybridization indicating bacteria within aggregated macrophages. (B) An acellular granuloma example in a TR1 morphant embryo indicating bacteria found primarily outside of macrophages. (C) The results of a blind scoring of 82 granulomas in 22 WT embryos and 78 granulomas in 22 TR1 morphant embryos for cellularity indicates that TR1 granulomas were twice as likely to contain 1/3 or fewer fms-positive cells than to WT granulomas (p<0.0001) as analyzed by Fisher’s exact test of a contingency table. Embryos were 7 dpf and infected with 267 ± 28 CFU.
TNF has been shown to modulate cell death in inflammation by both pro and anti-apoptotic effects, and in vitro studies have suggested that TNF mediates apoptosis of Mycobacterium-infected macrophages which in turn results in death of the resident bacteria (Fratazzi et al., 1999; Stenger, 2005). Since the TR1 morphants had increased cell death associated with exuberant bacterial growth, we wondered if there was due to a reduction in apoptosis caused by the absence of TNF signaling that channels infected cells into a default pathway of necrotic death. Necrosis of infected cells, in contrast to apoptotic death, is postulated to allow the bacteria to survive (Fratazzi et al., 1999). We have previously shown that some infected macrophages within granulomas undergo apoptosis as indicated by the terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labeling (TUNEL) assay that labels double strand DNA breaks (Volkman et al., 2004). We found that several of these TUNEL-positive macrophages express TNF as judged by dual fluorescent labeling of whole embryos using in situ hybridization for TNF expression and TUNEL labeling for apoptotic cells (Clay, 2005)(Figure 5A-D). This finding suggests that the TNF pathway could be modulating the apoptosis that occurs in granuloma macrophages. However we found no difference in the number of TUNEL-positive cells in like-sized granulomas of control and TR1 morphant embryos (Figure 5E). In contrast, we found the expected reduction in TUNEL positive cells in granulomas that formed as a result of RD1-deficient M. marinum (ΔRD1) infection (Figure 5E) (Volkman et al., 2004). Next, we scored granulomas for either the presence or absence of TUNEL-positive cells and compared TR1 morphant and control embryos for the percentage of total granulomas that had at least one TUNEL-positive cell and again found no difference between the two groups (Figure 5F). Recognizing that some forms of apoptosis do not lead to detectable double stranded DNA breaks (Huerta et al., 2007), these data suggest that TNF signaling does not modulate apoptotic macrophage death within granulomas. Furthermore, the increased cell death occurring in the absence of TNF signaling as evidenced by the acellular granulomas is independent of changes in TUNEL-positive apoptotic cell death.
Figure 5.

TNF signaling does not influence the rate of apoptosis of infected cells. DIC (A) and fluorescent (B-D) imaging of a granuloma. (B) Colocalization of TNF expression (red) and TUNEL-labeled double strand DNA breaks (green). Individual fluorescence channels for TNF expression (C) and TUNEL-labeling (D) are shown. Scale bar, 50 μm. (E) The average number of TUNEL-positive cells within granulomas in 4 dpi embryos is unchanged between control (n=16 granulomas) and TR1 morphant embryos (n=20). Infection of control embryos with RD1-deficient bacteria (ΔRD1) is used as a control (n=20). The number of TUNEL-positive cells in ΔRD1 granulomas is significantly less than control and TR1 morphant granulomas with wild-type bacteria (p<0.05 by Student’s unpaired t-test for both comparisons). Granulomas were selected to be between 40 and 50μm in diameter to normalize for total cell number. (F) The percentage of TUNEL-positive granulomas between control and TR1 morphant 4 dpi embryos is unchanged. Three separate experiments of pools of 20–40 granulomas were scored per condition and plotted as number of granulomas with TUNEL-positive cells over total granuloma number. Error bars represent standard error of the mean.
Extracellular corded mycobacteria accumulate in the TR1 morphants leading to accelerated bacterial growth and spread
On imaging live embryos, we had found that concomitant with the loss of granuloma cellularity, the mycobacteria took on a corded appearance. Cording morphology, where the bacteria are intertwined into serpentine rope-like structures (Koch, 1882), is a distinctive feature of M. tuberculosis grown in culture (Darzins and Fahr, 1956; Dubos and Pierce, 1956; Middlebrook et al., 1947). Genetic mutations in M. tuberculosis that abolish cording in vitro also result in reduced virulence (Bhatt et al., 2007; Glickman et al., 2000). However, while cording in vitro is tightly correlated to virulence, this phenotype has not been observed in vivo previously. In this light, we were first struck by the appearance of corded bacteria in the Pu.1 morphants (Clay et al., 2007) where the bacteria are always extracellular (Figure 6A). In contrast, bacteria within macrophages do not exhibit this phenotype, suggesting that cording is a property of extracellular bacteria regardless of whether they are in vitro or in vivo (Figure 6B). While not all extracellular bacteria demonstrate a cording phenotype, it serves as a conservative estimate of whether or not bacteria are growing extracellularly in vivo. We found that the percentage of embryos with corded bacteria was significantly higher in TR1 morphants than in control embryos from very early in infection (Figure 6C-G). These results correlate with our findings of increased macrophage death in the absence of TNF signaling. Taken together, these data indicate that the absence of TNF signaling leads to increased macrophage death and necrotic breakdown of granulomas with resultant exuberant growth of extracellular mycobacteria. Notably, corded bacteria were not always restricted to the acellular granulomas. Individual clumps of corded bacteria were found that likely represent those released from a single dead macrophage (Figure 6E). This result suggests that TNF pathway deficiency produces accelerated death of both individual and granuloma macrophages with release of extracellular bacteria.
Figure 6.
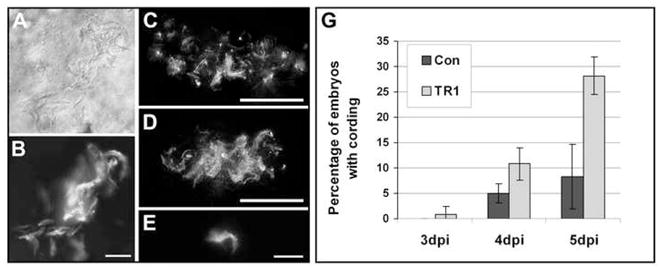
TR1 morphant embryos are more likely to have extracellular bacteria as indicated by cording. DIC (A) and fluorescence (B-E) imaging of bacteria in vivo. (AB) Extracellular bacteria in Pu.1 morphant embryos without macrophages displays the cording phenotype. Scale bar, 25 μm. (C-D) Flattened three-dimensional z-stacks of control (C) and TR1 morphant (D) granulomas demonstrate cording in TR1 morphant embryos. Scale bars, 50 μm. (E) Cording after what appears to be breakdown of an individual infected macrophage in infected TR1 morphant embryo. Scale bar, 25 μm. (G) Four pools of 30 embryos were injected with 18 ± 5 CFU and scored daily for the presence of extracellular bacteria as indicated by cording and plotted as the percentage of embryos with cording plus or minus the standard deviation. TR1 morphant embryos had significantly higher percentages of embryos with cording at all time points by Student’s unpaired t-test (p<0.05 for 3 and 4 dpi, p<0.01 for 5 dpi).
To further dissect the pathway of the accelerated cell death occurring in the absence of TNF, we again used the M. marinum ESX-1/RD1 mutant. Infection of macrophages with the ESX-1/RD1 virulence locus in vitro produces increased cell death (Guinn et al., 2004; Hsu et al., 2003; Kaku et al., 2007). In the zebrafish, infection with ΔRD1 M. marinum results in fewer granulomas that contain TUNEL-positive cells (Volkman et al., 2004). In order to test whether or not bacterial RD1 is required for the increased macrophage cell death seen in the absence of TNF signaling, we infected control and TR1 morphant embryos with ΔRD1 and looked for the presence of cording bacteria. Despite the fact that ΔRD1 M. marinum infection remains partially attenuated in TR1 morphant embryos presumably due to their deficiency in forming granulomas (Figure 2F and G), infection of these morphants with higher doses of this attenuated strain revealed increased levels of cording bacteria in TR1 morphant embryos (Supplementary Figure 4A). Consistent with the formation of poor granulomas, the majority of corded bacterial masses were smaller than those found during wild-type infection (Compare Supplementary Figure 4B and C to Figure 6D). This finding corroborated our observations with wild-type bacteria that individual macrophages with accelerated necrosis could be found outside of granulomas (Figure 6E). In wild-type infection, the accelerated formation of granulomas makes it more likely that macropahge necrosis occurs within granulomas. However, it is clear from these experiments that granuloma formation is not a prerequisite for macrophage necrosis. Taken together, these data indicate that increased cell death resulting from the lack of TNF signaling is not dependent on the ΔRD1 virulence locus or granuloma formation.
Additionally, time-lapse microscopy revealed several individual infected dead macrophages and even demonstrated that a single dead macrophage may provide a means for dissemination of infection (Supplemental Figure 5 and Movie 1). In this example, a single infected macrophage in a TR1 morphant embryo is phagocytosed by two separate uninfected macrophages, indicating that the death of single infected host cells is capable of spreading bacteria to multiple uninfected macrophages. Thus, infected macrophage cell death could accelerate infection both by giving rise to extracellular bacteria as well as spreading infection to new macrophages. The ability of bacteria to remain extracellular in later stages of infection likely reflects a saturation effect where uninfected macrophages available for rephagocytosis of extracellular bacteria are unable to keep up with the rate of bacterial growth and infected macrophage death.
Discussion
Our detailed stepwise dissection of the consequences of TNF Receptor loss to mycobacterial pathogenesis shows that an accelerated increase in bacterial numbers occurs from the very early stages of infection. Our prior work had demonstrated that innate macrophages are capable of limiting mycobacterial growth to a considerable extent and that TNF is induced in infected macrophages within 24 hours post infection (Clay et al., 2007). The finding that TR1 morphant embryos have an accelerated bacterial burden during this early phase of infection suggests that the TNF pathway plays a role in this early mycobacterial growth restriction by macrophages. In support of this conclusion, we find that the specific macrophage growth defect of a mycobacterial mutant is partially rescued during infection of TNF-deficient embryos. Our finding that TNF is protective in early tuberculosis confirms experimental infection data in adult mammalian infection (Stenger, 2005), but is the first formal demonstration that TNF signaling can exert its protective effects in vivo in the absence of adaptive immunity. By demonstrating that a key host protective determinant can exert its effects in the context of innate immunity alone, these results add to the growing appreciation for a substantial role for innate immunity as an autonomous mediator of tuberculosis control aside from serving as the effector arm of adaptive immunity (Berrington and Hawn, 2007; Clay et al., 2007; Pan et al., 2005; Tosh et al., 2006; van Crevel et al., 2002)
Consistent with other experimental models of mycobacterial infection, we find that loss of TNF signaling leads to an increase in necrotic lesions and a loss of properly formed granulomas (Algood et al., 2005; Bean et al., 1999; Chakravarty et al., 2008; Flynn et al., 1995; Kindler et al., 1989; Roach et al., 2002). Importantly, we demonstrate that granuloma breakdown can occur independently of T cell activity. The disorganized granulomas seen in the absence of TNF signaling have been attributed to defects in the primary formation of granulomas (Flynn and Chan, 2001; Lin et al., 2007; Stenger, 2005). Our ability to monitor the sequential steps of early infection has uncovered that the loss of TNF signaling during M. marinum infection of zebrafish embryos leads to altered kinetics of granuloma formation, with granulomas forming more rapidly. This may be because TNF signaling may impede granuloma initiation by restricting bacterial growth early in infection. Conversely, TNF signaling deficiency causes rapid and progressive disruption of granuloma structure due to increased granuloma expansion and the accelerated necrosis of participating macrophages. Our data are consistent with the previous finding that TNF receptor deficient mice infected with M. tuberculosis formed granulomas in equal numbers to control mice when observed at early time points, but that only the TNF deficient granulomas underwent subsequent necrosis (Flynn et al., 1995). Other studies using Mycobacterium avium have also noted that granulomas are formed, but that they rapidly disintegrate (Benini et al., 1999; Ehlers et al., 2000). Thus the deficiencies in granuloma formation noted in many studies may be explained by their structural instability rather than a primary deficit in early granuloma formation (Bean et al., 1999; Kindler et al., 1989; Roach et al., 2002). Alternatively, TNF may have additional effects on granuloma structure in the context of adaptive immunity by influencing processes such as T-cell trafficking and activation, and these changes may independently alter cell recruitment and structural organization in mature granulomas. This hypothesis is supported by recent work demonstrating that TNF is essential for retaining T cells, but not infected macrophages, within established granulomas (Egen et al., 2008).
Our data suggest a model wherein TNF deficiency first leads directly to impaired macrophage defenses that result in increased intracellular bacterial burdens (Figure 7). The macrophage effector mechanisms downstream of TNF signaling responsible for restricting mycobacterial growth are not well characterized. iNOS activity confers resistance to mycobacterial infection in mice (Adams et al., 1997), and TNF has been found to synergize with IFN-γ to activate macrophages to produce inducible nitric oxide synthase iNOS, facilitating macrophage killing of intracellular M. tuberculosis (Ding et al., 1988; Flesch et al., 1994). However, our results as well as previous reports (Bean et al., 1999; Bekker et al., 2000; Ehlers et al., 1999) indicate that TNF signaling mediates macrophage microbicidal activities in the context of innate immunity are likely not mediated by differences in nitric oxide production resulting from iNOS activity. Other known anti-mycobacterial mechanisms include phagolysosomal fusion (Armstrong and Hart, 1971), vitamin D-mediated bacterial killing (Liu et al., 2006) (Martineau et al., 2007), the production of reactive oxygen intermediates (Adams et al., 1997), and production and release of defensins (Miyakawa et al., 1996), small antimicrobial peptides thought to be responsible for disrupting bacterial cell wall integrity (Menendez and Brett Finlay, 2007).
Figure 7.
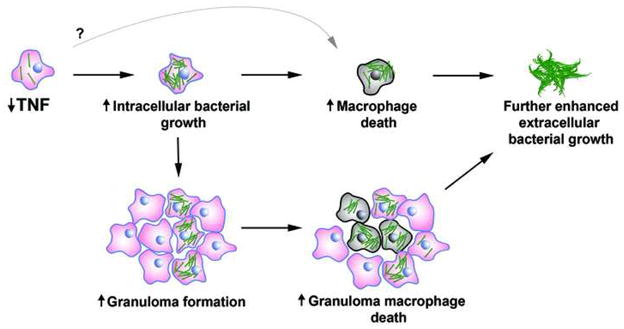
Conceptual overview of how TNF deficiency leads to changes during mycobacterial pathogenesis. TNF deficiency leads to increased bacterial growth and macrophage death, which in turn lead to the release of bacteria into the extracellular milieu and granuloma breakdown.
In addition to an increase in intracellular bacterial burdens in the absence of TNF signaling, there is also an accelerated necrotic death of infected macrophages both within and outside of granulomas. This increased cell death may be secondary to the accelerated increase in their bacterial burdens. The increased size of granulomas that form without TNF signaling suggests that increased bacterial burdens leads to increased numbers of heavily infected macrophages, and that this increased bacterial burden ultimately leads to macrophage death and depletion. Alternatively, it is possible that the loss of TNF signaling initiates cell death via other mechanisms. The macrophage necrosis we observe may well result from the induction of the lysosomal death pathway, a caspase-independent cell death pathway that resembles necrotic rather than apoptotic cell death (Lee et al., 2006; O'Sullivan et al., 2007). This pathway is induced by infection with M. tuberculosis only when bacterial burdens within macrophages are high, thus providing a potential link between the increased bacterial burdens and necrotic cell death that we observe (Lee et al., 2006; O'Sullivan et al., 2007). While high intramacrophage bacterial burdens in the absence of TNF signaling may be sufficient to induce the lysosomal death pathway, there are multiple places where TNF signaling and its downstream effectors could potentially intersect with and influence it (Broker et al., 2005; Liu et al., 2003).
Regardless of whether increased macrophage death is a primary or secondary effect of the loss of TNF signaling, it confers a further growth advantage to the bacteria by rendering them extracellular (Clay et al., 2007). Therefore in the absence of TNF signaling, bacterial growth is enhanced at two stages: first within macrophages whose microbicidal capacity is significantly but not completely abolished, and then by the death of the infected macrophages that leads to a further enhancement in bacterial replication by eliminating remaining macrophage defense mechanisms and mimicking the phenotype seen in Pu.1 morphant embryos, where bacterial growth in the extracellular milieu is unrestricted (Clay et al., 2007). In addition, the observed acceleration of granuloma formation and death of infected macrophages both have the potential to increase net bacterial burdens by increasing cell to cell spread (Volkman et al., 2004)(Supplementary Movie 1). Therefore our work has isolated a primary role of TNF in restricting bacteria within and to macrophages during early infection. The accelerated macrophage death that ensues further promotes bacterial growth and could also independently influence disease pathology at later stages by contributing directly to tissue destruction as well as by disrupting chemokine gradients and T-cell activation, as noted during mycobacterial infection of adult animals in the absence of TNF signaling (Algood et al., 2005; Roach et al., 2002; Smith et al., 2002; Zganiacz et al., 2004).
The importance of maintaining TNF signaling during mycobacterial infection has been dramatically demonstrated by the use of anti-TNF therapeutics during treatment of chronic autoimmune disorders such as rheumatoid arthritis and Crohn’s disease, which have demonstrated a particularly increased risk of tuberculosis (Gardam et al., 2003; Keane, 2005). Due to the kinetics of when patients develop symptoms of active tuberculosis, the use of anti-TNF therapeutics has been hypothesized to result from reactivation of latent disease rather than an increased susceptibility to primary infection (Gomez-Reino et al., 2007; Keane, 2005). Notably, a number of clinical case studies of reactivated tuberculosis under these conditions that have reported discrete well formed granulomas in patient biopsies (Garcia Vidal et al., 2005; Iliopoulos et al., 2006; Lange et al., 2007; Taylor et al., 2003; Vlachaki et al., 2005; Wagner et al., 2002). Consistent with our results that a loss of TNF signaling leads to increased cell death, caseous necrosis was also found to be a major histological feature of granulomas noted in these case studies. As the formation of caseum is a hallmark of active disease, these results suggest that the primary role for TNF signaling in human tuberculosis is also that of promoting macrophage survival. Our work has identified discrete steps of mycobacterial pathogenesis affected by this pleiotropic cytokine. Countering the specific deficits that occur during TNF blockade with downstream effectors has the potential to curtail the incidence of tuberculosis that occurs during TNF blockade. More importantly, a better understanding of TNF function may lead to a better understanding of how to enhance host defenses designed to control mycobacterial infection.
Experimental Procedures
Animal care and strains
Wild-type WIK zebrafish embryos were maintained and infected with bacteria as described (Cosma et al., 2006; Davis et al., 2002; Volkman et al., 2004). Survival curves were performed by separating embryos into 15mL plastic petri dishes (10-15 embryos per dish) with half water changes twice daily, with light cycles and feeding with paramecia starting on five dpf.
Bacterial strains
All bacterial strains are wild-type Mycobacterium marinum strain M unless otherwise indicated. Fluorescent wild-type bacteria was used and prepared as described (Davis et al., 2002). Erp mutant (Cosma et al., 2006) and ΔRD1 (Volkman et al., 2004) mutant strains were prepared as described.
Microscopy
Widefield microscopy was performed on a Nikon E600 equipped with DIC optics, a Nikon D-FL-E fluorescence unit with 100W Mercury lamp and MFC-1000 z-step controller (Applied Scientific Instrumentation). Objectives used included 10× Plan Fluor, 0.3 NA, 20× Plan Fluor, 0.5 NA, 40× Plan Fluor, 0.75 NA, and 60× water Fluor, 1.0 NA. Widefield fluorescence and DIC images were captured on a CoolSnap HQ CCD camera (Photometrics) using MetaMorph 7.1 (Molecular Devices).
Three-dimensional image processing
Where indicated, z-stacks were deconvolved using AutoDeblur Gold CWF, Version X1.4.1 (Media Cybernetics), with default settings for blind deconvolution.
Bacterial CFU enumeration
CFU counts were taken as described (Clay et al., 2007) with the following changes: embryos were dissociated in 100μL PBS by grinding with a microtube pestles (USA Scientific). Due to the increased susceptibility of the Erp mutant bacteria to the decontamination protocol used for plating fish lysates, bacterial levels could only be examined by fluorescence microscopy, which we have previously demonstrated correlate with bacterial loads (Clay et al., 2007).
Morpholinos
Morpholinos were obtained from Genetools and injected at the one to four cell stage. Control and Pu.1 morpholinos were used as described (Clay et al., 2007). TNF Receptor morpholinos were used as described (Bates et al., 2007). Morpholino controls used were wild-type WIK embryos (experiments for Figures 2B and 4, and Supplementary Figure 1), Genetool’s control morpholino (experiments for Figures 1, 2A, 3, 5, and 6, and Supplementary Figures 4 and 5), and the pbx-mutant morpholino (Hernandez et al., 2004) (experiments for Supplementary Figures 2 and 3).
In situ hybridization
Fluorescent in situ hybridization and TUNEL labeling was performed as described (Clay, 2005), with fms probe and colorometric detection as described (Parichy et al., 2000).
Antibody staining
Antibody staining for iNOS was performed with TSA detection as described (Clay, 2005) using a rabbit polyclonal antibody (BD Biosciences) shown previously to cross-react with zebrafish iNOS (Shin et al., 2000).
Statistics
Student’s unpaired t-tests and contingency table analysis were performed using In-Stat software (Graphpad Software, Inc). Kaplan-meier analysis was performed using Prism (Graphpad Software, Inc).
Supplementary Material
Supplementary Figure 1. Morpholino targeting of the TNF Receptor 1 (TR1) leads to complete splice blocking of transcript for 4 days. Two splice blocking morpholinos targeting the exon-intron boundaries of E2I2 and E4I4 were used in combination to maximize disruption of transcript. Control embryos at 2 days (d) and 4d post fertilization in comparison with TR1 morphant transcripts (2d-4d) show splice blocking of native transcript during this time period.
Supplementary Figure 2. TNF signaling is not required for recruitment of phagocytes to bacteria. Number of phagocytes recruited to the hindbrain ventricle 3 hours after the injection of 50 ± 10 CFU of M. marinum for control, TR1, and Pu.1 morphant embryos. Phagocytes are enumerated by scanning through the hindbrain ventricle and identified by their morphology. Pu.1 morphants are used as a negative control as they lack macrophages and have reduced numbers of neutrophils. Medians are indicated by bars. Control and TR1 morphant embryos recruited significantly more phagocytes than Pu.1 morphant embryos (p<0.0001). Number of recruited macrophages was not different between control and TR1 embryos.
Supplementary Figure 3. Inducible nitric oxide (iNOS) antibody staining is similar in infected control and TR1 morphant embryos. Fluorescent detection of iNOS antibody shows colocalization with fluorescent Mycobacterium marinum (MM). Scale bar, 50 μm.
Supplementary Figure 4. Increased cording seen in the absence of TNF signaling is not dependent on the RD1 virulence locus or granuloma formation. (A) Control (n = 23) and TR1 morphant (n = 9) embryos were infected with 353 ± 10 ΔRD1 bacteria and scored daily for the presence of extracellular bacteria as indicated by cording and plotted as the percentage of embryos with cording. A significantly higher percentage of TR1 morphant embryos display cording versus control embryos at 5 dpi (p<0.01) and 6 dpi (p<0.05) as analyzed by Fisher’s exact test of a contingency table. (B and C) Examples of cording found during infection of TR1 morphant embryos infected with ΔRD1 at 6 dpi. Scale bar, 25 μm.
Supplementary Figure 5. Macrophage death can lead to dissemination of infection. All images are stills taken from a two hour timelapse of an infected TR1 morphant fish at four dpi. A single dead infected macrophage is rephagocytosed by two separate macrophages. When available, flattened three-dimensional fluorescent stacks are provided to indicate position of bacteria (left column). Time is shown in minutes. The original macrophage is indicated by left-facing bracket, first incoming macrophage is indicated by upward-facing bracket, and seconding incoming macrophage is indicated by right-facing bracket.
Supplementary Movie 1. Macrophage death can lead to dissemination of infection to multiple uninfected macrophages. A two hour timelapse of a TR1 morphant fish was taken at 4 days post infection and shows the rephagocytosis of a single dead infected macrophage into two separate incoming macrophages. The original macrophage is indicated by left-facing bracket, first incoming macrophage is indicated by upward-facing bracket, and seconding incoming macrophage is indicated by right-facing bracket. Movie is paused at 14 and 104 minutes to demonstrate location of separate macrophages.
Acknowledgments
We thank Robin Lesley, Kevin Urdahl, and Paul Edelstein for invaluable discussions and review of the manuscript, Ferric Fang and Tony Richardson for advice and help with the nitric oxide studies, David Tobin for first identifying the cording phenotype as a measure of extracellular bacterial growth in vivo, Paul Edelstein for advice and help on statistical analyses of the data, Muse Davis for help with video assembly, Richard Burmeister for help with figure graphics, and Laura Swaim and Heather Wiedenhoft for managing the zebrafish facility. This work was funded by NIH grants RO1 AI036396 and RO1 AI54503 and a Burroughs Wellcome Fund Award to LR. HC was funded in part by PHS NRSA T32 GM07270 from the National Institute of General Medical Sciences. HEV was funded in part by an American Heart Association predoctoral fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams LB, Dinauer MC, Morgenstern DE, Krahenbuhl JL. Comparison of the roles of reactive oxygen and nitrogen intermediates in the host response to Mycobacterium tuberculosis using transgenic mice. Tuber Lung Dis. 1997;78:237–246. doi: 10.1016/s0962-8479(97)90004-6. [DOI] [PubMed] [Google Scholar]
- Algood HM, Lin PL, Flynn JL. Tumor necrosis factor and chemokine interactions in the formation and maintenance of granulomas in tuberculosis. Clin Infect Dis. 2005;41(Suppl 3):S189–193. doi: 10.1086/429994. [DOI] [PubMed] [Google Scholar]
- Appelberg R, Castro AG, Pedrosa J, Silva RA, Orme IM, Minoprio P. Role of gamma interferon and tumor necrosis factor alpha during T-cell-independent and -dependent phases of Mycobacterium avium infection. Infect Immun. 1994;62:3962–3971. doi: 10.1128/iai.62.9.3962-3971.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong JA, Hart PD. Response of cultured macrophages to Mycobacterium tuberculosis, with observations on fusion of lysosomes with phagosomes. J Exp Med. 1971;134:713–740. doi: 10.1084/jem.134.3.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates JM, Akerlund J, Mittge E, Guillemin K. Intestinal Alkaline Phosphatase Detoxifies Lipopolysaccharide and Prevents Inflammation in Zebrafish in Response to the Gut Microbiota. Cell Host & Microbe. 2007 doi: 10.1016/j.chom.2007.10.010. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean AG, Roach DR, Briscoe H, France MP, Korner H, Sedgwick JD, Britton WJ. Structural deficiencies in granuloma formation in TNF gene-targeted mice underlie the heightened susceptibility to aerosol Mycobacterium tuberculosis infection, which is not compensated for by lymphotoxin. J Immunol. 1999;162:3504–3511. [PubMed] [Google Scholar]
- Bekker LG, Moreira AL, Bergtold A, Freeman S, Ryffel B, Kaplan G. Immunopathologic effects of tumor necrosis factor alpha in murine mycobacterial infection are dose dependent. Infect Immun. 2000;68:6954–6961. doi: 10.1128/iai.68.12.6954-6961.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benini J, Ehlers EM, Ehlers S. Different types of pulmonary granuloma necrosis in immunocompetent vs. TNFRp55-gene-deficient mice aerogenically infected with highly virulent Mycobacterium avium. J Pathol. 1999;189:127–137. doi: 10.1002/(SICI)1096-9896(199909)189:1<127::AID-PATH398>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Berrington WR, Hawn TR. Mycobacterium tuberculosis, macrophages, and the innate immune response: does common variation matter? Immunol Rev. 2007;219:167–186. doi: 10.1111/j.1600-065X.2007.00545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt A, Fujiwara N, Bhatt K, Gurcha SS, Kremer L, Chen B, Chan J, Porcelli SA, Kobayashi K, Besra GS, Jacobs WR., Jr Deletion of kasB in Mycobacterium tuberculosis causes loss of acid-fastness and subclinical latent tuberculosis in immunocompetent mice. Proc Natl Acad Sci U S A. 2007;104:5157–5162. doi: 10.1073/pnas.0608654104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botha T, Ryffel B. Reactivation of latent tuberculosis infection in TNF-deficient mice. J Immunol. 2003;171:3110–3118. doi: 10.4049/jimmunol.171.6.3110. [DOI] [PubMed] [Google Scholar]
- Broker LE, Kruyt FA, Giaccone G. Cell death independent of caspases: a review. Clin Cancer Res. 2005;11:3155–3162. doi: 10.1158/1078-0432.CCR-04-2223. [DOI] [PubMed] [Google Scholar]
- Chakravarty SD, Zhu G, Tsai MC, Mohan VP, Marino S, Kirschner DE, Huang L, Flynn J, Chan J. Tumor necrosis factor blockade in chronic murine tuberculosis enhances granulomatous inflammation and disorganizes granulomas in the lungs. Infect Immun. 2008;76:916–926. doi: 10.1128/IAI.01011-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clay H, Davis JM, Beery D, Huttenlocher A, Lyons SE, Ramakrishnan L. Dichotomous Role of the Macrophage in Early Mycobacterium marinum Infection of the Zebrafish. Cell Host & Microbe. 2007;2:29–39. doi: 10.1016/j.chom.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clay H, Ramakrishnan L. Multiplex Fluorescent in situ hybridization in zebrafish embryos using tyramide signal amplification. Zebrafish. 2005;2:105–111. doi: 10.1089/zeb.2005.2.105. [DOI] [PubMed] [Google Scholar]
- Cosma CL, Klein K, Kim R, Beery D, Ramakrishnan L. Mycobacterium marinum Erp is a virulence determinant required for cell wall integrity and intracellular survival. Infect Immun. 2006;74:3125–3133. doi: 10.1128/IAI.02061-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosma CL, Sherman DR, Ramakrishnan L. The secret lives of the pathogenic mycobacteria. Annu Rev Microbiol. 2003;57:641–676. doi: 10.1146/annurev.micro.57.030502.091033. [DOI] [PubMed] [Google Scholar]
- Criscione LG, St Clair EW. Tumor necrosis factor-alpha antagonists for the treatment of rheumatic diseases. Curr Opin Rheumatol. 2002;14:204–211. doi: 10.1097/00002281-200205000-00002. [DOI] [PubMed] [Google Scholar]
- Dannenberg AM., Jr Immunopathogenesis of pulmonary tuberculosis. Hosp Pract (Off Ed) 1993;28:51–58. doi: 10.1080/21548331.1993.11442738. [DOI] [PubMed] [Google Scholar]
- Darzins E, Fahr G. Cord-forming property, lethality and pathogenicity of Mycobacteria. Dis Chest. 1956;30:642–648. doi: 10.1378/chest.30.6.642. [DOI] [PubMed] [Google Scholar]
- Davis JM, Clay H, Lewis JL, Ghori N, Herbomel P, Ramakrishnan L. Real-time visualization of mycobacterium-macrophage interactions leading to initiation of granuloma formation in zebrafish embryos. Immunity. 2002;17:693–702. doi: 10.1016/s1074-7613(02)00475-2. [DOI] [PubMed] [Google Scholar]
- Ding AH, Nathan CF, Stuehr DJ. Release of reactive nitrogen intermediates and reactive oxygen intermediates from mouse peritoneal macrophages. Comparison of activating cytokines and evidence for independent production. J Immunol. 1988;141:2407–2412. [PubMed] [Google Scholar]
- Draper BW, Morcos PA, Kimmel CB. Inhibition of zebrafish fgf8 pre-mRNA splicing with morpholino oligos: a quantifiable method for gene knockdown. Genesis. 2001;30:154–156. doi: 10.1002/gene.1053. [DOI] [PubMed] [Google Scholar]
- Dubos RJ, Pierce CH. Differential characteristics in vitro and in vivo of several substrains of BCG. II. Morphologic characteristics in vitro and in vivo. Am Rev Tuberc. 1956;74:667–682. doi: 10.1164/artpd.1956.74.5.667. [DOI] [PubMed] [Google Scholar]
- Egen JG, Rothfuchs AG, Feng CG, Winter N, Sher A, Germain RN. Macrophage and T cell dynamics during the development and disintegration of mycobacterial granulomas. Immunity. 2008;28:271–284. doi: 10.1016/j.immuni.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers S, Benini J, Kutsch S, Endres R, Rietschel ET, Pfeffer K. Fatal granuloma necrosis without exacerbated mycobacterial growth in tumor necrosis factor receptor p55 gene-deficient mice intravenously infected with Mycobacterium avium. Infect Immun. 1999;67:3571–3579. doi: 10.1128/iai.67.7.3571-3579.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers S, Kutsch S, Ehlers EM, Benini J, Pfeffer K. Lethal granuloma disintegration in mycobacteria-infected TNFRp55−/− mice is dependent on T cells and IL-12. J Immunol. 2000;165:483–492. doi: 10.4049/jimmunol.165.1.483. [DOI] [PubMed] [Google Scholar]
- Engele M, Stossel E, Castiglione K, Schwerdtner N, Wagner M, Bolcskei P, Rollinghoff M, Stenger S. Induction of TNF in human alveolar macrophages as a potential evasion mechanism of virulent Mycobacterium tuberculosis. J Immunol. 2002;168:1328–1337. doi: 10.4049/jimmunol.168.3.1328. [DOI] [PubMed] [Google Scholar]
- Flesch IE, Hess JH, Oswald IP, Kaufmann SH. Growth inhibition of Mycobacterium bovis by IFN-gamma stimulated macrophages: regulation by endogenous tumor necrosis factor-alpha and by IL-10. Int Immunol. 1994;6:693–700. doi: 10.1093/intimm/6.5.693. [DOI] [PubMed] [Google Scholar]
- Flesch IE, Kaufmann SH. Activation of tuberculostatic macrophage functions by gamma interferon, interleukin-4, and tumor necrosis factor. Infect Immun. 1990;58:2675–2677. doi: 10.1128/iai.58.8.2675-2677.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn JL, Chan J. Immunology of tuberculosis. Annu Rev Immunol. 2001;19:93–129. doi: 10.1146/annurev.immunol.19.1.93. [DOI] [PubMed] [Google Scholar]
- Flynn JL, Goldstein MM, Chan J, Triebold KJ, Pfeffer K, Lowenstein CJ, Schreiber R, Mak TW, Bloom BR. Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity. 1995;2:561–572. doi: 10.1016/1074-7613(95)90001-2. [DOI] [PubMed] [Google Scholar]
- Fratazzi C, Arbeit RD, Carini C, Balcewicz-Sablinska MK, Keane J, Kornfeld H, Remold HG. Macrophage apoptosis in mycobacterial infections. J Leukoc Biol. 1999;66:763–764. doi: 10.1002/jlb.66.5.763. [DOI] [PubMed] [Google Scholar]
- Garcia Vidal C, Rodriguez Fernandez S, Martinez Lacasa J, Salavert M, Vidal R, Rodriguez Carballeira M, Garau J. Paradoxical response to antituberculous therapy in infliximab-treated patients with disseminated tuberculosis. Clin Infect Dis. 2005;40:756–759. doi: 10.1086/427941. [DOI] [PubMed] [Google Scholar]
- Gardam MA, Keystone EC, Menzies R, Manners S, Skamene E, Long R, Vinh DC. Anti-tumour necrosis factor agents and tuberculosis risk: mechanisms of action and clinical management. Lancet Infect Dis. 2003;3:148–155. doi: 10.1016/s1473-3099(03)00545-0. [DOI] [PubMed] [Google Scholar]
- Glickman MS, Cox JS, Jacobs WR., Jr A novel mycolic acid cyclopropane synthetase is required for cording, persistence, and virulence of Mycobacterium tuberculosis. Mol Cell. 2000;5:717–727. doi: 10.1016/s1097-2765(00)80250-6. [DOI] [PubMed] [Google Scholar]
- Gomez-Reino JJ, Carmona L, Angel Descalzo M. Risk of tuberculosis in patients treated with tumor necrosis factor antagonists due to incomplete prevention of reactivation of latent infection. Arthritis Rheum. 2007;57:756–761. doi: 10.1002/art.22768. [DOI] [PubMed] [Google Scholar]
- Guinn KM, Hickey MJ, Mathur SK, Zakel KL, Grotzke JE, Lewinsohn DM, Smith S, Sherman DR. Individual RD1-region genes are required for export of ESAT-6/CFP-10 and for virulence of Mycobacterium tuberculosis. Mol Microbiol. 2004;51:359–370. doi: 10.1046/j.1365-2958.2003.03844.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbomel P, Thisse B, Thisse C. Ontogeny and behaviour of early macrophages in the zebrafish embryo. Development. 1999;126:3735–3745. doi: 10.1242/dev.126.17.3735. [DOI] [PubMed] [Google Scholar]
- Herbomel P, Thisse B, Thisse C. Zebrafish early macrophages colonize cephalic mesenchyme and developing brain, retina, and epidermis through a M-CSF receptor-dependent invasive process. Dev Biol. 2001;238:274–288. doi: 10.1006/dbio.2001.0393. [DOI] [PubMed] [Google Scholar]
- Hernandez RE, Rikhof HA, Bachmann R, Moens CB. vhnf1 integrates global RA patterning and local FGF signals to direct posterior hindbrain development in zebrafish. Development. 2004;131:4511–4520. doi: 10.1242/dev.01297. [DOI] [PubMed] [Google Scholar]
- Hsu T, Hingley-Wilson SM, Chen B, Chen M, Dai AZ, Morin PM, Marks CB, Padiyar J, Goulding C, Gingery M, et al. The primary mechanism of attenuation of bacillus Calmette-Guerin is a loss of secreted lytic function required for invasion of lung interstitial tissue. Proc Natl Acad Sci U S A. 2003;100:12420–12425. doi: 10.1073/pnas.1635213100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta S, Goulet EJ, Huerta-Yepez S, Livingston EH. Screening and detection of apoptosis. J Surg Res. 2007;139:143–156. doi: 10.1016/j.jss.2006.07.034. [DOI] [PubMed] [Google Scholar]
- Iliopoulos A, Psathakis K, Aslanidis S, Skagias L, Sfikakis PP. Tuberculosis and granuloma formation in patients receiving anti-TNF therapy. Int J Tuberc Lung Dis. 2006;10:588–590. [PubMed] [Google Scholar]
- Kaku T, Kawamura I, Uchiyama R, Kurenuma T, Mitsuyama M. RD1 region in mycobacterial genome is involved in the induction of necrosis in infected RAW264 cells via mitochondrial membrane damage and ATP depletion. FEMS Microbiol Lett. 2007;274:189–195. doi: 10.1111/j.1574-6968.2007.00838.x. [DOI] [PubMed] [Google Scholar]
- Kaneko H, Yamada H, Mizuno S, Udagawa T, Kazumi Y, Sekikawa K, Sugawara I. Role of tumor necrosis factor-alpha in Mycobacterium-induced granuloma formation in tumor necrosis factor-alpha-deficient mice. Lab Invest. 1999;79:379–386. [PubMed] [Google Scholar]
- Kaufmann SH. Is the development of a new tuberculosis vaccine possible? Nat Med. 2000;6:955–960. doi: 10.1038/79631. [DOI] [PubMed] [Google Scholar]
- Keane J. TNF-blocking agents and tuberculosis: new drugs illuminate an old topic. Rheumatology (Oxford) 2005;44:714–720. doi: 10.1093/rheumatology/keh567. [DOI] [PubMed] [Google Scholar]
- Kindler V, Sappino AP, Grau GE, Piguet PF, Vassalli P. The inducing role of tumor necrosis factor in the development of bactericidal granulomas during BCG infection. Cell. 1989;56:731–740. doi: 10.1016/0092-8674(89)90676-4. [DOI] [PubMed] [Google Scholar]
- Koch R. The Aetiology of Tuberculosis. New York: National Tuberculosis Association; 1882. [Google Scholar]
- Kwon OJ. The role of nitric oxide in the immune response of tuberculosis. J Korean Med Sci. 1997;12:481–487. doi: 10.3346/jkms.1997.12.6.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange C, Hellmich B, Ernst M, Ehlers S. Rapid immunodiagnosis of tuberculosis in a woman receiving anti-TNF therapy. Nat Clin Pract Rheumatol. 2007;3:528–534. doi: 10.1038/ncprheum0571. [DOI] [PubMed] [Google Scholar]
- Lawn SD, Butera ST, Shinnick TM. Tuberculosis unleashed: the impact of human immunodeficiency virus infection on the host granulomatous response to Mycobacterium tuberculosis. Microbes Infect. 2002;4:635–646. doi: 10.1016/s1286-4579(02)01582-4. [DOI] [PubMed] [Google Scholar]
- Lee J, Remold HG, Ieong MH, Kornfeld H. Macrophage apoptosis in response to high intracellular burden of Mycobacterium tuberculosis is mediated by a novel caspase-independent pathway. J Immunol. 2006;176:4267–4274. doi: 10.4049/jimmunol.176.7.4267. [DOI] [PubMed] [Google Scholar]
- Lin PL, Plessner HL, Voitenok NN, Flynn JL. Tumor necrosis factor and tuberculosis. J Investig Dermatol Symp Proc. 2007;12:22–25. doi: 10.1038/sj.jidsymp.5650027. [DOI] [PubMed] [Google Scholar]
- Liu N, Raja SM, Zazzeroni F, Metkar SS, Shah R, Zhang M, Wang Y, Bromme D, Russin WA, Lee JC, et al. NF-kappaB protects from the lysosomal pathway of cell death. EMBO J. 2003;22:5313–5322. doi: 10.1093/emboj/cdg510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu PT, Stenger S, Li H, Wenzel L, Tan BH, Krutzik SR, Ochoa MT, Schauber J, Wu K, Meinken C, et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science. 2006;311:1770–1773. doi: 10.1126/science.1123933. [DOI] [PubMed] [Google Scholar]
- Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104:487–501. doi: 10.1016/s0092-8674(01)00237-9. [DOI] [PubMed] [Google Scholar]
- Martineau AR, Wilkinson KA, Newton SM, Floto RA, Norman AW, Skolimowska K, Davidson RN, Sorensen OE, Kampmann B, Griffiths CJ, Wilkinson RJ. IFN-gamma- and TNF-independent vitamin D-inducible human suppression of mycobacteria: the role of cathelicidin LL-37. J Immunol. 2007;178:7190–7198. doi: 10.4049/jimmunol.178.11.7190. [DOI] [PubMed] [Google Scholar]
- Menendez A, Brett Finlay B. Defensins in the immunology of bacterial infections. Curr Opin Immunol. 2007;19:385–391. doi: 10.1016/j.coi.2007.06.008. [DOI] [PubMed] [Google Scholar]
- Middlebrook G, Dubos RJ, Pierce C. Virulence and morphological characteristics of mammalian tubercle bacilli. J Exp Med. 1947;86:175–184. doi: 10.1084/jem.86.2.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyakawa Y, Ratnakar P, Rao AG, Costello ML, Mathieu-Costello O, Lehrer RI, Catanzaro A. In vitro activity of the antimicrobial peptides human and rabbit defensins and porcine leukocyte protegrin against Mycobacterium tuberculosis. Infect Immun. 1996;64:926–932. doi: 10.1128/iai.64.3.926-932.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan VP, Scanga CA, Yu K, Scott HM, Tanaka KE, Tsang E, Tsai MM, Flynn JL, Chan J. Effects of tumor necrosis factor alpha on host immune response in chronic persistent tuberculosis: possible role for limiting pathology. Infect Immun. 2001;69:1847–1855. doi: 10.1128/IAI.69.3.1847-1855.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan MP, O’Leary S, Kelly DM, Keane J. A caspase-independent pathway mediates macrophage cell death in response to Mycobacterium tuberculosis infection. Infect Immun. 2007;75:1984–1993. doi: 10.1128/IAI.01107-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan H, Yan BS, Rojas M, Shebzukhov YV, Zhou H, Kobzik L, Higgins DE, Daly MJ, Bloom BR, Kramnik I. Ipr1 gene mediates innate immunity to tuberculosis. Nature. 2005;434:767–772. doi: 10.1038/nature03419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parichy DM, Ransom DG, Paw B, Zon LI, Johnson SL. An orthologue of the kit-related gene fms is required for development of neural crest-derived xanthophores and a subpopulation of adult melanocytes in the zebrafish, Danio rerio. Development. 2000;127:3031–3044. doi: 10.1242/dev.127.14.3031. [DOI] [PubMed] [Google Scholar]
- Pressley ME, Phelan PE, 3rd, Witten PE, Mellon MT, Kim CH. Pathogenesis and inflammatory response to Edwardsiella tarda infection in the zebrafish. Dev Comp Immunol. 2005;29:501–513. doi: 10.1016/j.dci.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Roach DR, Bean AG, Demangel C, France MP, Briscoe H, Britton WJ. TNF regulates chemokine induction essential for cell recruitment, granuloma formation, and clearance of mycobacterial infection. J Immunol. 2002;168:4620–4627. doi: 10.4049/jimmunol.168.9.4620. [DOI] [PubMed] [Google Scholar]
- Rojo I, de Ilarduya OM, Estonba A, Pardo MA. Innate immune gene expression in individual zebrafish after Listonella anguillarum inoculation. Fish Shellfish Immunol. 2007;23:1285–1293. doi: 10.1016/j.fsi.2007.07.002. [DOI] [PubMed] [Google Scholar]
- Shin DH, Lim HS, Cho SK, Lee HY, Lee HW, Lee KH, Chung YH, Cho SS, Ik Cha C, Hwang DH. Immunocytochemical localization of neuronal and inducible nitric oxide synthase in the retina of zebrafish, Brachydanio rerio. Neurosci Lett. 2000;292:220–222. doi: 10.1016/s0304-3940(00)01407-5. [DOI] [PubMed] [Google Scholar]
- Smith S, Liggitt D, Jeromsky E, Tan X, Skerrett SJ, Wilson CB. Local role for tumor necrosis factor alpha in the pulmonary inflammatory response to Mycobacterium tuberculosis infection. Infect Immun. 2002;70:2082–2089. doi: 10.1128/IAI.70.4.2082-2089.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamm LM, Morisaki JH, Gao LY, Jeng RL, McDonald KL, Roth R, Takeshita S, Heuser J, Welch MD, Brown EJ. Mycobacterium marinum escapes from phagosomes and is propelled by actin-based motility. J Exp Med. 2003;198:1361–1368. doi: 10.1084/jem.20031072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenger S. Immunological control of tuberculosis: role of tumour necrosis factor and more. Ann Rheum Dis. 2005;64(Suppl 4):iv24–28. doi: 10.1136/ard.2005.042531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stinear TP, Seemann T, Harrison PF, Jenkin GA, Davies JK, Johnson PD, Abdellah Z, Arrowsmith C, Chillingworth T, Churcher C, et al. Insights from the complete genome sequence of Mycobacterium marinum on the evolution of Mycobacterium tuberculosis. Genome Res. 2008 doi: 10.1101/gr.075069.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaim LE, Connolly LE, Volkman HE, Humbert O, Born DE, Ramakrishnan L. Mycobacterium marinum infection of adult zebrafish causes caseating granulomatous tuberculosis and is moderated by adaptive immunity. Infect Immun. 2006;74:6108–6117. doi: 10.1128/IAI.00887-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JC, Orkin R, Lanham J. Tuberculosis following therapy with infliximab may be refractory to antibiotic therapy. Rheumatology (Oxford) 2003;42:901–902. doi: 10.1093/rheumatology/keg158. [DOI] [PubMed] [Google Scholar]
- Tobin DM, Ramakrishnan L. Comparative pathogenesis of Mycobacterium marinum and Mycobacterium tuberculosis. Cell Microbiol. 2008 doi: 10.1111/j.1462-5822.2008.01133.x. [DOI] [PubMed] [Google Scholar]
- Tosh K, Campbell SJ, Fielding K, Sillah J, Bah B, Gustafson P, Manneh K, Lisse I, Sirugo G, Bennett S, et al. Variants in the SP110 gene are associated with genetic susceptibility to tuberculosis in West Africa. Proc Natl Acad Sci U S A. 2006;103:10364–10368. doi: 10.1073/pnas.0603340103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traver D, Herbomel P, Patton EE, Murphey RD, Yoder JA, Litman GW, Catic A, Amemiya CT, Zon LI, Trede NS. The zebrafish as a model organism to study development of the immune system. Adv Immunol. 2003;81:253–330. [PubMed] [Google Scholar]
- Trede NS, Langenau DM, Traver D, Look AT, Zon LI. The use of zebrafish to understand immunity. Immunity. 2004;20:367–379. doi: 10.1016/s1074-7613(04)00084-6. [DOI] [PubMed] [Google Scholar]
- Ulrichs T, Kaufmann SH. New insights into the function of granulomas in human tuberculosis. J Pathol. 2006;208:261–269. doi: 10.1002/path.1906. [DOI] [PubMed] [Google Scholar]
- van Crevel R, Ottenhoff TH, van der Meer JW. Innate immunity to Mycobacterium tuberculosis. Clin Microbiol Rev. 2002;15:294–309. doi: 10.1128/CMR.15.2.294-309.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassalli P. The pathophysiology of tumor necrosis factors. Annu Rev Immunol. 1992;10:411–452. doi: 10.1146/annurev.iy.10.040192.002211. [DOI] [PubMed] [Google Scholar]
- Vlachaki E, Psathakis K, Tsintiris K, Iliopoulos A. Delayed response to anti-tuberculosis treatment in a patient on infliximab. Respir Med. 2005;99:648–652. doi: 10.1016/j.rmed.2004.10.006. [DOI] [PubMed] [Google Scholar]
- Volkman HE, Clay H, Beery D, Chang JC, Sherman DR, Ramakrishnan L. Tuberculous granuloma formation is enhanced by a mycobacterium virulence determinant. PLoS Biol. 2004;2:e367. doi: 10.1371/journal.pbio.0020367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner TE, Huseby ES, Huseby JS. Exacerbation of Mycobacterium tuberculosis enteritis masquerading as Crohn’s disease after treatment with a tumor necrosis factor-alpha inhibitor. Am J Med. 2002;112:67–69. doi: 10.1016/s0002-9343(01)01035-x. [DOI] [PubMed] [Google Scholar]
- Zganiacz A, Santosuosso M, Wang J, Yang T, Chen L, Anzulovic M, Alexander S, Gicquel B, Wan Y, Bramson J, et al. TNF-alpha is a critical negative regulator of type 1 immune activation during intracellular bacterial infection. J Clin Invest. 2004;113:401–413. doi: 10.1172/JCI18991. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Morpholino targeting of the TNF Receptor 1 (TR1) leads to complete splice blocking of transcript for 4 days. Two splice blocking morpholinos targeting the exon-intron boundaries of E2I2 and E4I4 were used in combination to maximize disruption of transcript. Control embryos at 2 days (d) and 4d post fertilization in comparison with TR1 morphant transcripts (2d-4d) show splice blocking of native transcript during this time period.
Supplementary Figure 2. TNF signaling is not required for recruitment of phagocytes to bacteria. Number of phagocytes recruited to the hindbrain ventricle 3 hours after the injection of 50 ± 10 CFU of M. marinum for control, TR1, and Pu.1 morphant embryos. Phagocytes are enumerated by scanning through the hindbrain ventricle and identified by their morphology. Pu.1 morphants are used as a negative control as they lack macrophages and have reduced numbers of neutrophils. Medians are indicated by bars. Control and TR1 morphant embryos recruited significantly more phagocytes than Pu.1 morphant embryos (p<0.0001). Number of recruited macrophages was not different between control and TR1 embryos.
Supplementary Figure 3. Inducible nitric oxide (iNOS) antibody staining is similar in infected control and TR1 morphant embryos. Fluorescent detection of iNOS antibody shows colocalization with fluorescent Mycobacterium marinum (MM). Scale bar, 50 μm.
Supplementary Figure 4. Increased cording seen in the absence of TNF signaling is not dependent on the RD1 virulence locus or granuloma formation. (A) Control (n = 23) and TR1 morphant (n = 9) embryos were infected with 353 ± 10 ΔRD1 bacteria and scored daily for the presence of extracellular bacteria as indicated by cording and plotted as the percentage of embryos with cording. A significantly higher percentage of TR1 morphant embryos display cording versus control embryos at 5 dpi (p<0.01) and 6 dpi (p<0.05) as analyzed by Fisher’s exact test of a contingency table. (B and C) Examples of cording found during infection of TR1 morphant embryos infected with ΔRD1 at 6 dpi. Scale bar, 25 μm.
Supplementary Figure 5. Macrophage death can lead to dissemination of infection. All images are stills taken from a two hour timelapse of an infected TR1 morphant fish at four dpi. A single dead infected macrophage is rephagocytosed by two separate macrophages. When available, flattened three-dimensional fluorescent stacks are provided to indicate position of bacteria (left column). Time is shown in minutes. The original macrophage is indicated by left-facing bracket, first incoming macrophage is indicated by upward-facing bracket, and seconding incoming macrophage is indicated by right-facing bracket.
Supplementary Movie 1. Macrophage death can lead to dissemination of infection to multiple uninfected macrophages. A two hour timelapse of a TR1 morphant fish was taken at 4 days post infection and shows the rephagocytosis of a single dead infected macrophage into two separate incoming macrophages. The original macrophage is indicated by left-facing bracket, first incoming macrophage is indicated by upward-facing bracket, and seconding incoming macrophage is indicated by right-facing bracket. Movie is paused at 14 and 104 minutes to demonstrate location of separate macrophages.