

Abstract
Obesity is currently considered a serious public health issue due to its strong impact on health, economy, and quality of life. It is considered a chronic low-grade inflammation state and is directly involved in the genesis of metabolic disturbances, such as insulin resistance and dyslipidemia, which are well-known risk factors for cardiovascular disease. Furthermore, there is evidence that genetic variation that predisposes to inflammation and metabolic disturbances could interact with environmental factors, such as diet, modulating individual susceptibility to developing these conditions. This paper aims to review the possible interactions between diet and single-nucleotide polymorphisms (SNPs) in genes implicated on the inflammatory response, lipoprotein metabolism, and oxidative status. Therefore, the impact of genetic variants of the peroxisome proliferator-activated receptor-(PPAR-)gamma, tumor necrosis factor-(TNF-)alpha, interleukin (IL)-1, IL-6, apolipoprotein (Apo) A1, Apo A2, Apo A5, Apo E, glutathione peroxidases 1, 2, and 4, and selenoprotein P exposed to variations on diet composition is described.
1. Introduction
Obesity is a global epidemic [1] and its prevalence is growing worldwide [2]. In 2007, prevalence in North America was 26.4% for men and 24.8% for women [3]. In developing countries like Brazil, overweight (body mass index (BMI) between 25 and 30 kg/m2) reaches approximately 50% of adults and obesity (BMI ≥ 30 kg/m2) occurs on 16.9% of women and 12.4% of men [4]. One billion people in the world overweight and 300 million are considered obese. Estimates for 2030 are that 2 billion people will be overweight and 1.12 billion obese around the world [5]. This scenario may reduce life expectancy for this century [6], since obesity is a major condition triggering several diseases. Particularly accumulation of visceral fat elevates the risk of type 2 diabetes (T2DM), dyslipidemia, and hypertension, which contribute for cardiovascular diseases, reduction in quality of life, increase in costs of medical care, and for premature mortality [3, 7].
Excessive energy intake combined to low-energy expenditure induces lipid accumulation in adipose tissue but also in liver, muscle, and other internal organs, predisposing to the development of insulin resistance (IR) and metabolic disturbances [8]. In parallel to impairment of insulin action in tissues and organs, also insulin secretion is affected by diet. It is known that high saturated fatty acids (SFAs) intake provokes a deleterious lipid profile and that the elevated plasma-free fatty acid concentration induces apoptosis of β cells, contributing to dysfunctional insulin secretion [9, 10]. This condition of high cardiometabolic risk has been incriminated in the mortality of people living in developed and in developing countries [11–13]. Statistics indicates precocity of cardiovascular disease, already reaching 4% of Americans adolescents [14] being more prevalent among girls.
The cluster of cardiovascular risk factors led to the description of the metabolic syndrome (MS), in which IR represents a link between hemodynamic and metabolic disturbances [15]. Visceral fat accumulation, commonly present in MS, contributes to a proinflammatory and pro-oxidant state and to deterioration of glucose and lipid metabolism. High amounts of free fatty acids and several adipocytokines released from visceral adipose tissue to portal and systemic circulation are implicated in the genesis of IR [7, 16, 17]. Tumor necrosis factor alpha (TNF-α) is a proinflammatory adipocytokine whose production is increased in obese humans and its neutralization resulted in amelioration of IR [18, 19]. In vitro studies, in both rodents and humans, showed that TNF-α stimulates adipocyte lipolysis [19, 20] through downregulation of the expression of the perilipin, a lipid droplet-associated protein which is thought to modulate the access of hormone-sensitive lipase to the surface of the fat droplet [20]. Several evidences indicated that obesity may generate IR via inflammation [21, 22]. The relationship between obesity and subclinical inflammation was firstly described by Hotamisligil in 2003 [8], who demonstrated a positive correlation between adipose mass and the expression of the gene coding for TNF-α. Further, the link between obesity and inflammation was reinforced by the findings of increased concentrations of other inflammatory biomarkers in obese individuals, like interleukin 6 (IL-6) and interleukin 8 (IL-8) [21, 23] and acute phase proteins such as C reactive protein (CRP) [6].
In addition, augmented release of TNF-α, nonesterified fatty acids and angiotensinogen, found in visceral obesity, increases oxidative stress, which furthers contribute to IR [24]. In this context, it has been reported that angiotensinogen-II-induced reactive oxygen species (ROS) upregulation affects several parts of the intracellular insulin signalling pathways [25]. In vitro, ROS impairs insulin receptor substrate-1 (IRS-1) phosphorylation and IRS-1-induced phosphatidylinositol 3-kinase (PI3-kinase) activation in cultured adipocytes, leading to the impaired translocation of glucose transporter 4 (GLUT-4) into the membrane, resulting in IR [26].
Modern lifestyle—characterized by high food intake and low physical activity—has been considered the main determinant of increased adiposity in mankind. Not only the amount of calories contributes to the deleterious effects of obesity, since certain dietary patterns are related to cardiovascular risk or protection. Morbidity and mortality of Mediterranean populations are shown to be lower than those observed in population exposed to the typical Western diet, rich in SFA [27]. Animal studies have helped to understand the role of dietary fat in disturbances of lipid and glucose metabolism [28, 29]. SFA have shown to stimulate intracellular pathways, which result in proinflammatory gene expression and/or IR [9, 10, 30, 31]. The underlying mechanisms for deterioration of glucose and lipid metabolism include (a) accumulation of diacylglycerol and ceramide; (b) activation of nuclear factor-kB (NFkB), protein kinase C, and mitogen-activated protein kinases, and subsequent induction of inflammatory genes in white adipose tissue, immune cells, and myotubes; (c) decreased peroxisome proliferator-activated receptor gamma (PPAR-γ) coactivator-1 a/b activation and adiponectin production, which decreases the oxidation of glucose and fatty acids; (d) recruitment of immune cells like macrophages, neutrophils, and bone-marrow-derived dendritic cells to white adipose tissue and muscle [32, 33].
Reducing consumption of foods rich in SFA and increasing consumption of whole grains, fruits, vegetables, lean meats and poultry, fish, low-fat dairy products, and oils containing oleic acid are expected to reduce the incidence of metabolic diseases [10, 33]. In fact, this dietary pattern, which has similarities with the Mediterranean diet, has been associated with better metabolic and inflammatory profiles in some clinical trials [34].
The expression of genes is highly dependent on, and regulated by, nutrients and dietary bioactive compounds found in food. A variety of dietary components can alter gene expression, and thus significantly influence health. At the same time, the genetic makeup of an individual may coordinate its response to diet [35]. Investigations of gene-environment interactions have identified genetic polymorphisms associated with individual susceptibility to obesity, inflammation, dyslipidemia, and oxidative stress. In this context, unbalanced diets may shift the balance between healthy and diseased conditions, increasing the risk of these metabolic and immune disturbances, particularly in genetic-predisposed subjects (Figure 1).
Figure 1.
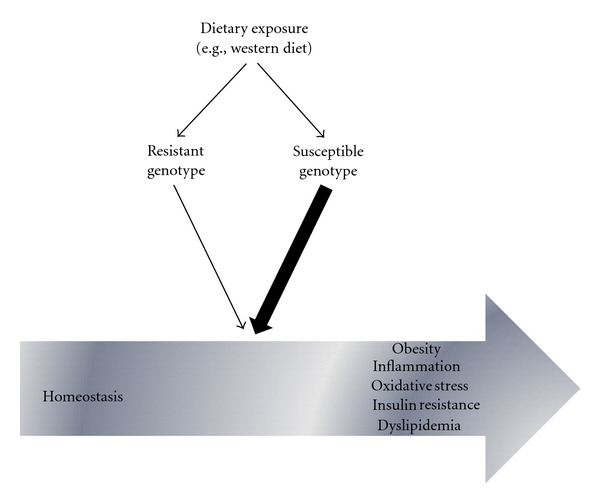
The association of gene variants with differential responses to diet and its impact on the shift between homeostasis and metabolic/immune disturbances.
The assessment of the interactions between nutrients, dietary bioactive compounds, and genotypes may pave the way for more targeted prevention strategies, and thereby a better success in the prevention and treatment [36]. In this context, there is increasing evidence that supports a role for genotype-nutrient interactions in obesity and its associated disorders [37, 38]. The present study aimed to review the relation between diet and SNPs that impact on inflammation (PPAR-γ, Tumor Necrosis Factor alpha (TNF-α), interleukin (IL)-1, IL-6), oxidative status (glutathione peroxidases (GPX) 1, 2 and 4, and selenoprotein P) and lipoprotein metabolism (apolipoprotein (Apo) A1, Apo A2, Apo A5, Apo E).
2. Peroxisome Proliferator: Activated Receptor Gamma
The PPARγ is a nuclear hormone receptor that serves as a master regulator of adipocytes-specific genes contributing to adipocytes differentiation, susceptibility to obesity, and insulin sensitivity. Furthermore, this induces the expression of genes that promote entrance of lipids in the cells, synthesis triglycerides, fatty acid oxidation, and extracellular metabolism of lipids (reverse cholesterol transport through regulation cholesterol-efflux from macrophages) [39–42].
Several SNPs variants related to PPARγ gene have been identified, like 161C/T, 1431C/T, and 162L/V, but the most prevalent is the Pro12Ala (substitution of proline to alanine at codon 12 of this gene, rs1801282), which is associated with T2DM, obesity, and other clinical disorders [43–47]. However, the prevalence of this polymorphism varies according to the ethnic background of the population [43–46, 48–54].
The relationship of SNP in the PPARγ gene and obesity or MS is controversial. A recent research showed that the Pro12Ala SNP was associated with obesity in Iranian individuals, and the presence of the Ala allele predicted a higher BMI [55]. In European descent Brazilian men [56], and in White men from Italy [57], the Ala allele interacts with gender and contributes to the susceptibility to obesity. Also in children of Greek origin, adiposity through measures of skinfold triceps and subscapular was shown to be influenced by Pro12Ala in a gender specific manner [58].
Nevertheless, a study with childhood and adult obesity in the French Caucasian population showed that the SNP Pro12Ala was not associated with obesity, but is confirmed a contribution in the genetic risk for T2DM especially in obese subjects, in which this allele worsens IR and increases fasting insulin levels [59]. Another study among native Javaneses did not show an association between the Pro12Ala polymorphism and T2DM; a weak association with obesity was found [38]. In Poland obese individuals with 10-year history of T2DM, there was no association between Pro12Ala SNP and body mass changes observed during the disease course, neither differences in IR and incidence and progression of T2DM complications [60].
In another study, fasting and postprandial triglycerides and insulin levels as well as homeostasis model assessment of insulin resistance (HOMA-IR) were significantly lower in the Ala12Ala group than in the Pro12Pro group after the mixed meal [61]. Others confirmed an association between the Ala allele and reduced incidence of obesity in prepubertal children and found strong associations between this SNP and percentage body fat [62]. Pro12Ala SNP does not seem to be associated with BMI in postmenopausal Polish women, although the Ala allele seems to predispose to a less favorable lipid profile in this population [63]. In Sweden, an intervention study included lifestyle modifications metformin or placebo. Metformin and lifestyle groups, Ala12 carriers had greater weight loss [64].
In addition to the association of Ala allele with reduced risk of T2DM [65], its presence resulted in fat oxidation increase and higher satiety suggesting benefits in food intake control [66]. Concordantly, a study conducted in Spanish obese woman showed that Pro12Ala SNP resulted in increased fat oxidation [67].
Nutrients and bioactive compounds of food are able to interfere with the genome by highly complex forms. Studies have shown that effect of Pro12Ala may be mediated by the dietary fat [61], particularly by the proportion of dietary polyunsaturated fatty acid (PUFA) and SFA. A population-based cohort study, including 592 nondiabetic Caucasian, showed that fasting insulin concentration was negatively associated with the dietary PUFA-to-SFA ratio and a strong interaction was evident between this ratio and the Pro12Ala polymorphism for both, BMI and fasting insulin, suggesting that when the dietary ratio is low, the BMI in Ala carriers is greater than that in Pro homozygotes, but when the dietary ratio is high, the opposite is seen [68]. In a Spanish population similar results were found; the intake of monounsaturated fatty acid (MUFA) mainly oleic acid, contributed to the variance of the HOMA-IR. These findings suggest the existence of an interaction between Pro12Ala and dietary MUFA, such that obese people with the Ala-12 allele have higher HOMA-IR values, especially if their MUFA intake is low [69]. Studying the P12Ala with Glu27Glu polymorphism of beta 2-adrenergic receptor, Rosado et al., [67], showed that individuals carrying both polymorphisms Pro12Ala and Gln27Glu alleles had high PUFA intake and greater body weight loss. Similar results were shown in study with children. Pro12Ala may be associated with higher insulin sensitivity and higher long-chain PUFAs, particularly n-3, levels in plasma phospholipids of 140 Italian normolipidemic obese children [70]. This gene-nutrient interaction emphasizes the difficulty of examining the effect of common polymorphisms in the absence of data about environment exposures.
Selected studies on the main interactions of polymorphisms with dietary factors are in Table 1.
Table 1.
Summary of studies evaluating interaction between diet and variants of genes involved in inflammation and oxidant status.
| Gene | Variant | Population [reference] | Frequency | Design | Main findings |
|---|---|---|---|---|---|
| 708 men from Germany [61] | Pro: 85% Ala: 15% |
Metabolic tolerance test: oral glucose tolerance test and oral metabolic tolerance test (mixed meal containing 51,6 kJ% fat, 29,6 kJ% carbohydrates, 11,9 kJ% protein, with a total of 4406 kJ. | Fasting and postprandial serum TG, insulin levels and HOMA-IR were significantly lower in the Ala12Ala group than in Pro12Pro group after the metabolic tolerance test. | ||
| PPAR-gamma | Pro12Ala (rs1801282) | Total of 3,356 individuals from Diabetes Prevention Program, USA [64] | Ala frequencies: European Americans: 11,7% African Americans: 4% Hispanics: 9% American Indians: 19,6% Asian: 8,6% |
Randomized clinical trial treatment with metformin, troglitazone, or lifestyle modification versus placebo for T2DM prevention in high-risk individuals. | In Pro homozygous, VAT was reduced to a similar degree irrespective of the level of polyunsaturated/saturated fatty acid ratio (P : S ratio) in the diet. Ala12 allele carriers where consuming low P : S ratio diets tended to gain VAT mass, whereas those who consumed high P : S ratio diets tended to lose VAT. In metformin and lifestyle groups, Ala12 carriers had greater weight loss. |
| 592 nondiabetic Caucasian from UK [68] | Pro/Pro: 79,1% Ala/Ala: 2% |
Cross-sectional | When the amount of SFA in the diet is greater than the amount of PUFA, the BMI of Ala carriers is high. When the amount of PUFA is greater than the SFA, the opposite effect is observed. Strong interaction was evident between PUFA/SFA ratio in the diet and Pro12Ala for both BMI and serum fasting insulin concentration. | ||
| 538 subjects from southern Spain [69] | Pro12Pro: 85,8 Pro12Ala: 13,4% Ala12Ala: 0,8% |
Cross-sectional | Subjects with the Ala allele had a lower risk for T2DM (OR = 0.30). Obese people with the Ala allele have higher HOMA-IR values, especially if their MUFA intake is low. | ||
| Healthy men aged 28 ± 8 yrs (n = 111) [71] | GG: 68,0% GA: 30,0% AA: 2,0% |
Clinical trial: supplementation with 6 g/day of fish oil (1.8 g DHA+EPA) for 12 weeks. |
The suppressive effect of fish oil supplementation on TNF-α production was greater among individuals in the highest tertile of presupplementation TNF-α when carrying −308A compared to the wild-type individuals. | ||
| TNF | −308A (rs1800629) | Elderly men and women (n = 110) [72] | GG: 72,5% AG: 21,5% AA: 6,0% |
Randomized, double-blind, placebo-controlled trial: supplementation with 182 mg α-tocopherol/day for one year. | Participants with the A/A and A/G genotype, treated with vitamin E, had lower TNF-α production in LPS-stimulated whole blood cell culture than those with the A allele treated with placebo. |
| Obese nondiabetic individuals (n = 203) [73] | GG: 75,5% GA/AA: 24,5% |
Randomized clinical trial: ingestion of energy-restricted diets (1,500 kcal/day) for 2 months. | Contrary to the wild genotype individuals, obese subjects carrying the −308A allele had no improvement in plasma glucose, insulin, TG, TC and LDL-c levels and blood pressure following the intervention. | ||
| Normal weight (n = 68) and obese (n = 70) women aged 18 ± 45 y [74] | GG: 70,0% GA+AA: 30,0% |
Cross-sectional | When the dietary fat intake was 30 to 35% of energy, the odds of being obese with the TNFA GA+AA genotype was lower than of that with GG. However, increasing intake of dietary fat was associated with a significantly faster rate of increase in obesity risk in women with the TNFA GA+AA genotype compared with those with the GG genotype (P = .036). For individuals with the GA+AA genotype, the obesity OR was 3.02 and 9.12 for total dietary fat intakes of 35 and 40 (%E), respectively, compared with 30%E, indicating that individuals carrying A allele were significantly more responsive to an increase in dietary fat intake in their risk of being obese compared with normal weight. | ||
| 722 obese subjects from 8 European centers [75]. | — | Metabolic tolerance test: a high fat load containing 95% energy from fat. | Presence of −174G/C SNP gives higher ability to increase fat oxidation after a high fat load (P = .01). | ||
| −174G/C (rs1800795) | 737 individuals with high cardiovascular risk from Spain [76]. | C allele: 0,39 | Clinical trial: each participant was placed in one of three diets: low-fat diet; Mediterranean diet supplemented with virgin olive oil; Mediterranean diet supplemented with nuts. | After a 3-year intervention with a Mediterranean-style diet CC individuals were predicted to have the greatest reduction in body weight. At baseline, these individuals had the highest body weight and BMI. | |
| IL-6 | 32 healthy Caucasian origin subjects from Spain [77] | CC: 34% GC+CC: 66% |
Metabolic tolerance test: plasma free fatty acids suppression was evaluated in the fasting state and 120 minutes after an oral glucosee tolerance test. | Individuals with the G alelle presented twice the concentration of serum TG (P = .01) and VLDL-c (P = .002), higher fasting (P = .01), and postglucose load-free fatty acids (P = .03), slightly lower HDL-c (P = .058) than the ones with C allele. | |
| −174G/C (rs1800795) and PPAR Pro12Ala (rs1801282) | 67 obese subjects from Spain [78] | C allele: 40,2% | Clinical trial: volunteers were enrolled in a 10-week dietary intervention programme with a balanced low-energy diet, followed by dietitians. They were contacted again 1 year after the end of this period. | The C allele was more frequently observed (P = .032) in individuals with successful weight maintenance (<10% weight regain). The presence of the Ala allele of PPAR-γ together with the C allele of the IL-6 strengthens this protection (OR 0.19; P = .043). | |
| Haplotype (IL-1B):6054G/A, 3966C/T, 231T/C, 2511G/A and 21473G/C | Caucasian men and women aged 49 ± 16 yrs from the GOLDN Study (n = 1,120) [79] | Haplotype 11222: 25% | Cross-sectional | The prevalence of metabolic syndrome for haplotype 11222 was significantly higher than for haplotype 21111 among those with low DHA+EPA membrane content. | |
| IL-1A IL-1B |
−31T (IL-1B) | Japaneses aged 39–70 yrs (n = 200 men and 425 women) [80] | CC: 22,4% CT: 51,8% TT: 25,8% |
Cross-sectional | The association of the IL-1B −31C/T polymorphism with hypertension was weak in women with high serum β-carotene levels, but, in those with low β-carotene levels, the TT genotype increased the prevalence of hypertension. |
|
IL-1B: −511T +3954T IL-1A: +4845T |
Healthy adults [81] | — | Randomized placebo-controlled trial: supplementation with a botanical extract (1,200 mg of rose hips extract, 165 mg of blackberry powder, 330 mg of blueberry powder, and 40 mg of grapevine extract per day) for 12 weeks. | IL-1 risk genotype individuals receiving the formulation experienced a greater reduction in IL-1B gene expression from LPS-stimulated peripheral blood mononuclear cells and in plasma CRP levels, being the risk genotype defined by the presence of the following 3 genotypes: (1) homozygous for the common allele (C) at IL1B (−511); (2) carrying 2 copies of the less common allele (T) at IL1A (+4845); or (3) carrying one copy of the less common allele at IL1A (+4845) plus at least one copy of the less common allele (T) at IL1B (+3954). | |
| GPx1 | Pro198Leu | 37 morbidly obese women from Brazil [82] | Pro/Pro: 48,7% Pro/Leu: 37,8% Leu/Leu: 13,5% |
Randomized trial: consumed one Brazililian nut (290 μg of Se/day) for 8 weeks. | At baseline, 100% of the subjects were Se deficient, and after the supplementation, there was an improvement in plasma Se concentration (P < .001 for Pro/Pro and Pro/Leu, P < .05 for Leu/Leu), erythrocyte Se concentration (P = .00 for Pro/Pro and Pro/Leu, P < .05 for Leu/Leu), and GPx activity (P = .00 for Pro/Pro, P < .00001 for Pro/Leu, P < .001 for Leu/Leu). In addition, the Pro/Pro group showed a decrease in DNA damage after Brazil nut consumption compared with baseline (P < .005), and those levels were higher in Leu/Leu subjects compared with those with the wild-type genotype (P < .05). |
| GPx4 | 718 T/C (rs713041) | 40 nonsmokers subjects from United Kingdom [83] | CC: 55% TT: 45% |
A clinical trial: 6-week selenium supplementation (100 μg Se/day). | Both lymphocyte GPx1 protein concentrations and plasma GPx3 activity increased significantly in CC but not TT participants. After Se withdrawal, there was a significant fall in both lymphocyte GPx4 protein concentration and GPx4 activity in TT, but not in CC participants; females had higher concentrations than did males. |
| SeP gene | Ala234Thr (rs3877899) and 25191G/A (rs7579) in 3′UTR | 121 nonsmokers subjects from European, Indian, and Chinese ethnic origins [84]. | Ala234Thr in Caucasian: GG: 46% GA: 47% AA: 7%25191G/A in Caucasian: GG: 47,8% GA: 45,6% AA: 6,4% |
A clinical trial: 6-week selenium supplementation (100 μg Se/day) as sodium selenite. |
Plasma Se, SeP, and GPx3 levels increased after selenium supplementation. Presupplementation SeP concentration was associated with gender and genotype at SNP 24731 and postsupplementation concentration with SNP 25191. Both SNPs and gender were associated with differences in GPx3 activity, plasma, and erythrocyte thioredoxin reductase 1 concentrations and lymphocyte glutathione peroxidase 1 and 4 activities and concentrations. |
BMI: body mass index; CRP: C-reactive protein; DHA+EPA: eicosapentaenoic acid plus docosahexaenoic acid; GOLDN Study: Genetics of Lipid-Lowering Drugs and Diet Network (GOLDN) Study; GPx: glutathione peroxidase; HDL-c high-density lipoprotein cholesterol; HOMA-IR: homeostasis model assessment of insulin resistance; IL: interleukin; LDL-c: low-density lipoprotein cholesterol; LPS: lipopolysaccharide; MUFA: monounsaturated fat acid; OR: odds ratio; PPAR: peroxisome proliferator-activated receptor; P : S ratio: polyunsaturated to saturated fatty acid ratio; PUFA: polyunsaturated fatty acid; Se: selenium; SeP: selenoprotein P; SFA: saturated fatty acid; SNP: single nucleotide polymorphism; T2DM: Type 2 diabetes mellitus; TC: total cholesterol; TG: triglycerides; TNF-α: tumor necrosis factor-α; VAT: visceral adipose tissue; VLDL-c: very low-density lipoprotein cholesterol.
3. Tumor Necrosis Factor Alpha
TNF-α is a pleiotropic cytokine with a central role in inflammation. Obesity leads to an increased production of TNF-α, which is a crucial mediator of IR states and other related comorbidities [85–87].
TNF-α production in whole blood cell culture of healthy individuals shows a great, though stable, variation, making it possible to identify high and low producer phenotypes in the population [88, 89]. This fact points to a substantial genetic contribution to TNF-α regulation [90, 91]. The TNF gene is located on human chromosome 6p21.3 in the class III region of the major histocompatibility complex [92].
Eight SNPs were described within the TNF promoter, at positions −1031T/C, −863C/A, −857C/T, −575G/A, −376G/A, −308G/A, −244G/A, and −238G/A [91, 92]. Regulating the transcription of TNF gene could potentially blunt the deleterious effects of dysregulated TNF-α synthesis, as seen in obesity-induced inflammation. As such, genetic variations in TNF gene regulatory region may affect TNF-α transcription and expression, influencing the development of conditions associated with excessive TNF-α production, such as rheumatoid arthritis, inflammatory bowel disease and IR [93].
Wilson et al. [94] identified the polymorphism located at position −308 in the TNF promoter (rs1800629), which defined the TNF1 (−308G) and TNF2 (−308A) alleles. The presence of TNF2 allele seems to be associated with a greater risk of diseases, mainly infectious and autoimmune diseases [95]. However, results concerning the possible functional role of −308 TNF polymorphism are conflicting [96]. Gene reporter assays have been employed to investigate whether the TNF 2 allele could influence TNF-α gene transcription. Some studies show significant differences in transcriptional activity between TNF1 and TNF2 alleles [97–99], while others could not confirm these findings [99, 100]. Some of the variables that might explain the mixed results of these experiments are differences in cell type used for transfection, cell origin (human or nonhuman), type of stimulus (lipopolysaccharide, phorbol myristate acetate, and TNF-α), length of the promoter sequence used and the presence or absence of the 3 untranslated regions of the mRNA (3′UTR).
Nonobese healthy individuals carrying TNF2 allele showed an increase in circulating inflammatory biomarkers such as CRP in some [101, 102] but not all studies [103]. It is noteworthy that this relationship was conditioned by age, gender, and ethnicity [101, 102]. A meta-analysis aiming to investigate whether −308G/A TNF-α gene variant was associated with MS phenotype found that carriers of the TNF2 allele had higher systolic blood pressure and fasting insulin, which appeared to be dependent of age group and ethnic background, however, there was no significant association of TNF-α genotypes with plasma fasting glucose and leptin, T2DM and hypertension [104]. Concerning cardiovascular events, another meta-analysis failed to find an association of −308G/A polymorphism with ischemic heart disease and ischemic stroke, except for Asians in which carrying −308 variant seemed to be protective [105].
Some studies reported that the presence of −308G/A TNF-α polymorphism may affect the individuals' response to supplementation of dietary components such as fish oil and vitamin E [71, 72]. In this context, Grimble et al. [71] reported that supplementing fish oil (6 g/day for 12 weeks) in healthy men decreased TNF-α production by lipopolysaccharides-stimulated peripheral blood mononuclear cells only in individuals with high TNF-α producer phenotype, irrespective of genotype. Nevertheless, the suppressive effect of fish oil supplementation on TNF-α production was greater among individuals in the highest tertile of presupplementation with the TNF1/TNF2 genotype compared to the wild-type individuals.
In a randomized, double-blind, placebo-controlled trial, elderly men and women received vitamin E supplementation (182 mg α-tocopherol) for one year. The ex vivo production of interleukin 1 β (IL-β), IL-6, and TNF-α was determined in whole blood samples incubated with lipopolysaccharide (LPS) (either 1.0 or 0.01 mg/L) for 24 h. Although there were no overall differences with respect to treatment group for any of the cytokines, when genotype was taken into account, participants with the A/A and A/G genotype at TNF −308G/A, treated with vitamin E, had lower TNF-α production than those with the A allele treated with placebo [72].
There is also some evidence that carrying TNF2 allele may influence diet response in obese individuals. Obese nondiabetic individuals were randomly allocated to 2 types of energy-restricted diets for 2 months: a diet lower in fat (LF: 1500 kcal/day, 52% carbohydrates, 20% proteins, and 27% fats) and a diet lower in carbohydrates (LC: 1507 kcal/day, 38% carbohydrates, 26% proteins, and 36% fats). The results of this study indicated that obese subjects carrying the TNF2 allele responded differently to the diets since, contrary to the wild genotype individuals, no improvement in plasma glucose, insulin, triglyceride, total cholesterol, low density lipoprotein (LDL), and blood pressure was observed. Despite the fact that both genotype groups have lost fat mass following the interventions, it is important to emphasize that such decrease in fat mass was slightly smaller in −308G/A individuals (LF diet: 5.0% versus 6.5%; LC diet: 4.9% versus 7.3%) [73].
Another study [74] reported significant interactions between dietary fat intake and TNFA −308G/A polymorphism affecting obesity risk in a sub-Saharan women population. When the dietary fat intake was 30 (% energy), the odds of being obese with the TNF-α GA+AA genotype was only 12% of that with GG, and at 35 (% energy) it was 33%. However, increasing intake of dietary fat (% energy) was associated with a significantly faster rate of increase in obesity risk in women with the TNF-α GA+AA genotype compared with those with the GG genotype (P = .036). For individuals with the GG genotype, the obesity OR was 1.12 and 1.26 for total dietary fat intakes of 35 and 40 (%E), respectively, compared with 30 (% energy). On the other hand, for individuals with the GA+AA genotype, the obesity OR was 3.02 and 9.12 for total dietary fat intakes of 35 and 40 (%E), respectively, compared with 30%E, indicating that individuals carrying A allele were significantly more responsive to an increase in dietary fat intake in their risk of being obese compared with normal weight.
Inflammatory status is characterized by the production of a wide range of mediators that work in a complex network. Thus, studying only one gene is not sufficient to understand the complex nutrient-gene interactions that take place in obesity-induced inflammation. In this context, a case-control study evaluated the additive effect of polymorphisms in TNF-α, lymphotoxin-α (LTA), and IL-6 genes in MS. The risk genotype, characterized by the combination of the LTA rs915654 A allele, the TNF GG, and the IL-6 rs1800797 GG genotype were associated with an increased risk of MS when compared to noncarriers (OR 2.10, 95% P < .005). Interestingly, such association was affected by plasma fatty acid profile. Among carriers of the 3 risk genotypes with the lowest 50th percentile of PUFA/SFA, MS risk was increased more than 4-fold compared with noncarriers (OR 4.40, P < .005). Similarly, a low PUFA/SFA exacerbated the risk of fasting hyperglycemia, high systolic blood pressure, abdominal obesity, and high plasma complement component 3 (C3) levels in these individuals. Also, MS risk was more than 5-fold higher in the risk genotype carriers with the highest SFA levels compared with noncarriers (OR 5.20, P < .005) [106].
Another TNF-α genetic variant that has received attention in the context of MS and chronic diseases is the TNF 238G/A polymorphism (rs 361525), which is located within a TNF gene repressor site whose effect on transcription activity still remains unknown [92]. The association between TNF 238G/A polymorphism and T2DM in case-control studies has generated conflicting results. A recent meta-analysis indicated that the TNF 238G/A polymorphism did not increase the risk of T2DM.
It has been suggested that there is linkage disequilibrium between the −238, −308, and −376 (rs1800750) polymorphisms. The −376A allele preferentially segregates with the −308G and the −238A alleles in most Africans and in Europeans [92, 122]. This fact has allowed the study of haplotypes, groups of SNPs that are inherited together in blocks. Using haplotype approaches to identify functional loci is interesting since multiple polymorphic loci may have combined effects on expression [97]. In the case of TNF-α gene, the haplotypes −238G, −308A, and −376G are associated with higher TNF-α production, while −238A, −308G, and −376A are associated with lower TNF-α production, which may depend on the racial group [92]. Unfortunately, at present, no study investigated the effect of diet or its components on obesity-induced inflammation according to individuals' TNF-α haplotype.
Selected studies on the main interactions of polymorphisms with dietary factors are in Table 1.
4. Interleukin-6
IL-6 is secreted by a wide variety of cells such as endothelial cells, keratinocytes, osteoblasts, myocytes, adipocytes, β pancreatic cells, monocytes, macrophages, and a number of other tissues, including a few tumors. This cytokine is essential in reducing the inflammatory process by promoting the synthesis of anti-inflammatory cytokines and by negatively regulating inflammatory targets. Therefore, this protein has been classified as both a pro- and anti-inflammatory; at certain level it acts as a defense mechanism but in chronic inflammation it has rather proinflammatory properties [123–125]
In humans, higher circulating IL-6 levels have been associated with obesity and visceral fat deposition [126–128], increased risk of impaired glucose tolerance, T2DM [126, 129–131], and high blood pressure [132, 133]. IL-6 is a central mediator of the acute-phase response and a primary determinant of hepatic production of CRP [134, 135]. Visceral adipose tissue secretes about two to three times more IL-6 than subcutaneous tissue, secreting also other molecules that stimulate further IL-6 expression.
The associations between common variations in the IL-6 gene and obesity have been examined in many studies [136–141]. The human gene of IL-6 is located on the short arm of chromosome 7 (7p21). In the last years many different SNPs in the promoter region of this gene were reported, like −572G/C, −373A(n)T(n), −597G/A (rs10242595), and −174G/C (rs1800795), the latter being the most prevalent and of greatest biological importance [142].
In a population-based cross-sectional study in Sweden, SNP −597G/A (minor allele frequency = 29%) 3′ of the IL6 gene was negatively associated with the primary outcome total body fat mass (BFM) (effect size −0.11 SD per A allele, P = .02). When results from three cohorts were combined (n = 8,927 Caucasians), allele A showed a negative association with total BFM (effect size −0.05 SD per A allele, P < .0002). Furthermore, the allele A was associated with lower BMI (effect size −0.03, P < .001) and smaller regional BFM [143].
As far as the SNP −174G/C (rs1800795) is concerned, three genotypes G/G, G/C, and C/C seem to affect the IL-6 transcription (116,119). Genotype G/C is more frequently reported by most studies [142]. In a population-based study including 9,960 Americans, it was shown frequencies of this SNP of 0.57 in Caucasians and 0.93 in Afro-Americans [144].
The presence of C allele has been related with a higher risk of obesity, high BMI, high waist circumference (WC), high leptin levels, and high BFM [145, 146]. Both studies with a Swedish and Canadian population reported association of the C allele with obesity indices [136]. Carriers of the CC genotype had lower energy expenditure and insulin sensitivity, hence implying a causative role in IR and obesity [77, 147]. The C allele was associated with higher BMI in T2DM individuals, but not amongst healthy subjects [134]. Studies relate G allele with an increase production of IL-6 [142]. The GG genotype in Spanish Caucasian was associated with T2DM [148]. In a Spanish population, the G allele was associated with hyperglycaemia with a reduction in insulin sensitivity [77] and with lipid profile disturbance [149]. Contradicting others findings, a recent study with 228 patients with T2DM and 300 healthy controls in Tunisia, the SNP −174G/C was not associated with T2DM or risk for overweight (P = .86). Bonferroni correction showed that the association of the SNP with T2DM susceptibility was restricted to overweight individuals and may be likely to be a random result [150]. On the other hand, it was described that the individuals with GG genotype lost weight significantly after aerobic exercises training. This effect was not observed in heterozygous neither the homozygous CC individuals who did not reduced the fat mass and insulin levels after the physical activity [146]. In addition we observed a higher incidence of G allele in subjects with normal weight (BMI < 25 kg/m2) [136].
Whereas several studies indicate that C allele is associated with higher plasma levels of IL-6, particularly in inflammatory situations [151, 152], in study including obese men, this SNP was associated with variation of plasma CRP concentration after weight loss [153]. A study with 504 Spanish adolescents shows no differences between genotypes observed in anthropometric values, body composition measurements, and plasma markers concentration. Physical activity level differs between genotypes with individuals carrying the C allele polymorphism being significantly more active than GG ones. The association between body fat mass and plasma glucose was influenced by this SNP. Those carrying the C allele of the mutation seem to have higher values of lipoproteins and CRP as their percentage of BFM increases [154]. Nevertheless, the C allele was more frequently observed (P = .032) in individuals with successful weight maintenance (<10% weight regain). Moreover, the presence of the Ala allele of the PPAR-γ together with the C allele of the IL-6 strengthens this protection (OR 0.19; P = .043), suggesting a synergetic effect of both variants on weight maintenance after a diet to lose weight [78].
Fernández–Real et al. [77], studying the same SNP, observed in healthy individuals that those with the G alelle presented concentration two times bigger of triglycerides than the ones with C allele, suggesting that the associated to a bigger secretion of IL-6 can predispose disturbs of lipid metabolism. Berthier et al. reported that GG allele was more common among lean men, and those with lower plasma concentrations of insulin and glucose following an oral glucose tolerance test [155].
Recent findings show that individuals with certain polymorphism can respond differently to diet than those without polymorphism. The association between variation in fat oxidation rates among obese subjects and genotype was studied for 42 common SNPs. Subjects with GC phenotype of SNP −174 had higher ability to increase fat oxidation after a high fat load (P = .01) [75]. In a Spanish study, after a 3-year intervention with a Mediterranean-style diet conducted in a high cardiovascular risk population, CC individuals with the −174G/C polymorphism were predicted to have the greatest reduction in body weight. At baseline these individuals had the highest body weight and BMI [76].
Therefore, the body of evidence suggests that variation in IL-6 gene is associated with metabolic and cytokine modulation, and subsequently may play an important role in impaired glucose and lipid homeostasis, cardiometabolic risk and seems to influence how the individual responds to fat intake. Results are still controversial and further studies on this issue are needed.
Selected studies on the main interactions of polymorphisms with dietary factors are in Table 1.
5. Interleukin-1β
Together with TNF-α and IL-6, IL-1β also plays a central role in the regulation of the immune responses and inflammatory process. IL-1β regulates the production of a variety of inflammatory mediators, such as IL-6, intercellular adhesion molecule (ICAM)-1, and E-selectin [156, 157], and it has been associated with metabolic disturbances present in obese subjects such as dyslipidemia and IR [158, 159].
The IL-1 family of genes comprises 9 genes, including the IL-1A, IL-1B, and IL-1 receptor antagonist gene (IL-1RN), which are all located on the long arm of human chromosome 2. Of the gene products, IL-1α and IL-1β are agonists, whereas IL-1 receptor antagonist (IL-1Ra) is a competitive antagonist of the IL-1 receptor and is, therefore, a negative regulator of inflammation [160, 161].
Current data indicate that some SNPs in IL-1B gene are associated with increased levels of IL-1β and other inflammatory markers such as CRP, suggesting a relationship between IL-1 gene variants and systemic inflammation. In patients with established coronary heart disease, IL-1B (+3954)T allele correlated with increased CRP levels, even after adjusting for smoking status, BMI, total cholesterol, and the presence of DM [162]. In a larger study performed in 454 subjects undergoing coronary angiography, individuals homozygous for IL-1B (+3954)T allele had 2- to 3-fold higher CRP levels than wild-type individuals, which remained significant after adjusting for gender, smoking, and age [163]. Another study reported that the presence of −511 T (rs1143627) decreased IL-1β production in LPS-stimulated mononuclear cells and that homozygous subjects for T genotype showed a decreased risk of myocardial infarction (OR 0.36) and stroke (OR 0.32) [164].
Given the role of IL-1RA in modulating the biological activity of IL-1, polymorphic variants in IL-1RN gene may affect IL-1RA production, influencing the inflammatory response. In fact, there is evidence that minor alleles of IL1RN 1018 (rs4251961) and 13888 (rs2232354) are associated with increased levels of inflammatory biomarkers, such as IL-1β, CRP, fibrinogen, IL-6, interferon-γ, and α-2 macroglobulin (a hepatic acute phase protein). Also, these polymorphisms are associated with lower IL-1RA concentration and ex vivo cellular production in response to an inflammatory stimulus [79, 165].
In cross-sectional study with a predominantly Caucasian population, adjusted odds ratio for MS was significantly greater in subjects carrying 6054G allele, while it was significantly lower in individuals carrying −511G and −1473G IL-1β allele. This study also provided information on the interaction between genotype and membrane fatty acid content, a surrogate of habitual dietary fatty acid intake, in determining MS risk. Among individuals in the lowest 50th percentile of total PUFA or total n-3 PUFA—eicosapentaenoic acid + docosahexaenoic acid (EPA+DHA)—carriers of 6054G had a higher prevalence of MS when compared to wild-type individuals; such association was not found in the highest 50th percentile of total PUFA or total EPA+DHA membrane content, which suggests that EPA+DHA may be helpful in preventing MS in 6054G genetic predisposed individuals. The authors also examined a haplotype consisting of 5 SNPs: 6054G/A, 3966C/T, 231T/C, 2511G/A, and 21473G/C and found that individuals with haplotype 11222 had higher prevalence of MS than haplotype 21111. Among those with low DHA+EPA membrane content, there was significant haplotype association with MS, being OR of MS for haplotype 11222 significantly higher than for haplotype 21111. However, there was no significant association among individuals with high membrane DHA+EPA content [166].
Another study found an association between the prevalence of hypertension and the presence of −31C allele, which could be modified by plasma β-carotene levels. Male CC carriers with high serum β-carotene levels had a significantly lower OR (OR 0.25, P < .05) for hypertension relative to those with low serum β-carotene levels. The association of the IL-1B −31C/T polymorphism with hypertension was weak in women with high β-carotene circulating levels, but, in those with low β-carotene levels, the TT genotype clearly increased the prevalence of hypertension (OR 2.47, P < .05). The results were not adjusted to some known risk factors for hypertension as smoking/drinking habits, menopause, and diet [80].
A randomized placebo-controlled trial was carried out in healthy adults to evaluate the effect of 12-week supplementation with a formulated botanical extract rich in dehydroascorbate, anthocyanins, and all-trans-resveratrol (1200 mg/d of rose hips extract, 165 mg/d of blackberry powder, 330 mg/d of blueberry powder, and 40 mg/d of grapevine extract) on inflammation in individuals with genetic variations that predispose to overexpression of IL-1β. Individuals were stratified by IL-1 genotype prior to randomization. They were classified as IL-1 positive genotype if they had any of the following 3 genotypes: (1) homozygous for the common allele (C) at IL1B (−511); (2) carrying 2 copies of the less common allele (T) at IL1A (+4845); or (3) carrying one copy of the less common allele at IL1A (+4845) plus at least one copy of the less common allele (T) at IL1B (+3954). IL-1 positive genotype individuals receiving the formulation experienced a greater reduction in IL-1B gene expression and in plasma CRP levels [81].
Current data is clearly insufficient to draw definite conclusions; however, it seems likely that some polymorphisms in IL-1 gene may affect diet response in some proinflammatory conditions.
Selected studies on the main interactions of polymorphisms with dietary factors are in Table 1.
6. Apolipoprotein A1
Apo A1 is the major protein component (roughly 70%) of high-density lipoproteins (HDL), which is synthesized primarily in the liver and small intestine, and acts in the transport of cholesterol from the peripheral tissues to the liver. Apo A1 plays a crucial role in the regulation of reverse cholesterol transport [167–169]. Upon secretion into plasma, Apo A1 is lapidated through ATP-binding cassette transporter-(ABC-)A1 that mediated efflux of free cholesterol and phospholipids, resulting in the formation of nascent disc shaped HDL particles [170].
More than a dozen functionally significant mutations of the Apo A1 gene have been described, including gene disruptions, nonsense mutations, frameshifts, missense mutations, chromosomal aberrations or deletions, and inversion of the APOA1/APOA3/APOA4 gene cluster; these are typically associated with decreased plasma HDL concentration [171–174].
Overexpression of the human Apo A1 gene in mice increased plasma HDL concentration and protected the animals from the development of high-fat diet-(HFD-)induced atherosclerosis [175]. Conversely, Apo A1 knockout mice exhibited decreased plasma HDL concentration and developed atherosclerotic lesions [176].
Apo A1 missense mutations are a type of nonsynonymous mutation that causes a single nucleotide change, resulting in a codon that codes for a amino acid, which turns the protein without function. This kind of structural variation have also been found and, although there are exceptions [177], most of these do not change plasma HDL concentration. However, a well-known variant of the Apo A1, called A-I Milano (apoA-IM), is associated with reduced HDL and LDL levels [178, 179]. This polymorphism is a variant form of Apo A1 that contains a cysteine replaced by arginine at amino acid 173 (R173C) [180]. Moreover, Calabresi et al. [181] showed that carriers of the apoA-IM mutation have an increased postprandial lipemic response. Another polymorphism called PstI (rs12721026) is located in the 30 flanking region of Apo A1 gene and has been associated with the development of cardiovascular disease and decreased plasma HDL concentration [182, 183].
A common G to A substitution in the promoter area at position −76 bp (−76G/A) of the Apo A1 gene has been extensively studied and some researchers have observed that carriers of this polymorphism presented an increase in the promoter activity in vitro and plasma HDL concentration [184, 185]. Also, Marín et al. [186] observed that carriers of this mutation have a greater postprandial increase in large triglyceride-rich lipoproteins and a smaller decline in LDL and Apo B, after the ingestion of a dietary fat load than carriers of the G/G genotype.
The minor allele of MspI polymorphism, consisting of a G to A transition at position −75 bp (rs799837), has been associated in a number of studies with higher Apo A1 and HDL plasma levels [187], although others were unable to confirm this relation [188]. Moreover, such polymorphism may contribute to variability in postprandial lipid metabolism and in the lipoprotein response to dietary changes in healthy subjects [107].
Some dietary factors have been extensively studied and related to the mutation of Apo A1 gene. The total dietary fat and the type of fat are the main cited interactions resulting in serum lipids alterations. Gomez et al. [107] investigated whether the presence of the −76G/A SNP in the Apo A1 gene interacts with diet to determine changes in LDL particle size and their susceptibility to oxidative modifications. In a second step, they examined these effects by analyzing the contribution of the combination of the Apo A1 −76G/A and Apo A4 Thr347Ser SNP. Each of 97 healthy volunteers consumed 3 types of diet for 4 weeks: a high-saturated fat diet (38% fat, being 20% SFA), 12% monounsaturated fatty acids (MUFA), and 6% polyunsaturated (PUFA) followed by a low-fat and high-carbohydrate (CHO) diets (30% fat, 55% CHO) or a MUFA diet (38% fat, 22% MUFA). After consuming the CHO diet, there was a significant decrease in LDL size with respect to high-fat diets in GG homozygotes for the Apo A1 −76G/A SNP. However, LDL size did not differ in GA carriers among participants consuming the 3 diets. Carriers of the A allele for this polymorphism had smaller LDL size as well as increased susceptibility to oxidation after the SFA diet than the GG homozygous.
Ordovas et al. [108] examined whether dietary fat modulates the association between the −75G/A of Apo A1 polymorphism and plasma HDL concentration. Participants (755 men and 822 women) of the Framingham Offspring Study were divided into low (<4% of energy), medium (4–8% of energy), and high (>8% of energy) PUFA intake. Women who were carriers of the A allele submitted to high PUFA intake increased plasma HDL concentration significantly, but in homozygous women for the G allele an inverse relation was found. This meant that when PUFA intake provided <4% of energy, women who were homozygous for the G allele had almost 14% higher plasma HDL concentration than A allele carriers and when PUFA intake provided >8% of energy, plasma HDL concentration in carriers of the A allele were 13% higher than those of G/G individuals. Thereby, a significant interaction in terms of plasma HDL concentration was observed between Apo A1 genotype and PUFA intake. No significant association was detected in men.
The soy protein has been associated with improvement on lipid levels and the mechanisms by which it acts include several metabolic pathways regulated by different transcription factors. Animals who consumed a long-term of soy protein had a decrease in insulin/glucagon ratio, due mainly to a reduction in insulin secretion [189] and an increase in glucagon concentration, that leads to an expression reduction in the transcriptional factor sterol regulatory element-binding protein (SREBP)-1 [190], which is involved in fatty acid synthesis and triglyceride (TG) esterification [191].
The effectiveness of a dietary portfolio consisting of soy protein (25 g/day) and soluble fiber mix (Fibregum—100% made of gum acacia, which has 90% of soluble dietary fiber—15 g/day), integrated in a low SFA diet, on blood lipids and the association between this diet and the Apo E, Apo A1, and ABCG5/8 polymorphisms were investigated in hyperlipidemic individuals [192]. Significant decreases in plasma total cholesterol and TG concentrations was observed; 51% of the individuals had >20% reduction in plasma total cholesterol concentration, and 77% had a reduction >20% in plasma TG concentration. Among hypercholesterolemic individuals, 14% had the ABCG8 (+52G/C) polymorphism, 65% had the ABCG5 (+1950 C/G and G/G) polymorphism, 53.5% the Apo A1 (−75G/A and A/A) polymorphism, and 23.3% had the Apo E (ε4) polymorphism. The presence of ABCG5/8 has been associated with reduction of absorption of plant sterols and cholesterol from the diet by effluxing these sterols from the enterocyte back into the intestinal lumen and by facilitating efficient secretion of plant sterols and cholesterol from hepatocytes into the bile [193]. However, independently of genotype, the combination of cholesterol-lowering foods in a low-saturated fat diet improved lipid profile. Another interesting study examined the effect of the dietary fat saturation when −75G/A mutation was present. Fifty men and women were first fed with a saturated fat diet (17% SFA) for 28 days, followed by a MUFA diet (22% MUFA) for 35 days and a PUFA diet (13% PUFA) for 35 days. The allele frequency for the A allele was 0.13 and individuals who carry this allele had higher cholesterol, LDL, and TG levels than those with G/G. In women with the A allele, the PUFA diet was compared to the saturated-fat-diet-induced significant decreases in total and LDL concentrations. These results suggested that the G/A polymorphism appears to have a small but significant effect on plasma LDL responsiveness to changes in dietary fat saturation specially in women [109].
Selected studies on the main interactions of Apo polymorphisms with dietary factors are in Table 2.
Table 2.
Summary of studies evaluating interaction between diet and variants of genes involved in lipoprotein metabolism.
| Gene | Variant | Population [reference] | Frequency | Design | Main findings |
|---|---|---|---|---|---|
| −76G/A (rs 1799837) | 97 subjects recruited among students of the University of Cordoba at age-range 18–49 years [107] | G allele: 41,2% A allele: 58,8% |
Clinical trial followed by a randomized crossoverThe subjects consumed three diets for four weeks: SFA diet, CHO diet or MUFA diet. | After the participants consumed the CHO diet, there was a decrease in LDL size with respect to high-fat diets in GG homozygotes for the carriers of −76G/A (P = .05). LDL size did not differ in GA carriers. Carriers of the A allele for this polymorphism had smaller LDL size as well as increased susceptibility to oxidation after the SFA diet than the GG homozygous (P = .045). | |
| Apo A1 | −75G/A (rs 670) | 1,577 subjects from the Framingham Offspring Study [108] | G allele: 83,5% A allele: 16,5% |
Cross-sectional | When PUFA intake was across <4% of energy, the G allele carriers had ~14% higher plasma HDL-c than did carriers of the A allele (P = .05). However, when PUFA intake was >8%, HDL-c levels in the A allele carriers were 13% higher than those of G/G subjects (P = .05). These interactions were not significant in men |
| 50 subjects voluntaries members of two urban religious communities with 47,1 medium age [109] | G allele: 87% A allele: 13% |
Clinical Trial Subjects were first fed a SFA diet for 28 days, followed by a MUFA diet for 35 days and a PUFA diet for 35 days. | The A allele carriers had higher plasma CT, LDL-c, and TG levels than the G homozygous allele (P < .05). PUFA diet-induced significantly greater CT (P = ,003) and LDL-c decreases (P = ,001) in G/A women than in G/G as compared to SFA diet. The variability in LDL-c response from the SFA diet to the PUFA diet in women was associated with LDL-c (55,1%), waist hip ratio (11,4%), and the G/A polymorphism (10%). | ||
| 3,093 French Caucasian subjects with T2DM [110] | In normal glycemia T: 63%, C: 62% In T2DM subjects T: 37%, C: 38%. |
Case-control study | Marginally associated with CT levels (P = .026) and waist-to-hip ratio (P = .029). Not associated with T2DM. | ||
| Apo A2 | −256T/C (rs 5082) | 3,462 subjects from Framingham Offspring Study (1,454 Whites), The GOLDN Study (1,078 Whites) and BPR Study (930 Hispanics of Caribbean origin) [111] | CC subjects between Framingham and GOLDN: 15%; in BPR study: 10,5%. |
Cross-sectional, follow-up (20 years), and case-control analyses. | No significant association with HDL-c. When SFA intake is low (≤22 g/d), the SNP does not affect BMI. When SFA intake is high (≥22 g/d), SNP is associated with BMI and obesity: a mean increase of 6.2% BMI (P < .05). CC genotype was associated with higher obesity prevalence in all populations only in the high-SFA intake stratum. |
| 4,602 subjects from two independent populations: –high–cardiovascular risk Mediterranean –multiethnic Asian population including Chinese, Malays and Asian Indians) [112] | Frequency of CC subjects differed strongly among Chinese, Malays and Asian Indians (1–15%). | Cross-sectional study: analyzed gene-diet interactions between the APOA2 −265T>C and SFA intake on BMI and obesity. | In Mediterranean individuals, the CC genotype was associated with a 6.8% greater BMI in those consuming a high-SFA diet (≥22 g/d), (P = .018), but not a low (≤22 g/d), (P = .316) SFA diet. CC genotype was associated with higher obesity prevalence in Chinese and Asian Indians only with a high-SFA intake (P = .036). | ||
| 88 normolipidemic young men from Spain [113] | CT+CC: 60% TT: 40% |
Clinical trial: subjects were given a fatty meal containing 1 g fat and 7 mg cholesterol/kg weight and capsules containing 60,000 IU vitamin A. Postprandial lipemia was assessed during the 11 h following the meal. | Carriers of the C allele have significantly lower postprandial increases in plasma total TG and chylomicron TG, suggesting a protective effect against cardiovascular disease. | ||
| 2,148 subjects from the Framingham Offspring Study with 50,45 medium age [114] | T allele: 86,6% C allele: 13,4% C allele: 88,3% G allele: 11,7% |
Cross-sectional | Significant interactions between the −1131T/C and PUFA intake was found (P = .001) in determining fasting plasma TG, RLP, and particle size. The same results could not be observed in 56 C/G polymorphism. In individuals who consumed a high PUFA diet (6% of the energy), the −1131T/C was associated with higher fasting TG and RLP concentration (P = .01). The size of VLDL increases and LDL decreases while PUFA intake increased in the −1131T/C carriers. The PUFA Apo A5 interactions were specific for dietary n-6 fatty acids. | ||
| Apo A5 | −1131T/C (rs 662799) 56C/G (rs 3135506) | 2,280 subjects from the Framingham Offspring Study with 54,2 medium age [115] | T allele: 87,15% C allele: 12,85% C allele: 88,95% G allele: 11,05% |
Cross-sectional | A significant interaction between −1131T/C and total fat in relation to the BMI (P = ,001) was found. The −1131C minor allele carriers had a lower obesity (P = .032) and overweight (P = .031) risk when compared with TT subjects in the high fat intake group, but not when fat intake was low (P = .48). The monounsaturated fatty acids intake showed the highest statistical significance for these interactions. |
| 1,020 of the Boston Puerto Rican Health Study at age 45–75 [115] | The pairwise LD coefficient R between the APOA5 −1131T/C and 56C/G was 0,016 | Cross-sectional | The 56C/G polymorphism was associated with HDL-c (P = .044). The minor allele carriers had lower HDL-c than those with common variant (P = .012). Both polymorphisms were associated with TG or other lipids. Associations of the −1131T/C with total fat energy intake was observed for TG (P = .032) and CT (P = .034). | ||
| −1131T/C (rs 662799) | 49 male subjects at age 28–55 years were recruited from volunteers who responded to an advertisement for a nutrition study conducted by the Clinical Nutrition Research Team at Yonsei University [116] | TT: 46,9% TC: 36,8% CC: 16,3% |
Clinical Trial The subjects were randomly assigned to consume one of two types of experimental enteral formulae (LF versus HF) with a seven-day interval. | Fasting total TG were higher in TC+CC men than TT men, but fasting chylomicron TG were not significantly different between TT men and C carriers. TT subject had no significant difference in postprandial responses of total TG and chylomicron TG between LF and HF meal. C carriers had delayed peak time of total TG compared to TT subject and higher postprandial response at HF meal when compared to LF meal. | |
| 299 healthy male at age 20–75 of the 5th Framework Program, [117] | TT: 84,51% TC: 15,16% CC: 0,34% |
Cross-sectional | Individual who had the C allele presented higher plasma TG, VLDL-C, and LDL-c levels. Plasma α-tocopherol was increased in C allele carriers compared with homozygote T allele carriers (P = .02). | ||
| Apo E | Apo E2 rs429358 (T) + rs7412 (T) Apo E3 rs429358 (T) + rs7412 (C) Apo E4 rs429358 (C) + rs7412 (C) | 22,915 subjects at age 45–75 years from Norfolk arm of the European Prospective Investigation of Cancer (EPIC) [118] | 0,6% ε2/ε2 12,4% ε2/ε3 2,6% ε2/ε4 58,6% ε3/ε3 23,5% ε3/ε4 2,3% ε4/ε4 |
Cross-sectional | Individuals who have ε4/ε4 genotype presented the highest serum CT and LDL-c and lowest HDL-c and TG (P = .001). There were positive associations between total and saturated fat from usual diet and serum total and LDL-c, and an inverse associations (P = .001) between polyunsaturated fat, dietary fiber, and lipid fractions overall. Associations were in the same direction for ε2, ε3, and ε4 expressing individuals with no significant interactions between diet and genotype group on blood lipids, except in those who expressed ε2/ε43% (P = .05). |
| 84 subjects from students at the University of Cordoba at age 21–55 years [119] | 9,5% ε4/ε3 78,6% ε3/ε3 11,9% ε3/ε2 |
Clinical trial: subjects consumed for 28 days a SFA-rich diet. After this, they were randomly assigned to one of two diet sequences: the first one received a MUFA-rich diet for 28 days, followed for more 28 days with CHO-rich diet. The other group consumed CHO diet before the MUFA diet. | Apo ε2 carriers presented the highest Apo E plasma levels, while Apo ε4 individuals showed the lowest concentration after the SFA, CHO, and MUFA diets (P = .023 for men and P = .034 for women). The Apo E was higher in women (P = .009) when compared with men after they consumed the SFA diet. In Apo ε3/ε2 and Apo ε3/ε3 carriers, the shift from the SFA to CHO or MUFA-rich diets decreased the Apo E concentration in women (P = .041). Sex and Apo E genotype determine the Apo E and plasma levels, but this effect is dependent on dietary fat. | ||
| Apo E2 rs429358 (T) + rs7412 (T) Apo E3 rs429358 (T) + rs7412 (C) Apo E4 rs429358 (C) + rs7412 (C) | 132 clinically healthy Caucasians subjects at age 40–69 years [120] | 0,8% ε2/ε2 14,4% ε2/ε3 3,0% ε2/ε4 64,4% ε3/ε3 17,4% ε3/ε4 |
Cross-sectional | A significant correlation between CT and energy intake derived from total (P = .025) and saturated fat (P = .046) was observed. Carriers of the Apo ε3/ε4 genotype displayed a stronger positive correlation between serum LDL-c level and percentage of energy derived from intake of saturated fat (P = .043). The individuals who carried the Apo ε2 allele demonstrated a positive correlation between alcohol consumption and serum HDL-c level (P = .01). | |
| 121 subjects recruited by advertising in Gothenburg's major newspaper at age-range 30–65 years [121] | 7% ε2/ε2 7,4% ε2/ε3 1,7% ε2/ε4 57% ε3/ε3 29,8% ε3/ε4 2,5% ε4/ε4 |
Clinical Trial: the subjects were submitted to a four intervention periods: 1 and 3 a coffee free period of three weeks, 2 and 4 600 mL coffee/day for four weeks. | The ApoE ε2 allele showed an inverse association with serum CT concentration (P = .01), but the Apo E polymorphisms do not influence the cholesterol-raising effect of coffee. |
BMI: body mass index; BPR: Boston-Puerto Rican Study; CHO: carbohydrate; CT: total cholesterol; GOLDN Study: Genetics of Lipid Lowering Drugs and Diet Network (GOLDN) Study; HDL-c: High density lipoprotein-cholesterol; HF: High fat; LDL-c: Low density lipoprotein-cholesterol; LF: Low fat; MUFA: Monounsaturated fatty acids; PUFA: Polyunsaturated fatty acids; RLP: Remnant-like particle; SFA: saturated fatty acids; SNP: single nucleotide polymorphism; T2DM: type 2 diabetes mellitus; TG: triglycerides; VLDL-c: Very low density lipoprotein-cholesterol.
7. Apolipoprotein A2
Synthesized in the liver, apolipoprotein A2 (Apo A2) is the second most abundant protein of HDL particles and can be found with the Apo A1 in subfractions of HDL. However, its function remains largely unknown [111]. Although some initial studies reported an inverse relationship between plasma Apo A2 concentration and cardiovascular risk [194, 195], findings of subsequent studies did not reveal significant associations or even suggest a proatherogenic role [196–198].
Several SNPs have been reported in Apo A2 gene, but only MSP-I and −265T/C have been associated with plasma lipid concentration [113]. MSP-1 was analyzed in a study of 1,102 individuals from the Pacific island of Kosrae, in which several other candidate genes for increased cardiovascular risk were also tested. The carriers of the less frequent MSP-1 allele had higher serum TG concentration and, interestingly, reduced blood pressure [199].
The most studied SNP in the literature is the −265 T/C (rs5082) that may affect element D of the Apo A2 promoter, reported to be functional in 2 independent studies, in which C allele was associated with decreased plasma Apo A2 concentration [200, 201]. In agreement, a meta-analysis of data from 12,387 subjects found no association of this SNP with T2DM [110]. The C allele was associated with waist circumference in men [200] and in white woman, but not in African-American woman [202]. In addition, plasma Apo A2 concentration was significantly lower in carriers of C allele, while postprandial Apo B100 in the large VLDL particles (Sf > 60) were lower only in those in homozygous for the C allele (P = .01) [200]. Discordantly, in other study carriers of the C allele had significantly lower postprandial increases in plasma total and chylomicron TG, suggesting a protective cardiovascular effect, which may be related to the lowered risk of postprandial hypertriglyceridemia [113].
A gene-diet interaction influencing BMI and obesity has been strongly and consistently replicated in many studies. Interactions between the −265T/C and SFA intake on BMI and obesity in 3,462 individuals from three American populations: Framingham Offspring Study: (FOS) (1,454 Whites), Genetics of Lipid Lowering Drugs and Diet Network: (GOLDN) Study (1,078 Whites), and Boston-Puerto Rican: (BPR) Study (930 Hispanics of Caribbean origin). Frequencies of CC individuals did not differ between FOS and GOLDN (15% in both), but this was lower in the BPR Study (10.5%). No significant association of the SNP with plasma HDL-c concentration was found among all the three populations. Also, statistically significant interactions between this SNP and SFA intake were detected: when SFA intake is low (≤22 g/d), the −265T>C SNP did not affect BMI. However, when SFA intake was high (≥22 g/d), this SNP is strongly associated with BMI and obesity: a mean increase of 6.2% BMI (4.3%–7.9%; P < .05). Moreover, the CC genotype was significantly associated with higher obesity prevalence in all populations only in the high-SFA intake stratum [111]. Similar results were showed in a cross-sectional study with 4,602 subjects from two independent populations (a high-cardiovascular risk Mediterranean and a multiethnic Asian population including Chinese, Malays, and Asian Indians). In this study, the frequency of CC subjects differed strongly among populations (1–15%) and an interaction of Apo A2 with saturated fat on body weight was confirmed: in Mediterranean subjects, the CC genotype was associated with a 6.8% greater BMI in those consuming a high (P = .018), but not a low (P = .316) SFA diet. Likewise, the CC genotype was significantly associated with higher obesity prevalence in Chinese and Asian Indians, only those with a high SFA intake (P = .036) [112].
Despite the evidences of the influence of the ApoA2 −265T/C polymorphism on body-weight-related measures, modulated by saturated fat consumption, in different populations, its role in the control of circulating lipoproteins and cardiovascular risk profile is not completely understood.
Selected studies on the main interactions of Apo polymorphisms with dietary factors are in Table 2.
8. Apolipoprotein A5
It was well known that the main genetic factor related to the plasma triglyceride determination is apolipoprotein C3, however trying to better understand the mechanisms, an intensive investigation of DNA sequence around the APOA1/APOC3/APOA4 gene cluster was conducted [203]. Therefore the available mice and human sequences (about 200000 bp) around this locus were completed by sequencing and compared, leading to the identification of evolutionary highly conserved sequence that contained a putative lipid-binding apolipoprotein gene, named the Apo A5 [204, 205]. The human Apo A5 gene consists of 4 exons and codes 369 aminoacid protein, which is expressed almost exclusively in the liver [204]. Apo A5 is located on triglyceride rich particles (chylomicrons and very low density lipoproteins—VLDL) and HDL particles. An inverse relationship between APO A5 and TG levels has been described in animal studies, in which knockout mice developed hypertriglyceridemia and transgenic mice overexpressing APO A5 reduces plasma TG levels [204].
In comparison to other apolipoproteins, the plasma concentration of Apo A5 is low in human—about 100 μg/L [206]. In addition, researches confirm that Apo A5 binds to and enhances the activity of lipoprotein lipase (LPL) enzyme and, consequently, reduces triglyceride levels in VLDL particles. Moreover, the treatment in mice with Apo A5 lead to a reduction of VLDL-triglyceride production rate, but the concentration of the VLDL particles was the same as in normal mice [206, 207]. These results confirmed that Apo A5 plays a role in the LPL activation.
To identify common polymorphisms, extensive sequencing of the Apo A5 interval in humans has been performed. A set of 4 common polymorphisms (SNPs1 through 4; also named 259T/C, IVS3+476G/A, −1131T/C, and −12,238T/C, resp.) were first identified within the human Apo A5. Statistical analysis indicated that the minor alleles SNPs 1 through 3 formed a relatively common haplotype that is found in approximately 15% of Whites [204, 208].
Subsequently, through direct DNA sequencing of the gene in 116 hyperlipidemic individuals, 9 additional SNPs were identified. One of the polymorphisms (−3A/G) was found to be in strong linkage disequilibrium with the minor alleles for SNPs 1 trough 3 and this haplotype was named APOA5*2. In addition, a second common polymorphism was also identified, which results in a C to G nonsynonymous substitution (56C/G) that changes codon 19 from serine to tryptophan. Further haplotype analysis in Whites indicated that the minor allele of this polymorphism defines a third common Apo A5 haplotype (APOA5 *3), and this one was also found in almost 15% of Whites. The remaining 7 polymorphisms from this study were either uncommon or not obviously associated with plasma triglycerides concentration [208].
Thus, polymorphism discovery and haplotype analysis in Whites defined 3 common haplotypes in the Apo A5 interval and provided detailed information for genetic association studies in humans. In these analyses, the −1131T/C allele (SNP3-rs662799) was used as a marker to define APOA5 *2, whereas the 56C/G allele (S19W-rs3135506) was used to define APOA5 *3 [208].
Carriers of the less frequent allele −1131T/C have higher concentrations of plasma triglycerides, both in fasting [209] and postprandial [210] states, and total cholesterol, lower blood HDL-c [211], smaller LDL particles [212], and are at higher cardiovascular risk [213]. Moreover, carriers of allele 56C/G show an increase in triglyceride levels, independently of the effects observed for the −1131T/C, as well as an increased risk of suffering from atherosclerosis [214].
In this context, it is most commonly found in the literature studies that related −1131T/C and 56C/G SNPs to dietary factors. Thus, Lai et al. [114] have proposed to investigate the interaction between Apo A5 gene variation and dietary fat in determining plasma fasting triglycerides, remnant-like particle (RLP) concentrations, and lipoprotein particle size in 1001 men and 1147 women who were Framingham Heart Study participants. They found a significant gene-diet interactions between the −1131T/C polymorphism and PUFA intake in determining fasting triglyceride, RLP concentrations, and particle size. However, these interactions were not observed for the 56C/G polymorphism. The −1131C allele was associated with higher fasting triglyceride and RLP concentrations (P < .01) only in the individuals consuming a high-PUFA diet (>6% of total energy). Similar interactions were found for the sizes of VLDL and LDL particles. Only in carriers of the −1131C allele did the size of these particles increase (VLDL) or decrease (LDL) as PUFA intake increased (P < .01). The authors further analyzed the effects of w-6 and w-3 fatty acids, and found that the interactions between PUFA and Apo A5 were specific for dietary n-6 fatty acids.
Another research involving the Framingham Heart Study participants concluded that Apo A5 gene variation modulates the effects of dietary fat intake on BMI and obesity risk. The authors investigated the interaction between the Apo A5 −1131T/C and 56C/G polymorphisms and the macronutrient intake (total fat, CHO, and protein) in their relation to the BMI and obesity risk. They found a significant interaction between the −1131T/C SNP and total fat intake for BMI. In individuals homozygous for the −1131T major allele, BMI increased as total fat intake increased. In contrast, this increase was not present in carriers of the −1131C minor allele. Significant interactions were found in determining obesity and overweight risks, since the Apo A5 −1131C minor allele carriers had a lower obesity risk (OR 0.61, P = .032) and overweight risk (OR 0.63, P = .031) compared to TT individuals in the high-fat intake group (≥30% of energy) but not when fat intake was low (OR 1.16, P = .47 and OR 1.15, P = .48) for obesity and overweight, respectively. When specific fatty acid groups were analyzed, MUFA showed the highest statistical significance for these interactions [215].
A more recent study [115] that aimed to determine the association of the same Apo A5 polymorphism previously mentioned with plasma lipids and markers of MS, alone and in interaction with total fat intake in Puerto Ricans (n = 802, 45–75 yrs) found interesting results. Apo A5 S19W was associated with HDL (P < .05). Neither polymorphism was associated with triglyceride or other lipids, but interaction of the −1131T/C SNP with total fat energy intake was observed for plasma triglyceride (P = .032) and total cholesterol (P = .034). Apo A5 56C/G interacted with total fat intake in association with systolic and diastolic blood pressure (P < .001).
As previously cited, Apo A5 mutations have been extensively related to the postprandial lipemic response. Kim et al. [116] compared low-fat (LF) meal and high-fat (HF) meal on the postprandial lipemic responses according to the −1131T/C polymorphism of the APOA5 gene. Fasting total triglycerides were higher in heterozygous for the C allele (TC) and homozygous for C allele (CC) men than homozygous for the T allele (TT) men, but fasting chylomicron TG were not significantly different between TT men and C carriers. TT individuals had no significant differences in postprandial responses of total TG and chylomicron TG and postprandial mean changes of chylomicron TG between LF and HF meal. On the other hand, C carriers had delayed peak time of total TG compared to TT individuals and higher postprandial response and mean changes of chylomicron TG at HF meal compared to LF meal. The authors hypothesized that these facts occur due to the limiting capacity to clear chylomicron TG or hydrolyze TG on HF diet in TC and CC men, resulting in higher postprandial triglyceridemia.
Vitamin E also has been related to Apo A5 polymorphisms since it is a lipophilic micronutrient that is also transported within lipoproteins. The effects of the Apo A5 −1131T/C gene variant according to vitamin E status (α-tocopherol, γ-tocopherol, buccal mucosa cells total vitamin E, LDL α-tocopherol, and LDL γ-tocopherol) and lipid profile (VLDL, HDL, intermediate density lipoprotein (IDL), and LDL) were investigated. C allele carriers showed significantly higher TG, VLDL, and LDL concentrations, higher cholesterol in VLDL and IDL, and higher plasma fatty acids. Plasma α-tocopherol was increased significantly in C allele carriers compared with homozygote T allele carriers (P = .02), suggesting that higher plasma lipids in the TC and CC genotypes were efficiently protected against lipid peroxidation by higher plasma α-tocopherol concentration [117].
Selected studies on the main interactions of Apo polymorphisms with dietary factors in Table 2.
9. Apolipoprotein E
Apo E was discovered in 1970 as a component of triglyceride-rich lipoproteins [216]. It is an amphipathic 299 amino acid glycoprotein of 34,145 KDa that is mainly secreted by hepatocytes [217], but expressed in the brain and liver [218].
The primary functional role of Apo E is to transport and deliver lipids mainly through the LDL-c receptor pathway. A secondary proposed pathway involves the heparin sulphate proteoglycan (HSPG)/LDL-C receptor-related protein pathway [219]. Apo E acting as a ligand for these receptors [220] plays a crucial role in determining the metabolic fate of plasma lipoproteins and consequently of cholesterol [220, 221], while its accumulation on the surface of lipoproteins can decrease the lipolysis rate of TG by lipase [222–224]. Furthermore, Apo E as a component of HDL influences the cholesterol influx and efflux of cells [225–227].
Plasma Apo E isoforms have two kinds of polymorphism, one genetically determined and one not. The former polymorphism is the result of three alleles, called epsilon (ε) alleles: ε2, ε3, and ε4 at a single gene locus and that differs in 112 e 158 position of the aminoacids (Apo E2: Cys112-Cys158; Apo E3: Cys112-Arg158; Apo E4: Arg112-Arg158). The relative frequency in white population of Apo E2, Apo E3, and Apo E4 alleles is 0,08, 0,77, and 0,15, respectively [228]. This alleles can form six genotypes ranking in order of most to least common (ε33, ε34, ε23, ε44, ε24, and ε22) [229]. The ε33 genotype, being the most common, is used as reference for all Apo-E-related functions [230]. The nongenetically determined polymorphism results from variable posttranslational sialyation of Apo E and accounts for 10%–20% of plasma Apo E [219]. In addition, Apo E isoforms are important determinants of postprandial lipemia [231]. It has been demonstrated that Apo E2 homozygous subjects have the lowest affinity for TRL remnant receptors, and this genotype is associated with delayed postprandial clearance. A recent Brazilian study indicates that carriers of Apo E4 had a positive association with higher total cholesterol (P < .001), LDL-C (P < .001), total-cholesterol/HDL-C ratio (P < .001), LDL/HDL-C ratio (P < .001), lower HDL-C values (P < .001), and higher risk to obesity (OR = 1.358, 95% CI = 1.019–1.811) [232]. Furthermore, patients with MS, with genotypes other than the Apo E3/3, are at greater risk of postprandial hypertriglyceridemia and hyperuricemia following the acute ingestion of a fat overload [233]. Several studies have shown interaction between the postprandial lipemic response and Apo E polymorphisms. Subjects who are carriers of the −219T allele, which is located in the promoter region of the Apo E gene, −219G/T, display lower levels of Apo E in plasma in both fasting and postprandial states, and this effect was associated with a stronger postprandial response, and a decrease in transcription activity [231].
Moreno et al. [119] evaluated whether the quantity and quality of dietary fat interacts with the Apo E genotype and sex modifying plasma Apo E levels in young healthy subjects. The participants were subjected to three dietary periods, each one of 4 weeks. The first was a SFA-enriched diet (38% fat, being 20% SFA, 12% MUFA, and 6% PUFA), which was followed by a CHO rich diet (55% CHO, <30% fat, being 10% SFA, 12% MUFA, and 6% PUFA) or a MUFA-rich diet (38% fat, being <10% SFA, 22% MUFA and 6% PUFA). Apo E2 carriers have the highest Apo E levels, whereas Apo E4 individuals show the lowest concentration after the saturated fat, CHO, and MUFA diets. Women had significantly higher Apo E concentration than men only after the consumption of the high-saturated fat diet. The SFA-enriched diet increased the Apo E plasma concentration when compared with the CHO-, and MUFA-rich diets in women, but not in men. In women, but not in men, the shift from the SFA- to CHO- or MUFA-rich diets significantly decreased the Apo E concentration in apoE3/2 and apoE3/3 subjects, whereas no differences were observed in women with the apoE4/3 genotype. They concluded that sex and Apo E genotype determine Apo E circulating levels; however, this effect is dependent on dietary fat.
Loktionov et al. [120] investigated the relationship between Apo E genotype and the effects of a CHD-promoting diet in free-living individuals. No relationship between dietary fats and cholesterol levels were observed in the E2 or E3 group when analyzed separately. However, the presence of the E4 allele resulted in positive associations between total and saturated fat intake with total cholesterol and LDL. When expressing fat and saturated fat intake as a percentage of total energy intake compared to absolute amounts, this relationship was stronger. The study suggested that individuals with the E4 allele may be more responsive to the dietary therapy. Calculations showed that cutting saturated fat consumption in half resulted in a 20% decrease in LDL in those with the E4 allele compared to just a 5% decrease in those without. This speculation is supported by the results of Dreon et al. [234] who found that that LDL-lowering effects of a low-fat diet were greater in men with the E4 allele than those without.
A prospective transversal cohort study including 22,915 participants of the Norfolk arm of the European Prospective Investigation of Cancer (EPIC Norfolk) investigated whether blood lipid response to dietary fat and fibers varied according to the Apo E gene locus. Significant (P < .001) differences in serum lipids according to genotype were found. Highest total and LDL and lowest HDL and TG levels were detected in ε4/ε4 individuals. Total dietary fat and SFA were associated with total and LDL cholesterol concentrations and significant inverse associations between PUFA and dietary fiber and lipid fractions were found. Associations were in the same direction for ε2, ε3, and ε4 expressing individuals with no significant interactions between diet and genotype group on blood lipids, except in the 3% individuals expressing ε2/ε4 (P < .05) in whom the associations were doubled [118].
Strandhagen et al. [121] hypothesized that the increase of plasma CT and TG stimulated by the ingestion of the coffee might be modulated by the Apo ε2/ε3/ε4. The subjects were submitted to four periods of intervention: two of them included three free weeks of coffee and the others two included the ingestion of 600 mL/day for four weeks. The cholesterol-raising effect of coffee was not influenced significantly by the Apo E allele carriers status, however individuals with Apo ε2 polymorphism had significantly lower total cholesterol concentration at baseline (P = .01).
Selected studies on the main interactions of Apo polymorphisms with dietary factors are in Table 2.
10. Glutathione Peroxidases
GPx1 and GPx2 have antioxidant functions, protecting cells from oxidative stress damage. Knockout mice lacking both GPx1 and 2 are more susceptible to an oxidative challenge [235]. Responses of transgenic mice lacking or overexpressing GPx1 have suggested roles for GPx1 in relation to both reactive oxygen species and reactive nitrogen species as well as a link to insulin-mediated effects [235]. GPx4 appears to have a complex range of functions in protection from oxidative stress, lipoxygenase metabolism, and sperm function [236, 237] and may play a role in regulation of leukotriene biosynthesis which in turn regulates the biosynthesis of leukotrienes, thromboxanes, and prostaglandins, thus modulating inflammatory. [238].
A recent research [82] investigated the associations between glutathione peroxidase-1 Pro198Leu polymorphism, selenium status, and DNA damage levels in obese women after consumption of Brazil nuts. Participants consumed one Brazil nut, which provided approximately 290 age of Se a day, for 8 weeks. At baseline, 100% of the subjects were Se deficient, and after the supplementation, there was an improvement in plasma Se (P < .001 for Pro/Pro and Pro/Leu, P < .05 for Leu/Leu), erythrocyte Se (P = .00 for Pro/Pro and Pro/Leu, P < .05 for Leu/Leu), and GPx activity (P = .00 for Pro/Pro, P < .00001 for Pro/Leu, P < .001 for Leu/Leu). In addition, the Pro/Pro group showed a decrease in DNA damage after Brazil nut consumption compared with baseline (P < .005), and those levels were higher in Leu/Leu individuals compared with those with the wild-type genotype (P < .05).
A study about the region of the GPX4 gene corresponding to the 3′UTR was scanned for mutations in a group of 66 volunteers Scotland. The data show a T/C variant at position 718, distribution was 34% CC, 25% TT, and 41% TC. Individuals of different genotypes exhibited significant differences in the plasma levels of 5-lipoxygenase total products in lymphocyte, with 718C showed increased levels of those products compared to 718T and 718T/C (36% and 44% increases, resp.). The data suggest that the SNP 718 has functional effects and support the hypothesis that GPX4 plays a regulatory role in leukotriene biosynthesis [239].
The same SNP 718 (rs713041) influenced the concentration of GPx4 and other selenoproteins in vivo. A selenium supplementation trial was carried out with both homozygote genotypes for this SNP. Blood samples were analyzed at baseline, after a 6 week supplementation with 100 g Seleniun (Se) as sodium selenite/day and during a 6 week washout period. Both lymphocyte GPx1 protein concentrations and plasma GPx3 activity increased significantly after Se supplementation in CC but not TT participants. After Se withdrawal, there was a significant fall in both lymphocyte GPx4 protein concentrations and GPx4 activity in TT but not in CC participants; this effect was modulated by sex [83].
Selected studies on the main interactions of polymorphisms with dietary factors are in Table 1.
11. Selenoprotein P
The physiological functions of Se result from its existence in a number of selenoproteins in which Se is present as the amino acid selenocysteine (Sec). Se was first shown to be an essential component of glutathione peroxidase and subsequently has been found in 25 mammalian selenoproteins [240–242]. In eukaryotes Se is incorporated into selenoproteins as the amino acid selenocysteine in a process requiring a stem loop within region 3′UTR of the mRNA [243]. These proteins and their genes, as well as the factors involved in their synthesis, provide a focus for both mechanistic and genetic studies.
Selenoprotein synthesis and functional activity will result from the combined influences of the genetic information in the genes encoding the selenome and dietary Se intake. Polymorphism in these genes could potentially influence the Se intake required to achieve a particular level of functional activity and thus individual requirements for Se [244].
Any functional effect of a single nucleotide change is likely to be relatively small and can be higher with a combination of different SNP(s) in selenoprotein-related gene(s), possibly in combination with dietary Se intake. When this is suboptimal, such an interaction may give rise to differences in the ability of selenoproteins to function at optimal capacity; thus, selenoprotein function in protection of cells from oxidative stress or inflammation could be compromised [244].
The best characterized selenoproteins are the glutathione peroxidases (GPxs), the thioredoxin reductases (TRs), and deiodinases (IDIs), and selenoprotein P (SeP) [244]. SeP is an extracellular glycoprotein and is expressed most abundantly in liver and at relatively high level in kidney and heart [245]. However, the expression and regulation of SeP in adipose tissue has not been previously reported [246, 247]. SeP represents the best Se marker, accounting for the majority of circulating Se in blood and responding over a broad range to Se intake [248]. In addition to the role as a selenium transporter, SeP has been postulated to possess antioxidant properties, for instance, SeP has the ability to bind epithelial cells and displays phospholipid hydroperoxide thiol peroxidase activity [248]. The overexpression of SeP1 suppressed hydrogen peroxide-induced activation of JNK and p38 in retinal pigment epithelial cells [249]. Another study showed that the activation of GPx activity is required for the protective role of SeP against oxidative damage in human endothelial cells and astrocytes [250, 251] indicating that SeP participates in the control of the cellular redox balance. Since oxidative stress in adipose tissue has been implicated in obesity and insulin resistance, it has been hypothesized that antioxidant SeP may likely be involved in cellular mechanisms of oxidative stress -linked inflammation and insulin resistance [252].
A study showed that SeP1 gene expression was significantly reduced in adipose tissue of ob/ob and high-fat-diet-induced obese mice, as well as in primary adipose cells isolated from Zucker obese rats. Interestingly, SeP1 gene silencing resulted in the reduction in GPX activity and the upregulation of inflammatory cytokines MCP-1 and IL-6 in preadipocytes, leading to the inhibition of adipogenesis and adipokine and lipogenic gene expression. SeP has an important role in adipocyte differentiation via modulating oxidative stress and inflammatory response [252].
A research studied two SNPs in the SeP gene, one in the coding region (position 24731, causing an Ala to Thr change) and one in 3′UTR (position 25191). Their frequency was similar in Caucasian, Chinese, and South Asian populations. Prospectively genotyped volunteers were supplemented for 6 wk with 100 ug sodium selenite/day. Blood samples were analyzed for plasma Se and selenoprotein biomarkers at baseline, after supplementation, and during a washout period. Plasma Se, SeP, and GPx3 levels increased with supplementation. Baseline plasma Se content depended on both SeP genotypes and BMI. Presupplementation SeP concentration was associated with gender and genotype at SNP 24731 and postsupplementation concentration with SNP 25191. The data revealed two common functional SNPs within the human SePP gene that may predict behavior of biomarkers of Se status and response to supplementation and thus susceptibility to disease [84].
Selected studies on the main interactions of polymorphisms with dietary factors are in Table 1.
Several SNPs have been identified in the genes for selenoproteins and some are linked to the occurrence of diseases. However, the current knowledge is limited and the further studies are needed to identify and assess the effect of these SNPs in the nutritional requirements of Se, your metabolism, and susceptibility to diseases.
12. Conclusion
There is evidence that genetic variation at genes involved in the etiology of inflammation and dyslipidemia could interact with environmental exposures, such as diet, to modulate individuals' susceptibility to developing these conditions. Present data support the notion that more tailored dietary recommendations may be helpful in the prevention of obesity-induced comorbidities. However, the relevance and magnitude of nutrient-gene interaction needs to be further elucidated. It is also important to emphasize that the study of the combined effect of SNPs in different genes, as well as haplotypes, may be promising since gene-nutrient interactions in obesity-induced metabolic disturbances are complex.
References
- 1.WHO. Obesity: Preventing and Managing the Global Epidemic. Geneva, Switzerland: World Health Organization; 2003. (WHO Technical Report Series, 894). [PubMed] [Google Scholar]
- 2.Ogden CL, Yanovski SZ, Carroll MD, Flegal KM. The epidemiology of obesity. Gastroenterology. 2007;132(6):2087–2102. doi: 10.1053/j.gastro.2007.03.052. [DOI] [PubMed] [Google Scholar]
- 3.Galuska DA, Gillespie C, Kuester SA, Mokdad AH, Cogswell ME, Philip CM. State-specific prevalence of obesity among adults—United States, 2007. Morbidity and Mortality Weekly Report. 2008;57(28):765–768. [PubMed] [Google Scholar]
- 4.Instituto Brasileiro de Geografia e Estatistica (IBGE) Diretoria de Pesquisas, Coordenaçã de Trabalho e Rendimento. Pesquisa de Orçamentos Familiares 2008–2009. Rio de Janeiro, Brazil: Instituto Brasileiro de Geografia e Estatistica; 2010. [Google Scholar]
- 5.Kelly T, Yang W, Chen CS, Reynolds K, He J. Global burden of obesity in 2005 and projections to 2030. International Journal of Obesity. 2008;32(9):1431–1437. doi: 10.1038/ijo.2008.102. [DOI] [PubMed] [Google Scholar]
- 6.de Lorenzo A, del Gobbo V, Premrov MG, Bigioni M, Galvano F, Di Renzo L. Normal-weight obese syndrome: early inflammation? The American Journal of Clinical Nutrition. 2007;85(1):40–45. doi: 10.1093/ajcn/85.1.40. [DOI] [PubMed] [Google Scholar]
- 7.Kopelman PG. Obesity as a medical problem. Nature. 2000;404(6778):635–643. doi: 10.1038/35007508. [DOI] [PubMed] [Google Scholar]
- 8.Hotamisligil GS. Inflammatory pathways and insulin action. International Journal of Obesity. 2003;27(supplement 3):S53–S55. doi: 10.1038/sj.ijo.0802502. [DOI] [PubMed] [Google Scholar]
- 9.Boden G, Shulman GI. Free fatty acids in obesity and type 2 diabetes: defining their role in the development of insulin resistance and β-cell dysfunction. European Journal of Clinical Investigation. 2002;32(supplement 3):14–23. doi: 10.1046/j.1365-2362.32.s3.3.x. [DOI] [PubMed] [Google Scholar]
- 10.Ortsäter H. Arachidonic acid fights palmitate: new insights into fatty acid toxicity in β-cells. Clinical Science. 2011;120(5):179–181. doi: 10.1042/CS20100521. [DOI] [PubMed] [Google Scholar]
- 11.Barroso SG, Abreu VG, Francischetti EA. The adipose tissue in the genesis of hypertension and atherosclerotic cardiovascular disease. An emerging concept. Arquivos Brasileiros de Cardiologia. 2002;78(6):618–630. doi: 10.1590/s0066-782x2002000600012. [DOI] [PubMed] [Google Scholar]
- 12.Popkin BM. Nutrition in transition: the changing global nutrition challenge. Asia Pacific Journal of Clinical Nutrition. 2001;10:S13–S18. [PubMed] [Google Scholar]
- 13.WHO. Diet, Nutrition and the Prevention of Chronic Diseases. Report of a Joint WHO/FAO Expert Consultation. Geneva, Switzerland: World Health Organization; 2003. (WHO Technical Report Series, 916). [PubMed] [Google Scholar]
- 14.Sinha R, Fisch G, Teague B, et al. Prevalence of impaired glucose tolerance among children and adolescents with marked obesity. The New England Journal of Medicine. 2002;346(11):802–810. doi: 10.1056/NEJMoa012578. [DOI] [PubMed] [Google Scholar]
- 15.Reaven GM. Role of insulin resistance in human disease. Diabetes. 1988;37(12):1595–1607. doi: 10.2337/diab.37.12.1595. [DOI] [PubMed] [Google Scholar]
- 16.Boden G. Fatty acid-induced inflammation and insulin resistance in skeletal muscle and liver. Current Diabetes Reports. 2006;6(3):177–181. doi: 10.1007/s11892-006-0031-x. [DOI] [PubMed] [Google Scholar]
- 17.Arner P. Insulin resistance in type 2 diabetes: role of fatty acids. Diabetes/Metabolism Research and Reviews. 2002;18(2):S5–S9. doi: 10.1002/dmrr.254. [DOI] [PubMed] [Google Scholar]
- 18.Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-α in human obesity and insulin resistance. The Journal of Clinical Investigation. 1995;95(5):2409–2415. doi: 10.1172/JCI117936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Green A, Dobias SB, Walters DJA, Brasier AR. Tumor necrosis factor increases the rate of lipolysis in primary cultures of adipocytes without altering levels of hormone-sensitive lipase. Endocrinology. 1994;134(6):2581–2588. doi: 10.1210/endo.134.6.8194485. [DOI] [PubMed] [Google Scholar]
- 20.Rydén M, Arvidsson E, Blomqvist L, Perbeck L, Dicker A, Arner P. Targets for TNF-α-induced lipolysis in human adipocytes. Biochemical and Biophysical Research Communications. 2004;318(1):168–175. doi: 10.1016/j.bbrc.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 21.Yudkin JS, Juhan-Vague I, Hawe E, et al. Low-grade inflammation may play a role in the etiology of the metabolic syndrome in patients with coronary heart disease: the HIFMECH study. Metabolism. 2004;53(7):852–857. doi: 10.1016/j.metabol.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 22.Wexler DJ, Hu FB, Manson JE, Rifai N, Meigs JB. Mediating effects of inflammatory biomarkers on insulin resistance associated with obesity. Obesity Research. 2005;13(10):1772–1783. doi: 10.1038/oby.2005.216. [DOI] [PubMed] [Google Scholar]
- 23.Fernández-Real JM, Ricart W. Insulin resistance and chronic cardiovascular inflammatory syndrome. Endocrine Reviews. 2003;24(3):278–301. doi: 10.1210/er.2002-0010. [DOI] [PubMed] [Google Scholar]
- 24.Ando K, Fujita T. Metabolic syndrome and oxidative stress. Free Radical Biology & Medicine. 2009;47(3):213–218. doi: 10.1016/j.freeradbiomed.2009.04.030. [DOI] [PubMed] [Google Scholar]
- 25.Gwathmey TM, Westwood BM, Pirro NT, et al. Nuclear angiotensin-(1-7) receptor is functionally coupled to the formation of nitric oxide. American Journal of Physiology. 2010;299(5):F983–F990. doi: 10.1152/ajprenal.00371.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ogihara T, Asano T, Katagiri H, et al. Oxidative stress induces insulin resistance by activating the nuclear factor-κB pathway and disrupting normal subcellular distribution of phosphatidylinositol 3-kinase. Diabetologia. 2004;47(5):794–805. doi: 10.1007/s00125-004-1391-x. [DOI] [PubMed] [Google Scholar]
- 27.Sofi F, Abbate R, Gensini GF, Casini A. Accruing evidence on benefits of adherence to the Mediterranean diet on health: an updated systematic review and meta-analysis. The American Journal of Clinical Nutrition. 2010;92(5):1189–1196. doi: 10.3945/ajcn.2010.29673. [DOI] [PubMed] [Google Scholar]
- 28.Sishi B, Loos B, Ellis B, Smith W, Du Toit EF, Engelbrecht A-M. Diet-induced obesity alters signalling pathways and induces atrophy and apoptosis in skeletal muscle in a prediabetic rat model. Experimental Physiology. 2011;96(2):179–193. doi: 10.1113/expphysiol.2010.054189. [DOI] [PubMed] [Google Scholar]
- 29.Figueras M, Olivan M, Busquets S, López-Soriano FJ, Argilés JM. Effects of eicosapentaenoic acid (EPA) treatment on insulin sensitivity in an animal model of diabetes: improvement of the inflammatory status. Obesity. 2011;19(2):362–369. doi: 10.1038/oby.2010.194. [DOI] [PubMed] [Google Scholar]
- 30.Riccardi G, Giacco R, Rivellese AA. Dietary fat, insulin sensitivity and the metabolic syndrome. Clinical Nutrition. 2004;23(4):447–456. doi: 10.1016/j.clnu.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 31.Bunn RC, Cockrell GE, Ou Y, Thrailkill KM, Lumpkin CK, Jr., Fowlkes JL. Palmitate and insulin synergistically induce IL-6 expression in human monocytes. Cardiovascular Diabetology. 2010;9, article 73 doi: 10.1186/1475-2840-9-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chavez JA, Summers SA. Characterizing the effects of saturated fatty acids on insulin signaling and ceramide and diacylglycerol accumulation in 3T3-L1 adipocytes and C2C12 myotubes. Archives of Biochemistry and Biophysics. 2003;419(2):101–109. doi: 10.1016/j.abb.2003.08.020. [DOI] [PubMed] [Google Scholar]
- 33.Kennedy A, Martinez K, Chuang CC, Lapoint K, Mcintosh M. Saturated fatty acid-mediated inflammation and insulin resistance in adipose tissue: mechanisms of action and implications. The Journal of Nutrition. 2009;139(1):1–4. doi: 10.3945/jn.108.098269. [DOI] [PubMed] [Google Scholar]
- 34.Esposito K, Marfella R, Ciotola M, et al. Effect of a Mediterranean-style diet on endothelial dysfunction and markers of vascular inflammation in the metabolic syndrome: a randomized trial. Journal of the American Medical Association. 2004;292(12):1440–1446. doi: 10.1001/jama.292.12.1440. [DOI] [PubMed] [Google Scholar]
- 35.Raqib R, Cravioto A. Nutrition, immunology, and genetics: future perspectives. Nutrition Reviews. 2009;67:S227–S236. doi: 10.1111/j.1753-4887.2009.00244.x. [DOI] [PubMed] [Google Scholar]
- 36.Gibney MJ, Gibney ER. Diet, genes and disease: implications for nutrition policy. Proceedings of the Nutrition Society. 2004;63(3):491–500. doi: 10.1079/pns2004369. [DOI] [PubMed] [Google Scholar]
- 37.Memisoglu A, Hu FB, Hankinson SE, et al. Interaction between a peroxisome proliferator-activated receptor γ gene polymorphism and dietary fat intake in relation to body mass. Human Molecular Genetics. 2003;12(22):2923–2929. doi: 10.1093/hmg/ddg318. [DOI] [PubMed] [Google Scholar]
- 38.Martínez JA, Corbalán MS, Sánchez-Villegas A, Forga L, Marti A, Martínez-González MA. Obesity risk is associated with carbohydrate intake in women carrying the Gln27Glu β-adrenoceptor polymorphism. The Journal of Nutrition. 2003;133(8):2549–2554. doi: 10.1093/jn/133.8.2549. [DOI] [PubMed] [Google Scholar]
- 39.Danawati CW, Nagata M, Moriyama H, et al. A possible association of Pro12Ala polymorphism in peroxisome proliferator-activated receptor γ 2 gene with obesity in native Javanese in Indonesia. Diabetes/Metabolism Research and Reviews. 2005;21(5):465–469. doi: 10.1002/dmrr.543. [DOI] [PubMed] [Google Scholar]
- 40.Jones AB. Peroxisome proliferator-activated receptor (PPAR) modulators: diabetes and beyond. Medicinal Research Reviews. 2001;21(6):540–552. doi: 10.1002/med.1025. [DOI] [PubMed] [Google Scholar]
- 41.Torra IP, Chinetti G, Duval C, Fruchart JC, Staels B. Peroxisome proliferator-activated receptors: from transcriptional control to clinical practice. Current Opinion in Lipidology. 2001;12(3):245–254. doi: 10.1097/00041433-200106000-00002. [DOI] [PubMed] [Google Scholar]
- 42.Aldhoon B, Zamrazilová H, Aldhoon Hainerová I, et al. Role of the PPARα Leu162Val and PPARγ2 Pro12Ala gene polymorphisms in weight change after 2.5-year follow-up in Czech obese women. Folia Biologica. 2010;56(3):116–123. doi: 10.14712/fb2010056030116. [DOI] [PubMed] [Google Scholar]
- 43.Deeb SS, Fajas L, Nemoto M, et al. A Pro12Ala substitution in PPARγ2 associated with decreased receptor activity, lower body mass index and improved insulin sensitivity. Nature Genetics. 1998;20(3):284–287. doi: 10.1038/3099. [DOI] [PubMed] [Google Scholar]
- 44.Frederiksen L, Brødbæk K, Fenger M, et al. Comment: Studies of the Pro12Ala polymorphism of the PPAR-γ gene in the Danish MONICA cohort: homozygosity of the Ala allele confers a decreased risk of the insulin resistance syndrome. The Journal of Clinical Endocrinology and Metabolism. 2002;87(8):3989–3992. doi: 10.1210/jcem.87.8.8732. [DOI] [PubMed] [Google Scholar]
- 45.Mori H, Ikegami H, Kawaguchi Y, et al. The Pro12→Ala substitution in PPAR-γ is associated with resistance to development of diabetes in the general population: possible involvement in impairment of insulin secretion in individuals with type 2 diabetes. Diabetes. 2001;50(4):891–894. doi: 10.2337/diabetes.50.4.891. [DOI] [PubMed] [Google Scholar]
- 46.Altshuler D, Hirschhorn JN, Klannemark M, et al. The common PPARγ Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nature Genetics. 2000;26(1):76–80. doi: 10.1038/79216. [DOI] [PubMed] [Google Scholar]
- 47.Lu Y, Ye X, Cao Y, et al. Genetic variants in peroxisome proliferator-activated receptor-γ and retinoid X receptor-α gene and type 2 diabetes risk: a case-control study of a Chinese han population. Diabetes Technology and Therapeutics. 2011;13(2):157–164. doi: 10.1089/dia.2010.0122. [DOI] [PubMed] [Google Scholar]
- 48.Masud S, Ye S. Effect of the peroxisome proliferates activated receptor-γ gene Pro12Ala variant on body mass index: a meta-analysis. Journal of Medical Genetics. 2003;40(10):773–780. doi: 10.1136/jmg.40.10.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beamer BA, Yen C-J, Andersen RE, et al. Association of the Pro12Ala variant in the peroxisome proliferator-activated receptor-γ2 gene with obesity in two Caucasian populations. Diabetes. 1998;47(11):1806–1808. doi: 10.2337/diabetes.47.11.1806. [DOI] [PubMed] [Google Scholar]
- 50.Tai ES, Corella D, Deurenberg-Yap M, et al. Differential effects of the C1431T and Pro12Ala PPARγ gene variants on plasma lipids and diabetes risk in an Asian population. Journal of Lipid Research. 2004;45(4):674–685. doi: 10.1194/jlr.M300363-JLR200. [DOI] [PubMed] [Google Scholar]
- 51.Oh EY, Min KM, Chung JH, et al. Significance of ProAla mutation in peroxisome proliferator-activated receptor-γ2 in Korean diabetic and obese subjects. The Journal of Clinical Endocrinology and Metabolism. 2000;85(5):1801–1804. doi: 10.1210/jcem.85.5.6499. [DOI] [PubMed] [Google Scholar]
- 52.Muller YL, Bogardus C, Beamer BA, Shuldiner AR, Baier LJ. A functional variant in the peroxisome proliferator—activated receptor γ2 promoter is associated with predictors of obesity and type 2 diabetes in Pima Indians. Diabetes. 2003;52(7):1864–1871. doi: 10.2337/diabetes.52.7.1864. [DOI] [PubMed] [Google Scholar]
- 53.Rosmond R, Chagnon M, Bouchard C. The Pro12Ala PPARγ2 gene missense mutation is associated with obesity and insulin resistance in Swedish middle-aged men. Diabetes/Metabolism Research and Reviews. 2003;19(2):159–163. doi: 10.1002/dmrr.371. [DOI] [PubMed] [Google Scholar]
- 54.González Sánchez JL, Serrano Ríos M, Fernández Pérez C, Laakso M, Martínez Larrad MT. Effect of the Pro12Ala polymorphism of the peroxisome proliferator-activated receptor γ-2 gene on adiposity, insulin sensitivity and lipid profile in the Spanish population. European Journal of Endocrinology. 2002;147(4):495–501. doi: 10.1530/eje.0.1470495. [DOI] [PubMed] [Google Scholar]
- 55.Mirzaei H, Akrami SM, Golmohammadi T, et al. Polymorphism of Pro12Ala in the peroxisome proliferator-activated receptor 2 gene in Iranian diabetic and obese subjects. Metabolic Syndrome and Related Disorders. 2009;7(5):453–458. doi: 10.1089/met.2008.0099. [DOI] [PubMed] [Google Scholar]
- 56.Mattevi VS, Zembrzuski VM, Hutz MH. Effects of a PPARG gene variant on obesity characteristics in Brazil. Brazilian Journal of Medical and Biological Research. 2007;40(7):927–932. doi: 10.1590/s0100-879x2006005000114. [DOI] [PubMed] [Google Scholar]
- 57.Morini E, Tassi V, Capponi D, et al. Interaction between PPARγ2 variants and gender on the modulation of body weight. Obesity. 2008;16(6):1467–1470. doi: 10.1038/oby.2008.225. [DOI] [PubMed] [Google Scholar]
- 58.Dedoussis GV, Vidra N, Butler J, et al. Peroxisome proliferator-activated receptor-γ (PPARγ) Pro12Ala polymorphism and risk for pediatric obesity. Clinical Chemistry and Laboratory Medicine. 2009;47(9):1047–1050. doi: 10.1515/CCLM.2009.242. [DOI] [PubMed] [Google Scholar]
- 59.Ghoussaini M, Meyre D, Lobbens S, et al. Implication of the Pro12Ala polymorphism of the PPAR-gamma 2 gene in type 2 diabetes and obesity in the French population. BMC Medical Genetics. 2005;6, article 11 doi: 10.1186/1471-2350-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stefanski A, Majkowska L, Ciechanowicz A, et al. Lack of association between the Pro12Ala polymorphism in PPAR-γ2 gene and body weight changes, insulin resistance and chronic diabetic complications in obese patients with type 2 diabetes. Archives of Medical Research. 2006;37(6):736–743. doi: 10.1016/j.arcmed.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 61.Helwig U, Rubin D, Kiosz J, et al. The minor allele of the PPARγ2 Pro12Ala polymorphism is associated with lower postprandial TAG and insulin levels in non-obese healthy men. British Journal of Nutrition. 2007;97(5):847–854. doi: 10.1017/S0007114507665179. [DOI] [PubMed] [Google Scholar]
- 62.Cecil JE, Watt P, Palmer CN, Hetherington M. Energy balance and food intake: the role of PPARγ gene polymorphisms. Physiology & Behavior. 2006;88(3):227–233. doi: 10.1016/j.physbeh.2006.05.028. [DOI] [PubMed] [Google Scholar]
- 63.Milewicz A, Tworowska-Bardziñska U, Dunajska K, Jêdrzejuk D, Lwow F. Relationship of PPARgamma2 polymorphism with obesity ana metabolic syndrome in postmenopausal Polish women. Experimental and Clinical Endocrinology and Diabetes. 2009;117(10):628–632. doi: 10.1055/s-0028-1112154. [DOI] [PubMed] [Google Scholar]
- 64.Franks PW, Jablonski KA, Delahanty L, et al. The Pro12Ala variant at the peroxisome proliferator-activated receptor γ gene and change in obesity-related traits in the Diabetes Prevention Program. Diabetologia. 2007;50(12):2451–2460. doi: 10.1007/s00125-007-0826-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Moon MK, Cho YM, Jung HS, et al. Genetic polymorphisms in peroxisome proliferator-activated receptor γ are associated with type 2 diabetes mellitus and obesity in the Korean population. Diabetic Medicine. 2005;22(9):1161–1166. doi: 10.1111/j.1464-5491.2005.01599.x. [DOI] [PubMed] [Google Scholar]
- 66.Rosado EL, Bressan J, Martins MF, Cecon PR, Martínez JA. Polymorphism in the PPARgamma2 and beta2-adrenergic genes and diet lipid effects on body composition, energy expenditure and eating behavior of obese women. Appetite. 2007;49(3):635–643. doi: 10.1016/j.appet.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 67.Rosado EL, Bressan J, Hernández JAM, Martins MF, Cecon PR. Effect of diet and PPARγ2 and β2-adrenergic receptor genes on energy metabolism and body composition in obese women. Nutricion Hospitalaria. 2006;21(3):317–331. [PubMed] [Google Scholar]
- 68.Luan J, Browne PO, Harding AH, et al. Evidence for gene-nutrient interaction at the PPARγ locus. Diabetes. 2001;50(3):686–689. doi: 10.2337/diabetes.50.3.686. [DOI] [PubMed] [Google Scholar]
- 69.Soriguer F, Morcillo S, Cardona F, et al. Pro12Ala polymorphism of the PPARG2 gene is associated with type 2 diabetes mellitus and peripheral insulin sensitivity in a population with a high intake of oleic acid. The Journal of Nutrition. 2006;136(9):2325–2330. doi: 10.1093/jn/136.9.2325. [DOI] [PubMed] [Google Scholar]
- 70.Scaglioni S, Verduci E, Salvioni M, et al. PPAR-γ2 Pro12Ala variant, insulin resistance and plasma long-chain polyunsaturated fatty acids in childhood obesity. Pediatric Research. 2006;60(4):485–489. doi: 10.1203/01.pdr.0000238259.41560.00. [DOI] [PubMed] [Google Scholar]
- 71.Grimble RF, Howell WM, O’Reilly G, et al. The ability of fish oil to suppress tumor necrosis factor α production by peripheral blood mononuclear cells in healthy men is associated with polymorphisms in genes that influence tumor necrosis factor α production. The American Journal of Clinical Nutrition. 2002;76(2):454–459. doi: 10.1093/ajcn/76.2.454. [DOI] [PubMed] [Google Scholar]
- 72.Belisle SE, Leka LS, Delgado-Lista J, Jacques PF, Ordovas JM, Meydani SN. Polymorphisms at cytokine genes may determine the effect of vitamin E on cytokine production in the elderly. The Journal of Nutrition. 2009;139(10):1855–1860. doi: 10.3945/jn.109.112268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.de Luis DA, Aller R, Izaola O, Sagrado MG, Conde R. Influence of G308A promoter variant of tumor necrosis factor-α gene on insulin resistance and weight loss secondary to two hypocaloric diets: a randomized clinical trial. Archives of Medical Research. 2009;40(1):36–41. doi: 10.1016/j.arcmed.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 74.Joffe YT, van der Merwe L, Carstens M, et al. Tumor necrosis factor-α gene -308 G/A polymorphism modulates the relationship between dietary fat intake, serum lipids, and obesity risk in black south African women. The Journal of Nutrition. 2010;140(5):901–907. doi: 10.3945/jn.109.109355. [DOI] [PubMed] [Google Scholar]
- 75.Corpeleijn E, Petersen L, Holst C, et al. Obesity-related polymorphisms and their associations with the ability to regulate fat oxidation in obese Europeans: the NUGENOB study. Obesity. 2010;18(7):1369–1377. doi: 10.1038/oby.2009.377. [DOI] [PubMed] [Google Scholar]
- 76.Razquin C, Martinez JA, Martinez-Gonzalez MA, Fernández-Crehuet J, Santos JM, Marti A. A mediterranean diet rich in virgin olive oil may reverse the effects of the-174g/c il6 gene variant on 3-year body weight change. Molecular Nutrition & Food Research. 2010;54(supplement 1):S75–S82. doi: 10.1002/mnfr.200900257. [DOI] [PubMed] [Google Scholar]
- 77.Fernández-Real JM, Broch M, Vendrell J, Richart C, Ricart W. Interleukin-6 gene polymorphism and lipid abnormalities in healthy subjects. The Journal of Clinical Endocrinology and Metabolism. 2000;85(3):1334–1339. doi: 10.1210/jcem.85.3.6555. [DOI] [PubMed] [Google Scholar]
- 78.Goyenechea E, Parra MD, Martínez JA. Weight regain after slimming induced by an energy-restricted diet depends on interleukin-6 and peroxisome-proliferator-activated-receptor-γ2 gene polymorphisms. British Journal of Nutrition. 2006;96(5):965–972. doi: 10.1017/bjn20061901. [DOI] [PubMed] [Google Scholar]
- 79.Reiner AP, Wurfel MM, Lange LA, et al. Polymorphisms of the IL1-receptor antagonist gene (IL1RN) are associated with multiple markers of systemic inflammation. Arteriosclerosis, Thrombosis, and Vascular Biology. 2008;28(7):1407–1412. doi: 10.1161/ATVBAHA.108.167437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yanagisawa A, Suzuki K, Kimura A, Ito Y, Hamajima N, Inoue T. Possible protective effect of serum β-carotene levels on the association between interleukin-1B C-31T polymorphism and hypertension in a Japanese population. Clinical Nutrition. 2009;28(2):198–202. doi: 10.1016/j.clnu.2009.01.020. [DOI] [PubMed] [Google Scholar]
- 81.Kornman K, Rogus J, Roh-Schmidt H, et al. Interleukin-1 genotype-selective inhibition of inflammatory mediators by a botanical: a nutrigenetics proof of concept. Nutrition. 2007;23(11-12):844–852. doi: 10.1016/j.nut.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 82.Cominetti C, de Bortoli MC, Purgatto E, et al. Associations between glutathione peroxidase-1 Pro198Leu polymorphism, selenium status, and DNA damage levels in obese women after consumption of Brazil nuts. doi: 10.1016/j.nut.2010.09.003. Nutrition. In press. [DOI] [PubMed] [Google Scholar]
- 83.Méplan C, Crosley LK, Nicol F, et al. Functional effects of a common single-nucleotide polymorphism (GPX4c718t) in the glutathione peroxidase 4 gene: interaction with sex. The American Journal of Clinical Nutrition. 2008;87(4):1019–1027. doi: 10.1093/ajcn/87.4.1019. [DOI] [PubMed] [Google Scholar]
- 84.Méplan C, Crosley LK, Nicol F, et al. Genetic polymorphisms in the human selenoprotein P gene determine the response of selenoprotein markers to selenium supplementation in a gender-specific manner (the SELGEN study) The FASEB Journal. 2007;21(12):3063–3074. doi: 10.1096/fj.07-8166com. [DOI] [PubMed] [Google Scholar]
- 85.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-α: direct role in obesity-linked insulin resistance. Science. 1993;259(5091):87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 86.Lumeng CN, Deyoung SM, Saltiel AR. Macrophages block insulin action in adipocytes by altering expression of signaling and glucose transport proteins. American Journal of Physiology. 2007;292(1):E166–E174. doi: 10.1152/ajpendo.00284.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Steinberg GR. Inflammation in obesity is the common link between defects in fatty acid metabolism and insulin resistance. Cell Cycle. 2007;6(8):888–894. doi: 10.4161/cc.6.8.4135. [DOI] [PubMed] [Google Scholar]
- 88.Louis E, Franchimont D, Piron A, et al. Tumour necrosis factor (TNF) gene polymorphism influences TNF-α production in lipopolysaccharide (LPS)-stimulated whole blood cell culture in healthy humans. Clinical and Experimental Immunology. 1998;113(3):401–406. doi: 10.1046/j.1365-2249.1998.00662.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.van der Linden MW, Huizinga TWJ, Stoeken DJ, Sturk A, Westendorp RGJ. Determination of tumour necrosis factor-α and interleukin-10 production in a whole blood stimulation system: assessment of laboratory error and individual variation. Journal of Immunological Methods. 1998;218(1-2):63–71. doi: 10.1016/s0022-1759(98)00108-2. [DOI] [PubMed] [Google Scholar]
- 90.Bayley JP, de Rooij H, van den Elsen PJ, Huizinga TWJ, Verweij CL. Functional analysis of linker-scan mutants spanning the -376, -308, -244, and -238 polymorphic sites of the TNF-α promoter. Cytokine. 2001;14(6):316–323. doi: 10.1006/cyto.2001.0902. [DOI] [PubMed] [Google Scholar]
- 91.Bayley JP, Ottenhoff THM, Verweij CL. Is there a future for TNF promoter polymorphisms? Genes & Immunity. 2004;5(5):315–329. doi: 10.1038/sj.gene.6364055. [DOI] [PubMed] [Google Scholar]
- 92.Hajeer AH, Hutchinson IV. TNF-α gene polymorphism: clinical and biological implications. Microscopy Research and Technique. 2000;50(3):216–228. doi: 10.1002/1097-0029(20000801)50:3<216::AID-JEMT5>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 93.Abraham LJ, Kroeger KM. Impact of the -308 TNF promoter polymorphism on the transcriptional regulation of the TNF gene: relevance to disease. Journal of Leukocyte Biology. 1999;66(4):562–566. doi: 10.1002/jlb.66.4.562. [DOI] [PubMed] [Google Scholar]
- 94.Wilson AG, di Giovine FS, Blakemore AIF, Duff GW. Single base polymorphism in the human tumour necrosis factor alpha (TNFα) gene detectable by NcoI restriction of PCR product. Human Molecular Genetics. 1992;1(5):p. 353. doi: 10.1093/hmg/1.5.353. [DOI] [PubMed] [Google Scholar]
- 95.Elahi MM, Asotra K, Matata BM, Mastana SS. Tumor necrosis factor alpha -308 gene locus promoter polymorphism: an analysis of association with health and disease. Biochimica et Biophysica Acta. 2009;1792(3):163–172. doi: 10.1016/j.bbadis.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 96.Smith AJP, Humphries SE. Cytokine and cytokine receptor gene polymorphisms and their functionality. Cytokine and Growth Factor Reviews. 2009;20(1):43–59. doi: 10.1016/j.cytogfr.2008.11.006. [DOI] [PubMed] [Google Scholar]
- 97.Kroeger KM, Carville KS, Abraham LJ. The -308 tumor necrosis factor-α promoter polymorphism effects transcription. Molecular Immunology. 1997;34(5):391–399. doi: 10.1016/s0161-5890(97)00052-7. [DOI] [PubMed] [Google Scholar]
- 98.Wilson AG, Symons JA, Mcdowell TL, Mcdevitt HO, Duff GW. Effects of a polymorphism in the human tumor necrosis factor α promoter on transcriptional activation. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(7):3195–3199. doi: 10.1073/pnas.94.7.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kroeger KM, Steer JH, Joyce DA, Abraham LJ. Effects of stimulus and cell type on the expression of the -308 tumour necrosis factor promoter polymorphism. Cytokine. 2000;12(2):110–119. doi: 10.1006/cyto.1999.0529. [DOI] [PubMed] [Google Scholar]
- 100.Brinkman BMN, Zuijdgeest D, Kaijzel EL, Breedveld FC, Verweij CL. Relevance of the tumor necrosis factor alpha (TNFα) -308 promoter polymorphism in TNFα gene regulation. Journal of Inflammation. 1996;46(1):32–41. [PubMed] [Google Scholar]
- 101.Araújo F, Pereira AC, Mota GF, Latorre MdoR, Krieger JE, Mansur AJ. The influence of tumor necrosis factor -308 and C-reactive protein G1059C gene variants on serum concentration of C-reactive protein: evidence for an age-dependent association. Clinica Chimica Acta. 2004;349(1-2):129–134. doi: 10.1016/j.cccn.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 102.Lakka HM, Lakka TA, Rankinen T, et al. The TNF-α G-308A polymorphism is associated with C-reactive protein levels: the HERITAGE Family Study. Vascular Pharmacology. 2006;44(5):377–383. doi: 10.1016/j.vph.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 103.Gander ML, Fischer JE, Maly FE, von Känel R. Effect of the G-308A polymorphism of the tumor necrosis factor (TNF)-α gene promoter site on plasma levels of TNF-α and C-reactive protein in smokers: a cross-sectional study. BMC Cardiovascular Disorders. 2004;4, article 17 doi: 10.1186/1471-2261-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sookoian SC, González C, Pirola CJ. Meta-analysis on the G-308A tumor necrosis factor α gene variant and phenotypes associated with the metabolic syndrome. Obesity Research. 2005;13(12):2122–2131. doi: 10.1038/oby.2005.263. [DOI] [PubMed] [Google Scholar]
- 105.Pereira TV, Rudnicki M, Franco RF, Pereira AC, Krieger JE. Effect of the G-308A polymorphism of the tumor necrosis factor α gene on the risk of ischemic heart disease and ischemic stroke: a meta-analysis. American Heart Journal. 2007;153(5):821–830. doi: 10.1016/j.ahj.2007.02.031. [DOI] [PubMed] [Google Scholar]
- 106.Phillips CM, Goumidi L, Bertrais S, et al. Additive effect of polymorphisms in the IL-6, LTA, and TNF-α genes and plasma fatty acid level modulate risk for the metabolic syndrome and its components. The Journal of Clinical Endocrinology and Metabolism. 2010;95(3):1386–1394. doi: 10.1210/jc.2009-1081. [DOI] [PubMed] [Google Scholar]
- 107.Gomez P, Perez-Martinez P, Marin C, et al. APOA1 and APOA4 gene polymorphisms influence the effects of dietary fat on LDL particle size and oxidation in healthy young adults. The Journal of Nutrition. 2010;140(4):773–778. doi: 10.3945/jn.109.115964. [DOI] [PubMed] [Google Scholar]
- 108.Ordovas JM, Corella D, Cupples LA, et al. Polyunsaturated fatty acids modulate the effects of the APOA1 G-A polymorphism on HDL-cholesterol concentrations in a sex-specific manner: the Framingham study. The American Journal of Clinical Nutrition. 2002;75(1):38–46. doi: 10.1093/ajcn/75.1.38. [DOI] [PubMed] [Google Scholar]
- 109.Mata P, Lopez-Miranda J, Pocovi M, et al. Human apolipoprotein A-I gene promoter mutation influences plasma low density lipoprotein cholesterol response to dietary fat saturation. Atherosclerosis. 1998;137(2):367–376. doi: 10.1016/s0021-9150(97)00265-7. [DOI] [PubMed] [Google Scholar]
- 110.Duesing K, Charpentier G, Marre M, et al. Evaluating the association of common APOA2 variants with type 2 diabetes. BMC Medical Genetics. 2009;10, article 13 doi: 10.1186/1471-2350-10-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Corella D, Peloso G, Arnett DK, et al. APOA2, dietary fat, and body mass index: replication of a gene-diet interaction in 3 independent populations. Archives of Internal Medicine. 2009;169(20):1897–1906. doi: 10.1001/archinternmed.2009.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Corella D, Tai ES, Sorlí JV, et al. Association between the APOA2 promoter polymorphism and body weight in Mediterranean and Asian populations: replication of a gene-saturated fat interaction. International Journal of Obesity. 2011;35(5):666–675. doi: 10.1038/ijo.2010.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Delgado-Lista J, Perez-Jimenez F, Tanaka T, et al. An apolipoprotein A-II polymorphism (-265T/C, rs5082) regulates postprandial response to a saturated fat overload in healthy men. The Journal of Nutrition. 2007;137(9):2024–2028. doi: 10.1093/jn/137.9.2024. [DOI] [PubMed] [Google Scholar]
- 114.Lai CQ, Corella D, Demissie S, et al. Dietary intake of n-6 fatty acids modulates effect of apolipoprotein A5 gene on plasma fasting triglycerides, remnant lipoprotein concentrations, and lipoprotein particle size: the Framingham Heart Study. Circulation. 2006;113(17):2062–2070. doi: 10.1161/CIRCULATIONAHA.105.577296. [DOI] [PubMed] [Google Scholar]
- 115.Mattei J, Demissie S, Tucker KL, Ordovas JM. Apolipoprotein A5 polymorphisms interact with total dietary fat intake in association with markers of metabolic syndrome in Puerto Rican older adults. The Journal of Nutrition. 2009;139(12):2301–2308. doi: 10.3945/jn.109.109900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kim JY, Kim OY, Koh SJ, et al. Comparison of low-fat meal and high-fat meal on postprandial lipemic response in non-obese men according to the -1131T>C polymorphism of the apolipoprotein A5 (APOA5) gene (randomized cross-over design) Journal of the American College of Nutrition. 2006;25(4):340–347. doi: 10.1080/07315724.2006.10719544. [DOI] [PubMed] [Google Scholar]
- 117.Sundl I, Guardiola M, Khoschsorur G, et al. Increased concentrations of circulating vitamin E in carriers of the apolipoprotein A5 gene -1131T>C variant and associations with plasma lipids and lipid peroxidation. Journal of Lipid Research. 2007;48(11):2506–2513. doi: 10.1194/jlr.M700285-JLR200. [DOI] [PubMed] [Google Scholar]
- 118.Wu K, Bowman R, Welch AA, et al. Apolipoprotein E polymorphisms, dietary fat and fibre, and serum lipids: the EPIC Norfolk study. European Heart Journal. 2007;28(23):2930–2936. doi: 10.1093/eurheartj/ehm482. [DOI] [PubMed] [Google Scholar]
- 119.Moreno JA, Pérez-Jimnéz F, Moreno-Luna R, et al. The effect of apoE genotype and sex on ApoE plasma concentration is determined by dietary fat in healthy subjects. British Journal of Nutrition. 2009;101(12):1745–1752. doi: 10.1017/S0007114508111515. [DOI] [PubMed] [Google Scholar]
- 120.Loktionov A, Scollen S, McKeown N, Bingham SA. Gene-nutrient interactions: dietary behaviour associated with high coronary heart disease risk particularly affects serum LDL cholesterol in apolipoprotein E ε4-carrying free-living individuals. British Journal of Nutrition. 2000;84(6):885–890. [PubMed] [Google Scholar]
- 121.Strandhagen E, Zetterberg H, Aires N, et al. The apolipoprotein E polymorphism and the cholesterol-raising effect of coffee. Lipids in Health and Disease. 2004;3, article 26 doi: 10.1186/1476-511X-3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Knight JC, Udalova I, Hill AVS, et al. A polymorphism that affects OCT-1 binding to the TNF promoter region is associated with severe malaria. Nature Genetics. 1999;22(2):145–150. doi: 10.1038/9649. [DOI] [PubMed] [Google Scholar]
- 123.Steensberg A, Fischer CP, Keller C, Møller K, Pedersen BK. IL-6 enhances plasma IL-1ra, IL-10, and cortisol in humans. American Journal of Physiology. 2003;285(2):E433–E437. doi: 10.1152/ajpendo.00074.2003. [DOI] [PubMed] [Google Scholar]
- 124.Starkie R, Ostrowski SR, Jauffred S, Febbraio M, Pedersen BK. Exercise and IL-6 infusion inhibit endotoxin-induced TNF-alpha production in humans. The FASEB Journal. 2003;17(8):884–886. doi: 10.1096/fj.02-0670fje. [DOI] [PubMed] [Google Scholar]
- 125.Ghazouani L, Abboud N, Khalifa SBH, et al. -174G>C interleukin-6 gene polymorphism in Tunisian patients with coronary artery disease. Annals of Saudi Medicine. 2011;31(1):40–44. doi: 10.4103/0256-4947.75777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Qi L, Zhang C, van Dam RM, Hu FB. Interleukin-6 genetic variability and adiposity: associations in two prospective cohorts and systematic review in 26,944 individuals. The Journal of Clinical Endocrinology and Metabolism. 2007;92(9):3618–3625. doi: 10.1210/jc.2007-0877. [DOI] [PubMed] [Google Scholar]
- 127.Rexrode KM, Pradhan A, Manson JE, Buring JE, Ridker PM. Relationship of total and abdominal adiposity with CRP and IL-6 in women. Annals of Epidemiology. 2003;13(10):674–682. doi: 10.1016/s1047-2797(03)00053-x. [DOI] [PubMed] [Google Scholar]
- 128.Vozarova B, Weyer C, Hanson K, Tataranni PA, Bogardus C, Pratley RE. Circulating interleukin-6 in relation to adiposity, insulin action, and insulin secretion. Obesity Research. 2001;9(7):414–417. doi: 10.1038/oby.2001.54. [DOI] [PubMed] [Google Scholar]
- 129.Stephens JW, Hurel SJ, Lowe GDO, Rumley A, Humphries SE. Association between plasma IL-6, the IL6 -174G>C gene variant and the metabolic syndrome in type 2 diabetes mellitus. Molecular Genetics and Metabolism. 2007;90(4):422–428. doi: 10.1016/j.ymgme.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 130.Sattar N, Perry CG, Petrie JR. Type 2 diabetes as an inflammatory disorder. British Journal of Diabetes and Vascular Disease. 2003;3(1):36–41. [Google Scholar]
- 131.Freeman DJ, Norrie J, Caslake MJ, et al. C-reactive protein is an independent predictor of risk for the development of diabetes in the west of Scotland coronary prevention study. Diabetes. 2002;51(5):1596–1600. doi: 10.2337/diabetes.51.5.1596. [DOI] [PubMed] [Google Scholar]
- 132.Humphries SE, Talmud PJ, Hawe E, Bolla M, Day INM, Miller GJ. Apolipoprotein E4 and coronary heart disease in middle-aged men who smoke: a prospective study. The Lancet. 2001;358(9276):115–119. doi: 10.1016/S0140-6736(01)05330-2. [DOI] [PubMed] [Google Scholar]
- 133.Fernandez-Real JM, Vayreda M, Richart C, et al. Circulating interleukin 6 levels, blood pressure, and insulin sensitivity in apparently healthy men and women. The Journal of Clinical Endocrinology and Metabolism. 2001;86(3):1154–1159. doi: 10.1210/jcem.86.3.7305. [DOI] [PubMed] [Google Scholar]
- 134.Stephens JW, Hurel SJ, Cooper JA, Acharya J, Miller GJ, Humphries SE. A common functional variant in the interleukin-6 gene is associated with increased body mass index in subjects with type 2 diabetes mellitus. Molecular Genetics and Metabolism. 2004;82(2):180–186. doi: 10.1016/j.ymgme.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 135.Heinrich PC, Castell JV, Andus T. Interleukin-6 and the acute phase response. Biochemical Journal. 1990;265(3):621–636. doi: 10.1042/bj2650621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Berthier MT, Paradis AM, Tchernof A, et al. The interleukin 6 -174G/C polymorphism is associated with indices of obesity in men. Journal of Human Genetics. 2003;48(1):14–19. doi: 10.1007/s100380300002. [DOI] [PubMed] [Google Scholar]
- 137.Cardellini M, Perego L, D’Adamo M, et al. C-174G polymorphism in the promoter of the interleukin-6 gene is associated with insulin resistance. Diabetes Care. 2005;28(8):2007–2012. doi: 10.2337/diacare.28.8.2007. [DOI] [PubMed] [Google Scholar]
- 138.Grallert H, Huth C, Kolz M, et al. IL-6 promoter polymorphisms and quantitative traits related to the metabolic syndrome in KORA S4. Experimental Gerontology. 2006;41(8):737–745. doi: 10.1016/j.exger.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 139.Hamid YH, Rose CS, Urhammer SA, et al. Variations of the interleukin-6 promoter are associated with features of the metabolic syndrome in Caucasian Danes. Diabetologia. 2005;48(2):251–260. doi: 10.1007/s00125-004-1623-0. [DOI] [PubMed] [Google Scholar]
- 140.Klipstein-Grobusch K, Möhlig M, Spranger J, et al. Interleukin-6 g.-174G>C promoter polymorphism is associated with obesity in the EPIC-potsdam study. Obesity. 2006;14(1):14–18. doi: 10.1038/oby.2006.3. [DOI] [PubMed] [Google Scholar]
- 141.Wernstedt I, Eriksson A-L, Berndtsson A, et al. A common polymorphism in the interleukin-6 gene promoter is associated with overweight. International Journal of Obesity. 2004;28(10):1272–1279. doi: 10.1038/sj.ijo.0802763. [DOI] [PubMed] [Google Scholar]
- 142.Goyenechea E, Parra MD, Hernández JAM. Role of IL-6 and its -174G>C polymorphism in weight management and in the metabolic comorbidities associated with obesityImplicación de la IL-6 y su polimorfismo -174G>C en el control del peso corporal y en las complicaciones metabólicas asociadas a la obesidad. Anales del Sistema Sanitario de Navarra. 2005;28(3):357–366. doi: 10.4321/s1137-66272005000500007. [DOI] [PubMed] [Google Scholar]
- 143.Andersson N, Strandberg L, Nilsson S, et al. A variant near the interleukin-6 gene is associated with fat mass in Caucasian men. International Journal of Obesity. 2010;34(6):1011–1019. doi: 10.1038/ijo.2010.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Huang HY, Thuita L, Strickland P, Hoffman SC, Comstock GW, Helzlsouer KJ. Frequencies of single nucleotide polymorphisms in genes regulating inflammatory responses in a community-based population. BMC Genetics. 2007;8, article 7 doi: 10.1186/1471-2156-8-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Poitou C, Lacorte J-M, Coupaye M, et al. Relationship between single nucleotide polymorphisms in leptin, IL6 and adiponectin genes and their circulating product in morbidly obese subjects before and after gastric banding surgery. Obesity Surgery. 2005;15(1):11–23. doi: 10.1381/0960892052993431. [DOI] [PubMed] [Google Scholar]
- 146.McKenzie JA, Weiss EP, Ghiu IA, et al. Influence of the interleukin-6 -174 G/C gene polymorphism on exercise training-induced changes in glucose tolerance indexes. Journal of Applied Physiology. 2004;97(4):1338–1342. doi: 10.1152/japplphysiol.00199.2004. [DOI] [PubMed] [Google Scholar]
- 147.Kubaszek A, Pihlajamäki J, Punnonen K, Karhapää P, Vauhkonen I, Laakso M. The C-174G promoter polymorphism of the IL-6 gene affects energy expenditure and insulin sensitivity. Diabetes. 2003;52(2):558–561. doi: 10.2337/diabetes.52.2.558. [DOI] [PubMed] [Google Scholar]
- 148.Vozarova B, Fernández-Real JM, Knowler WC, et al. The interleukin-6 (-174) G/C promoter polymorphism is associated with type-2 diabetes mellitus in Native Americans and Caucasians. Human Genetics. 2003;112(4):409–413. doi: 10.1007/s00439-003-0912-x. [DOI] [PubMed] [Google Scholar]
- 149.Fishman D, Faulds G, Jeffey R, et al. The effect of novel polymorphisms in the interleukin-6 (IL-6) gene on IL-6 transcription and plasma IL-6 levels, and an association with systemic-onset juvenile chronic arthritis. The Journal of Clinical Investigation. 1998;102(7):1369–1376. doi: 10.1172/JCI2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Bouhaha R, Baroudi T, Ennafaa H, et al. Study of TNFα-308G/A and IL6 -174G/C polymorphisms in type 2 diabetes and obesity risk in the Tunisian population. Clinical Biochemistry. 2010;43(6):549–552. doi: 10.1016/j.clinbiochem.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 151.Brull DJ, Montgomery HE, Sanders J, et al. Interleukin-6 gene -174G>C and -572G>C promoter polymorphisms are strong predictors of plasma interleukin-6 levels after coronary artery bypass surgery. Arteriosclerosis, Thrombosis, and Vascular Biology. 2001;21(9):1458–1463. doi: 10.1161/hq0901.094280. [DOI] [PubMed] [Google Scholar]
- 152.Jones KG, Brull DJ, Brown LC, et al. Interleukin-6 (IL-6) and the prognosis of abdominal aortic aneurysms. Circulation. 2001;103(18):2260–2265. doi: 10.1161/01.cir.103.18.2260. [DOI] [PubMed] [Google Scholar]
- 153.Eklund C, Nenonen A, Kukkonen-Harjula K, et al. Association of the IL6-174(G/C) polymorphism with C-reactive protein concentration after weight loss in obese men. European Cytokine Network. 2006;17(2):131–135. [PubMed] [Google Scholar]
- 154.Moleres A, Rendo-Urteaga T, Azcona C, et al. IL6 gene promoter polymorphism (-174G/C) influences the association between fat mass and cardiovascular risk factors. Journal of Physiology and Biochemistry. 2009;65(4):405–413. doi: 10.1007/BF03185936. [DOI] [PubMed] [Google Scholar]
- 155.Berthier MT, Houde A, Côté M, et al. Impact of adiponectin gene polymorphisms on plasma lipoprotein and adiponectin concentrations of viscerally obese men. Journal of Lipid Research. 2005;46(2):237–244. doi: 10.1194/jlr.M400135-JLR200. [DOI] [PubMed] [Google Scholar]
- 156.Ng SB, Tan YH, Guy GR. Differential induction of the interleukin-6 gene by tumor necrosis factor and interleukin-1. The Journal of Biological Chemistry. 1994;269(29):19021–19027. [PubMed] [Google Scholar]
- 157.Zhang D, Jiang SL, Rzewnicki D, Samols D, Kushner I. The effect of interleukin-1 on C-reactive protein expression in Hep3B cells is exerted at the transcriptional level. Biochemical Journal. 1995;310(1):143–148. doi: 10.1042/bj3100143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Khovidhunkit W, Kim MS, Memon RA, et al. Effects of infection and inflammation on lipid and lipoprotein metabolism: mechanisms and consequences to the host. Journal of Lipid Research. 2004;45(7):1169–1196. doi: 10.1194/jlr.R300019-JLR200. [DOI] [PubMed] [Google Scholar]
- 159.Jager J, Grémeaux T, Cormont M, Le Marchand-Brustel Y, Tanti J-F. Interleukin-1β-induced insulin resistance in adipocytes through down-regulation of insulin receptor substrate-1 expression. Endocrinology. 2007;148(1):241–251. doi: 10.1210/en.2006-0692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Nicklin MJH, Barton JL, Nguyen M, FitzGerald MG, Duff GW, Kornman K. A sequence-based map of the nine genes of the human interleukin-1 cluster. Genomics. 2002;79(5):718–725. doi: 10.1006/geno.2002.6751. [DOI] [PubMed] [Google Scholar]
- 161.Kornman KS. Interleukin 1 genetics, inflammatory mechanisms, and nutrigenetic opportunities to modulate diseases of aging. The American Journal of Clinical Nutrition. 2006;83(2):475S–483S. doi: 10.1093/ajcn/83.2.475S. [DOI] [PubMed] [Google Scholar]
- 162.Latkovskis G, Licis N, Kalnins U. C-reactive protein levels and common polymorphisms of the interleukin-1 gene cluster and interleukin-6 gene in patients with coronary heart disease. European Journal of Immunogenetics. 2004;31(5):207–213. doi: 10.1111/j.1365-2370.2004.00476.x. [DOI] [PubMed] [Google Scholar]
- 163.Berger P, McConnell JP, Nunn M, et al. C-reactive protein levels are influenced by common IL-1 gene variations. Cytokine. 2002;17(4):171–174. doi: 10.1006/cyto.2001.0974. [DOI] [PubMed] [Google Scholar]
- 164.Iacoviello L, Di Castelnuovo A, Gattone M, et al. Polymorphisms of the interleukin-1β gene affect the risk of myocardial infarction and ischemic stroke at young age and the response of mononuclear cells to stimulation in vitro. Arteriosclerosis, Thrombosis, and Vascular Biology. 2005;25(1):222–227. doi: 10.1161/01.ATV.0000150039.60906.02. [DOI] [PubMed] [Google Scholar]
- 165.Rafiq S, Stevens K, Hurst AJ, et al. Common genetic variation in the gene encoding interleukin-1-receptor antagonist (IL-1RA) is associated with altered circulating IL-1RA levels. Genes & Immunity. 2007;8(4):344–351. doi: 10.1038/sj.gene.6364393. [DOI] [PubMed] [Google Scholar]
- 166.Shen J, Arnett DK, Peacock JM, et al. Interleukin1β genetic polymorphisms interact with polyunsaturated fatty acids to modulate risk of the metabolic syndrome. The Journal of Nutrition. 2007;137(8):1846–1851. doi: 10.1093/jn/137.8.1846. [DOI] [PubMed] [Google Scholar]
- 167.Kuyl JM, Mendelsohn D. Observed relationship between ratios HDL-cholesterol/total cholesterol and apolipoprotein A1/apolipoprotein B. Clinical Biochemistry. 1992;25(5):313–316. doi: 10.1016/0009-9120(92)80004-z. [DOI] [PubMed] [Google Scholar]
- 168.Perez-Martinez P, Lopez-Miranda J, Perez-Jimenez F, Ordovas JM. Influence of genetic factors in the modulation of postprandial lipemia. Atherosclerosis Supplements. 2008;9(2):49–55. doi: 10.1016/j.atherosclerosissup.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 169.Sorci-Thomas M, Prack MM, Dashti N, Johnson F, Rudel LL, Williams DL. Differential effects of dietary fat on the tissue-specific expression of the apolipoprotein A-I gene: relationship to plasma concentration of high density lipoproteins. Journal of Lipid Research. 1989;30(9):1397–1403. [PubMed] [Google Scholar]
- 170.Marcil M, Brooks-Wilson A, Clee SM, et al. Mutations in the ABC1 gene in familial HDL deficiency with defective cholesterol efflux. The Lancet. 1999;354(9187):1341–1346. doi: 10.1016/s0140-6736(99)07026-9. [DOI] [PubMed] [Google Scholar]
- 171.Schaefer EJ. Familial lipoprotein disorders and premature coronary artery disease. The Medical Clinics of North America. 1994;78(1):21–39. doi: 10.1016/s0025-7125(16)30175-4. [DOI] [PubMed] [Google Scholar]
- 172.Pisciotta L, Miccoli R, Cantafora A, et al. Recurrent mutations of the apolipoprotein A-I gene in three kindreds with severe HDL deficiency. Atherosclerosis. 2003;167(2):335–345. doi: 10.1016/s0021-9150(03)00020-0. [DOI] [PubMed] [Google Scholar]
- 173.Ikewaki K, Matsunaga A, Han H, et al. A novel two nucleotide deletion in the apolipoprotein A-I gene, apoA-I Shinbashi, associated with high density lipoprotein deficiency, corneal opacities, planar xanthomas, and premature coronary artery disease. Atherosclerosis. 2004;172(1):39–45. doi: 10.1016/j.atherosclerosis.2003.09.024. [DOI] [PubMed] [Google Scholar]
- 174.Yokota H, Hashimoto Y, Okubo S, et al. Apolipoprotein A-I deficiency with accumulated risk for CHD but no symptoms of CHD. Atherosclerosis. 2002;162(2):399–407. doi: 10.1016/s0021-9150(01)00724-9. [DOI] [PubMed] [Google Scholar]
- 175.Rubin EM, Krauss RM, Spangler EA, Verstuyft JG, Clift SM. Inhibition of early atherogenesis in transgenic mice by human apolipoprotein A1. Nature. 1991;353(6341):265–267. doi: 10.1038/353265a0. [DOI] [PubMed] [Google Scholar]
- 176.Herbert PN, Assmann G, Grotton JAM, Fredrickson DS. Disorders of lipid and lipoprotein metabolism. In: Stanbury JB, Wyngaarden DS, Fredrickson DS, et al., editors. Metabolic Basis of Inherited Disease. New York, NY, USA: McGraw-Hill; 1999. pp. 589–651. [Google Scholar]
- 177.Dastani Z, Dangoisse C, Boucher B, et al. A novel nonsense apolipoprotein A-I mutation (apoA-I) causes low HDL cholesterol in French Canadians. Atherosclerosis. 2006;185(1):127–136. doi: 10.1016/j.atherosclerosis.2005.05.028. [DOI] [PubMed] [Google Scholar]
- 178.Franceschini G, Sirtori CR, Capurso A, et al. A-IMilano apoprotein. Decreased high density lipoprotein cholesterol levels with significant lipoprotein modifications and without clinical atherosclerosis in an Italian family. The Journal of Clinical Investigation. 1980;66(5):892–900. doi: 10.1172/JCI109956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Roma P, Gregg RE, Meng MS, et al. In vivo metabolism of a mutant form of apolipoprotein A-I, apo A-IMilano, associated with familial hypoalphalipoproteinemia. The Journal of Clinical Investigation. 1993;91(4):1445–1452. doi: 10.1172/JCI116349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Weisgraber KH, Rall SC, Jr., Bersot TP, et al. Apolipoprotein A-IMilano. Detection of normal A-I in affected subjects and evidence for a cysteine for arginine substitution in the variant A-I. The Journal of Biological Chemistry. 1983;258(4):2508–2513. [PubMed] [Google Scholar]
- 181.Calabresi L, Cassinotti M, Gianfranceschi G, et al. Increased postprandial lipemia in apo A-IMilano carriers. Arteriosclerosis and Thrombosis. 1993;13(4):521–528. doi: 10.1161/01.atv.13.4.521. [DOI] [PubMed] [Google Scholar]
- 182.Dan Q, Huang Y, Peng Z. DNA polymorphisms of the apolipoprotein AI-CIII-AIV gene cluster in coronary artery disease. Zhonghua Yi Xue Za Zhi. 1995;75(10):584–637. [PubMed] [Google Scholar]
- 183.Vavatsi NA, Kouidou SA, Geleris PN, et al. Increased frequency of the rare PstI allele (P2) in a population of CAD patients in Northern Greece. Clinical Genetics. 1995;47(1):22–26. doi: 10.1111/j.1399-0004.1995.tb03916.x. [DOI] [PubMed] [Google Scholar]
- 184.Jeenah M, Kessling A, Miller N, Humphries S. G to A substitution in the promoter region of the apolipoprotein AI gene is associated with elevated serum apolipoprotein AI and high density lipoprotein cholesterol concentrations. Molecular Biology and Medicine. 1990;7(3):233–241. [PubMed] [Google Scholar]
- 185.Pagani F, Sidoli A, Giudici GA, Barenghi L, Vergani C, Baralle FE. Human apolipoprotein A-I gene promoter polymorphism: association with hyperalphalipoproteinemia. Journal of Lipid Research. 1990;31(8):1371–1377. [PubMed] [Google Scholar]
- 186.Marín C, López-Miranda J, Gómez P, et al. Effects of the human apolipoprotein A-I promoter G-A mutation on postprandial lipoprotein metabolism. The American Journal of Clinical Nutrition. 2002;76(2):319–325. doi: 10.1093/ajcn/76.2.319. [DOI] [PubMed] [Google Scholar]
- 187.Saha N, Tay JSH, Low PS, Humphries SE. Guanidine to adenine (G/A) substitution in the promoter region of the apolipoprotein AI gene is associated with elevated serum apolipoprotein AI levels in Chinese non-smokers. Genetic Epidemiology. 1994;11(3):255–264. doi: 10.1002/gepi.1370110304. [DOI] [PubMed] [Google Scholar]
- 188.Groenendijk M, Cantor RM, de Bruin TWA, Dallinga-Thie GM. The apoAI-CIII-AIV gene cluster. Atherosclerosis. 2001;157(1):1–11. doi: 10.1016/s0021-9150(01)00539-1. [DOI] [PubMed] [Google Scholar]
- 189.Noriega-López L, Tovar AR, Gonzalez-Granillo M, et al. Pancreatic insulin secretion in rats fed a soy protein high fat diet depends on the interaction between the amino acid pattern and isoflavones. The Journal of Biological Chemistry. 2007;282(28):20657–20666. doi: 10.1074/jbc.M701045200. [DOI] [PubMed] [Google Scholar]
- 190.Ascencio C, Torres N, Isoard-Acosta F, Gómez-Pérez FJ, Hernández-Pando R, Tovar AR. Soy protein affects serum insulin and hepatic SREBP-1 mRNA and reduces fatty liver in rats. The Journal of Nutrition. 2004;134(3):522–529. doi: 10.1093/jn/134.3.522. [DOI] [PubMed] [Google Scholar]
- 191.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. The Journal of Clinical Investigation. 2002;109(9):1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Torres N, Guevara-Cruz M, Granados J, et al. Reduction of serum lipids by soy protein and soluble fiber is not associated with the ABCG5/G8, apolipoprotein E, and apolipoprotein A1 polymorphisms in a group of hyperlipidemic Mexican subjects. Nutrition Research. 2009;29(10):728–735. doi: 10.1016/j.nutres.2009.09.013. [DOI] [PubMed] [Google Scholar]
- 193.Berge KE, Tian H, Graf GA, et al. Accumulation of dietary cholesterol in sitosterolemia caused by mutations in adjacent ABC transporters. Science. 2000;290(5497):1771–1775. doi: 10.1126/science.290.5497.1771. [DOI] [PubMed] [Google Scholar]
- 194.Fager G, Wiklund O, Olofsson SO, Wilhelmsen L, Bondjers G. Multivariate analyses of serum apolipoproteins and risk factors in relation to acute myocardial infarction. Arteriosclerosis. 1981;1(4):273–279. doi: 10.1161/01.atv.1.4.273. [DOI] [PubMed] [Google Scholar]
- 195.Buring JE, O’Connor GT, Goldhaber SZ, et al. Decreased HDL and HDL cholesterol, apo A-I and apo A-II, and increased risk of myocardial infarction. Circulation. 1992;85(1):22–29. doi: 10.1161/01.cir.85.1.22. [DOI] [PubMed] [Google Scholar]
- 196.Ridker PM, Rifai N, Cook NR, Bradwin G, Buring JE. Non-HDL cholesterol, apolipoproteins A-I and B, standard lipid measures, lipid ratios, and CRP as risk factors for cardiovascular disease in women. Journal of the American Medical Association. 2005;294(3):326–333. doi: 10.1001/jama.294.3.326. [DOI] [PubMed] [Google Scholar]
- 197.Tailleux A, Duriez P, Fruchart JC, Clavey V. Apolipoprotein A-II, HDL metabolism and atherosclerosis. Atherosclerosis. 2002;164(1):1–13. doi: 10.1016/s0021-9150(01)00751-1. [DOI] [PubMed] [Google Scholar]
- 198.Escolá-Gil JC, Marzal-Casacuberta A, Julve-Gil J, et al. Human apolipoprotein A-II is a pro-atherogenic molecule when it is expressed in transgenic mice at a level similar to that in humans: evidence of a potentially relevant species-specific interaction with diet. Journal of Lipid Research. 1998;39(2):457–462. [PubMed] [Google Scholar]
- 199.Han Z, Heath SC, Shmulewitz D, et al. Candidate genes involved in cardiovascular risk factors by a family-based association study on the island of Kosrae, Federated States of Micronesia. American Journal of Medical Genetics. 2002;110(3):234–242. doi: 10.1002/ajmg.10445. [DOI] [PubMed] [Google Scholar]
- 200.Van’t Hooft FM, Ruotolo G, Boquist S, De Faire U, Eggertsen G, Hamsten A. Human evidence that the apolipoprotein A-II gene is implicated in visceral fat accumulation and metabolism of triglyceride-rich lipoproteins. Circulation. 2001;104(11):1223–1228. doi: 10.1161/hc3601.095709. [DOI] [PubMed] [Google Scholar]
- 201.Takada D, Emi M, Ezura Y, et al. Interaction between the LDL-receptor gene bearing a novel mutation and a variant in the apolipoprotein a-ii promoter: molecular study in a 1135-member familial hypercholesterolemia kindred. Journal of Human Genetics. 2002;47(12):656–664. doi: 10.1007/s100380200101. [DOI] [PubMed] [Google Scholar]
- 202.Lara-Castro C, Hunter GR, Lovejoy JC, Gower BA, Fernández JR. Apolipoprotein A-II polymorphism and visceral adiposity in African-American and white women. Obesity Research. 2005;13(3):507–512. doi: 10.1038/oby.2005.53. [DOI] [PubMed] [Google Scholar]
- 203.Talmud PJ, Humphries SE. Genetic polymorphisms, lipoproteins and coronary artery disease risk. Current Opinion in Lipidology. 2001;12(4):405–409. doi: 10.1097/00041433-200108000-00006. [DOI] [PubMed] [Google Scholar]
- 204.Pennacchio LA, Olivier M, Hubacek JA, et al. An apolipoprotein influencing triglycerides in humans and mice revealed by comparative sequencing. Science. 2001;294(5540):169–173. doi: 10.1126/science.1064852. [DOI] [PubMed] [Google Scholar]
- 205.van der Vliet HN, Sammels MG, Leegwater ACJ, et al. Apolipoprotein A-V: a novel apolipoprotein associated with an early phase of liver regeneration. The Journal of Biological Chemistry. 2001;276(48):44512–44520. doi: 10.1074/jbc.M106888200. [DOI] [PubMed] [Google Scholar]
- 206.Schaap FG, Rensen PCN, Voshol PJ, et al. ApoAV reduces plasma triglycerides by inhibiting very low density lipoprotein-triglycerides (VLDL-TG) production and stimulating lipoprotein lipase-mediated VLDL-TG hydrolysis. The Journal of Biological Chemistry. 2004;279(27):27941–27947. doi: 10.1074/jbc.M403240200. [DOI] [PubMed] [Google Scholar]
- 207.Fruchart-Najib J, Baugé E, Niculescu LS, et al. Mechanism of triglyceride lowering in mice expressing human apolipoprotein A5. Biochemical and Biophysical Research Communications. 2004;319(2):397–404. doi: 10.1016/j.bbrc.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 208.Pennacchio LA, Olivier M, Hubacek JA, Krauss RM, Rubin EM, Cohen JC. Two independent apolipoprotien A5 haplotypes influence human plasma triglyceride levels. Human Molecular Genetics. 2002;11(24):3031–3038. doi: 10.1093/hmg/11.24.3031. [DOI] [PubMed] [Google Scholar]
- 209.Aouizerat BE, Kulkarni M, Heilbron D, et al. Genetic analysis of a polymorphism in the human apoA-V gene: effect on plasma lipids. Journal of Lipid Research. 2003;44(6):1167–1173. doi: 10.1194/jlr.M200480-JLR200. [DOI] [PubMed] [Google Scholar]
- 210.Moreno R, Perez-Jimenez F, Marin C, et al. A single nucleotide polymorphism of the apolipoprotein A-V gene -1131T>C modulates postprandial lipoprotein metabolism. Atherosclerosis. 2006;189(1):163–168. doi: 10.1016/j.atherosclerosis.2005.11.029. [DOI] [PubMed] [Google Scholar]
- 211.Zhao T, Zhao J. Association of the apolipoprotein A5 gene -1131 T>C polymorphism with fasting blood lipids: a meta-analysis in 37859 subjects. BMC Medical Genetics. 2010;11(1, article 120):1–12. doi: 10.1186/1471-2350-11-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Austin MA, Talmud PJ, Farin FM, et al. Association of apolipoprotein A5 variants with LDL particle size and triglyceride in Japanese Americans. Biochimica et Biophysica Acta. 2004;1688(1):1–9. doi: 10.1016/j.bbadis.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 213.Szalai C, Keszei M, Duba J, et al. Polymorphism in the promoter region of the apolipoprotein A5 gene is associated with an increased susceptibility for coronary artery disease. Atherosclerosis. 2004;173(1):109–114. doi: 10.1016/j.atherosclerosis.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 214.Dallongeville J, Cottel D, Montaye M, Codron V, Amouyel P, Helbecque N. Impact of APOA5/A4/C3 genetic polymorphisms on lipid variables and cardiovascular disease risk in French men. International Journal of Cardiology. 2006;106(2):152–156. doi: 10.1016/j.ijcard.2004.10.065. [DOI] [PubMed] [Google Scholar]
- 215.Corella D, Lai CQ, Demissie S, et al. APOA5 gene variation modulates the effects of dietary fat intake on body mass index and obesity risk in the Framingham Heart Study. Journal of Molecular Medicine. 2007;85(2):119–128. doi: 10.1007/s00109-006-0147-0. [DOI] [PubMed] [Google Scholar]
- 216.Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988;240(4852):622–630. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- 217.Rall SC, Jr., Weisgraber KH, Mahley RW. Human apolipoprotein E. The complete amino acid sequence. The Journal of Biological Chemistry. 1982;257(8):4171–4178. [PubMed] [Google Scholar]
- 218.Mahley RW, Rall SC., Jr. Apolipoprotein E: far more than a lipid transport protein. Annual Review of Genomics and Human Genetics. 2000;1(2000):507–537. doi: 10.1146/annurev.genom.1.1.507. [DOI] [PubMed] [Google Scholar]
- 219.Mahley RW, Ji ZS. Remnant lipoprotein metabolism: key pathways involving cell-surface heparan sulfate proteoglycans and apolipoprotein E. Journal of Lipid Research. 1999;40(1):1–16. [PubMed] [Google Scholar]
- 220.Taira K, Bujo H, Hirayama S, et al. LR11, a mosaic LDL receptor family member, mediates the uptake of apoE-rich lipoproteins in vitro. Arteriosclerosis, Thrombosis, and Vascular Biology. 2001;21(9):1501–1506. doi: 10.1161/hq0901.094500. [DOI] [PubMed] [Google Scholar]
- 221.Xhignesse M, Lussier-Cacan S, Sing CF, Kessling AM, Davignon J. Influences of common variants of apolipoprotein E on measures of lipid metabolism in a sample selected for health. Arteriosclerosis and Thrombosis. 1991;11(4):1100–1110. doi: 10.1161/01.atv.11.4.1100. [DOI] [PubMed] [Google Scholar]
- 222.Huang Y, Liu XQ, Rail SC, Jr., Mahley RW. Apolipoprotein E2 reduces the low density lipoprotein level in transgenic mice by impairing lipoprotein lipase-mediated lipolysis of triglyceride-rich lipoproteins. The Journal of Biological Chemistry. 1998;273(28):17483–17490. doi: 10.1074/jbc.273.28.17483. [DOI] [PubMed] [Google Scholar]
- 223.Huang Y, Liu XQ, Rall SC, Jr., et al. Overexpression and accumulation of apolipoprotein E as a cause of hypertriglyceridemia. The Journal of Biological Chemistry. 1998;273(41):26388–26393. doi: 10.1074/jbc.273.41.26388. [DOI] [PubMed] [Google Scholar]
- 224.Rensen PCN, van Berkel TJC. Apolipoprotein E effectively inhibits lipoprotein lipase-mediated lipolysis of chylomicron-like triglyceride-rich lipid emulsions in vitro and in vivo. The Journal of Biological Chemistry. 1996;271(25):14791–14799. doi: 10.1074/jbc.271.25.14791. [DOI] [PubMed] [Google Scholar]
- 225.Huang Y, von Eckardstein A, Wu S, Maeda N, Assmann G. A plasma lipoprotein containing only apolipoprotein E and with γ mobility on electrophoresis releases cholesterol from cells. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(5):1834–1838. doi: 10.1073/pnas.91.5.1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Hui DY, Harmony JA. Inhibition of Ca2+ accumulation in mitogen-activated lymphocytes: role of membrane-bound plasma lipoproteins. Proceedings of the National Academy of Sciences of the United States of America. 1980;77(8):4764–4768. doi: 10.1073/pnas.77.8.4764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Miettinen TA, Gylling H, Vanhanen H, Ollus A. Cholesterol absorption, elimination, and synthesis related to LDL kinetics during varying fat intake in men with different apoprotein E phenotypes. Arteriosclerosis and Thrombosis. 1992;12(9):1044–1052. doi: 10.1161/01.atv.12.9.1044. [DOI] [PubMed] [Google Scholar]
- 228.Loktionov A, Vorster H, O'Neill IK, et al. Apolipoprotein E and methylenetetrahydrofolate reductase genetic polymorphisms in relation to other risk factors for cardiovascular disease in UK Caucasians and Black South Africans. Atherosclerosis. 1999;145(1):125–135. doi: 10.1016/s0021-9150(99)00022-2. [DOI] [PubMed] [Google Scholar]
- 229.Davignon J, Gregg RE, Sing CF. Apolipoprotein E polymorphism and atherosclerosis. Arteriosclerosis. 1988;8(1):1–21. doi: 10.1161/01.atv.8.1.1. [DOI] [PubMed] [Google Scholar]
- 230.Utermann G, Vogelberg KH, Steinmetz A, et al. Polymorphism of apolipoprotein E. II. Genetics of hyperlipoproteinemia type III. Clinical Genetics. 1979;15(1):37–62. [PubMed] [Google Scholar]
- 231.Moreno JA, López-Miranda J, Marín C, et al. The influence of the apolipoprotein E gene promoter (-219G/T) polymorphism on postprandial lipoprotein metabolism in young normolipemic males. Journal of Lipid Research. 2003;44(11):2059–2064. doi: 10.1194/jlr.M300124-JLR200. [DOI] [PubMed] [Google Scholar]
- 232.Alvim RO, Freitas SR, Ferreira NE, et al. APOE polymorphism is associated with lipid profile, but not with arterial stiffness in the general population. Lipids in Health and Disease. 2010;9, article 128:1–7. doi: 10.1186/1476-511X-9-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Cardona F, Morcillo S, Gonzalo-Marin M, Tinahones FJ. The apolipoprotein E genotype predicts postprandial hypertriglyceridemia in patients with the metabolic syndrome. The Journal of Clinical Endocrinology and Metabolism. 2005;90(5):2972–2975. doi: 10.1210/jc.2004-1912. [DOI] [PubMed] [Google Scholar]
- 234.Dreon DM, Fernstrom HA, Miller B, Krauss RM. Apolipoprotein E isoform phenotype and LDL subclass response to a reduced-fat diet. Arteriosclerosis, Thrombosis, and Vascular Biology. 1995;15(1):105–111. doi: 10.1161/01.atv.15.1.105. [DOI] [PubMed] [Google Scholar]
- 235.Chu FF, Esworthy RS, Chu PG, et al. Bacteria-induced intestinal cancer in mice with disrupted Gpx1 and Gpx2 genes. Cancer Research. 2004;64(3):962–968. doi: 10.1158/0008-5472.can-03-2272. [DOI] [PubMed] [Google Scholar]
- 236.Brigelius-Flohé R. Tissue-specific functions of individual glutathione peroxidases. Free Radical Biology & Medicine. 1999;27(9-10):951–965. doi: 10.1016/s0891-5849(99)00173-2. [DOI] [PubMed] [Google Scholar]
- 237.Ran Q, Liang H, Gu M, et al. Transgenic mice overexpressing glutathione peroxidase 4 are protected against oxidative stress-induced apoptosis. The Journal of Biological Chemistry. 2004;279(53):55137–55146. doi: 10.1074/jbc.M410387200. [DOI] [PubMed] [Google Scholar]
- 238.Imai H, Narashima K, Arai M, Sakamoto H, Chiba N, Nakagawa Y. Suppression of leukotriene formation in RBL-2H3 cells that overexpressed phospholipid hydroperoxide glutathione peroxidase. The Journal of Biological Chemistry. 1998;273(4):1990–1997. doi: 10.1074/jbc.273.4.1990. [DOI] [PubMed] [Google Scholar]
- 239.Villette S, Kyle JAM, Brown KM, et al. A novel single nucleotide polymorphism in the 3′ untranslated region of human glutathione peroxidase 4 influences lipoxygenase metabolism. Blood Cells, Molecules, & Diseases. 2002;29(2):174–178. doi: 10.1006/bcmd.2002.0556. [DOI] [PubMed] [Google Scholar]
- 240.Stadtman TC. Selenocysteine. Annual Review of Biochemistry. 1996;65:83–100. doi: 10.1146/annurev.bi.65.070196.000503. [DOI] [PubMed] [Google Scholar]
- 241.Hatfield DL, Gladyshev VN. How selenium has altered our understanding of the genetic code. Molecular and Cellular Biology. 2002;22(11):3565–3576. doi: 10.1128/MCB.22.11.3565-3576.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Kryukov GV, Castellano S, Novoselov SV, et al. Characterization of mammalian selenoproteomes. Science. 2003;300(5624):1439–1443. doi: 10.1126/science.1083516. [DOI] [PubMed] [Google Scholar]
- 243.Berry MJ, Banu L, Chen Y, et al. Recognition of UGA as a selenocysteine codon in type I deiodinase requires sequences in the 3’ untranslated region. Nature. 1991;353(6341):273–276. doi: 10.1038/353273a0. [DOI] [PubMed] [Google Scholar]
- 244.Hesketh J. Nutrigenomics and selenium: gene expression patterns, physiological targets, and genetics. Annual Review of Nutrition. 2008;28:157–177. doi: 10.1146/annurev.nutr.28.061807.155446. [DOI] [PubMed] [Google Scholar]
- 245.Burk RF, Hill KE. Selenoprotein P. A selenium-rich extracellular glycoprotein. The Journal of Nutrition. 1994;124(10):1891–1897. doi: 10.1093/jn/124.10.1891. [DOI] [PubMed] [Google Scholar]
- 246.Hill KE, Zhou J, McMahan WJ, et al. Deletion of selenoprotein P alters distribution of selenium in the mouse. The Journal of Biological Chemistry. 2003;278(16):13640–13646. doi: 10.1074/jbc.M300755200. [DOI] [PubMed] [Google Scholar]
- 247.Schomburg L, Schweizer U, Holtmann B, Flohé L, Sendtner M, Köhrle J. Gene disruption discloses role of selenoprotein P in selenium delivery to target tissues. Biochemical Journal. 2003;370(2):397–402. doi: 10.1042/BJ20021853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Burk RF, Hill KE. Selenoprotein P: an extracellular protein with unique physical characteristics and a role in selenium homeostasis. Annual Review of Nutrition. 2005;25:215–235. doi: 10.1146/annurev.nutr.24.012003.132120. [DOI] [PubMed] [Google Scholar]
- 249.Kabuyama Y, Oshima K, Kitamura T, et al. Involvement of selenoprotein P in the regulation of redox balance and myofibroblast viability in idiopathic pulmonary fibrosis. Genes to Cells. 2007;12(11):1235–1244. doi: 10.1111/j.1365-2443.2007.01127.x. [DOI] [PubMed] [Google Scholar]
- 250.Steinbrenner H, Alili L, Bilgic E, Sies H, Brenneisen P. Involvement of selenoprotein P in protection of human astrocytes from oxidative damage. Free Radical Biology & Medicine. 2006;40(9):1513–1523. doi: 10.1016/j.freeradbiomed.2005.12.022. [DOI] [PubMed] [Google Scholar]
- 251.Steinbrenner H, Bilgic E, Alili L, Sies H, Brenneisen P. Selenoprotein P protects endothelial cells from oxidative damage by stimulation of glutathione peroxidase expression and activity. Free Radical Research. 2006;40(9):936–943. doi: 10.1080/10715760600806248. [DOI] [PubMed] [Google Scholar]
- 252.Zhang Y, Chen X. Reducing selenoprotein P expression suppresses adipocyte differentiation as a result of increased preadipocyte inflammation. American Journal of Physiology. 2011;300(1):E77–E85. doi: 10.1152/ajpendo.00380.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]