

Abstract
Inactivation of β-lactams by the action of β-lactamase enzymes is the most common mode of resistance to these drugs among Gram-negative organisms. The genomes of some key clinical pathogens such as Enterobacter and Pseudomonas encode AmpC, an inducible chromosomal β-lactamase. The potent activity of AmpC against broad-spectrum β-lactams complicates treatment of organisms with this gene. Antibiotic exposure can select for mutants expressing high levels of this enzyme, leading to the emergence of resistant isolates and failure of therapy, even when the initial isolate is fully susceptible. The risk of selecting for resistant organisms varies according to the particular β-lactam used for treatment. This article reviews the microbiology of these enzymes, summarizes clinical data on the frequency emergence of resistance, and discusses considerations for antimicrobial treatment of these organisms.
Keywords: β-lactamases, drug resistance, microbial, Enterobacter, Pseudomonas
INTRODUCTION
β-Lactams (i.e., penicillins, cephalosporins, monobactams, and carbapenems) are the most widely used class of antibacterial agents, especially in children.1 Resistance to β-lactams among Gram-negative organisms is primarily mediated through the elaboration of β-lactamases, hundreds of different varieties of which have been characterized.2 The effect of β-lactamases is generally reflected by the standard susceptibility tests that microbiology laboratories perform on clinical isolates. Thus, clinicians do not require an exhaustive knowledge of β-lactamase activity to choose an appropriate agent. However, the expression of certain β-lactamases can result in resistance not reflected in the in vitro susceptibility measured at the time of isolation. This can lead to the development of resistance in an originally susceptible isolate and failure of therapy. Resistance to β-lactams is particularly problematic in pediatrics, as many non–β-lactams have safety concerns or limited experience in children. In this article we review the microbiology of organisms capable of expressing chromosomal β-lactamase (AmpC)-type β-lactamases and the implications for antibacterial therapy for clinicians, with an emphasis on the treatment of Enterobacter and Pseudomonas species.
MICROBIOLOGY AND EPIDEMIOLOGY
β-Lactamases of the AmpC type are enzymes that can rapidly hydrolyze penicillins, cephalosporins, and monobactams.3 They are not significantly inhibited by the action of clinically used β-lactamase inhibitors (i.e., clavulanate, sulbactam, tazobactam). The genes encoding these β-lactamases are found in the chromosomes of organisms such as Serratia, Pseudomonas, Acinetobacter, Citrobacter, and Enterobacter (often grouped by clinicians as the “SPACE” organisms). What determines the degree of β-lactam resistance conferred by these enzymes is their expression level.4 For example, the ampC gene is present in Escherichia coli; however, E coli lacks the necessary mechanisms for expressing the gene at a high enough level to cause clinical resistance.5 Less commonly, the ampC gene can be present on plasmids and contribute to resistance in organisms that do not harbor the gene chromosomally.6 In any of these organisms, secondary β-lactamases may be present that can contribute to β-lactam resistance. These are particularly common in Acinetobacter; thus, this organism is not a focus of this review.7
The expression of ampC as a chromosomal gene has been largely worked out for Enterobacter and is thought to be similar in mechanism in other organisms. The Figure 1 is a simplified illustration of the putative regulation of ampC expression. For the major wild-type Enterobacter species (aerogenes and cloacae), ampC displays inducible expression.8 With inducible expression, the action of the protein repressing transcription of gene for AmpC (AmpR) reduces expression of the AmpC β-lactamase to very low levels.5 The repression of ampC via AmpR can be disabled by the binding of certain cell wall degradation products to the AmpR protein, leading to transcription of ampC.9 The cell wall precursor recycling protein (AmpD) cleaves certain residues off of these degradation products.10 These shortened peptides can be “recycled” into the cell wall synthesis pathway. The AmpD-cleaved peptides, unlike the original degradation products, are not able to bind to and disable AmpR, leading to “de-repression” of ampC.11 The action of β-lactams on the cell wall increases the production of degradation products that bind to AmpR.9 Table 1 illustrates the relationship between degree of induction and susceptibility of the substrate to hydrolysis by AmpC: Antibacterials that are strong inducers and good substrates are inactive against wild-type inducible strains, whereas being either a weak inducer or poor substrate (or both) allows for clinically useful activity.3,4 The variation in inducing activity between β-lactams may be due to binding to different penicillin-binding proteins, the inactivation of which may lead to degradation products that are more or less likely to contribute to the activation of ampC.12
Figure 1.

Regulation of AmpC β-lactamase expression. Simplified depiction of the regulation of ampC. Left, the wild-type inducible phenotype: increased transcription of ampC results when β-lactam exposure increases the rate of formation of degradation products beyond the capacity of AmpD to cleave them to a length that does not bind AmpR. Right, mutant displaying stable de-repression: inactivating mutation of ampD leads to high-level expression of ampC in the presence or absence of β-lactams.
Table 1.
ampC Induction Profile of Various Antibacterials

Thus, although clinicians frequently believe that the “inducibility” of the AmpC β-lactamase is a reason to avoid cephalosporin use in SPACE organisms, inducibility per se generally only explains the wild-type susceptibility profile of these organisms. What clinicians should be more concerned about is the potential for stable de-repression of the ampC gene. This state comes about primarily through mutations that affect the copy number or function of AmpD.10 Without a functioning AmpD protein an excess of cell wall degradation products of the length capable of binding to AmpR builds up, leading to perpetual binding of these products to AmpR and constant de-repression of ampC. The subsequent constant high-level expression of AmpC is generally sufficient to lead to clinical resistance to all β-lactams, with the exception of carbapenems (and, in some organisms, cefepime), as illustrated in Table 2.13 Although constant high-level expression of ampC likely has a fitness cost to the organism, in the face of selective pressure through β-lactam exposure in an individual patient or the hospital environment, this phenotype may be sustained.10 Thus, through eliminating the susceptible (non–de-repressed) organisms within a bacterial population, β-lactam therapy can select for resistant mutants, leading to clinical and microbiologic failure and isolation of organisms resistant to the β-lactam used for therapy. This may occur more commonly in more severe infections (where organism burden is high) or in immunosuppressed patients, whose immune system would be unable to perform the “mop-up” work of killing off the resistant subpopulation.
Table 2.
Predicted Drug Susceptibility for ampC-inducible and ampC de-repressed Enterobacter and Pseudomonas
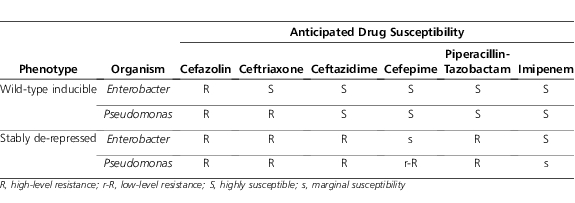
Infections due to Pseudomonas aeruginosa can be even more challenging than those due to Enterobacter. In Pseudomonas, multiple ampD genes control ampC expression, allowing for a broader range of expression of ampC and mutants with partially de-repressed phenotypes with greater retention of fitness and virulence.14 In addition to the potential for resistance from AmpC hyperexpression, Pseudomonas possesses other resistance mechanisms that enhance its resistance to β-lactams.15 Even among susceptible isolates of Pseudomonas, wild-type minimum inhibitor concentrations (MICs) are typically several-fold higher than other Gram-negative agents. For example, in 1 large surveillance study, the MIC for 50% of isolates of Pseudomonas to ceftazidime was 2 mcg/mL, compared with 0.25 for Enterobacter.16 This baseline level of reduced susceptibility is thought to be due to reduced access for β-lactams across the outer bacterial membrane to their target sites on the bacterial cell wall, active efflux of β-lactams out of the periplasmic space, and/or limited permeation through porin channels.15 Porin channels are proteins that allow passive diffusion of substrates through the bacterial outer membrane; ability to transit across porin channels is determined by molecular size and charge. Various β-lactams use certain porin channels to access their targets on the bacterial cell wall. Mutants deficient in the OprD porin channel protein display imipenem resistance, while those displaying upregulation of efflux pumps may be meropenem- and doripenem-resistant.17 Resistance to the carbapenems can occur independently of or concurrently with resistance to cephalosporins and penicillins. Upregulation of efflux likely also contributes to cefepime resistance, negating the advantage of cefepime's greater stability to AmpC hydrolysis.18
There are tests that allow for presumptive detection of AmpC-mediated resistance.19 However, they are generally too complex and time consuming for performance in the clinical microbiology laboratory. For organisms that harbor an inducible chromosomal AmpC, there is little value in detecting the presence of this enzyme. There may be some role for AmpC testing in differentiating β-lactamase resistance due to presence of a (usually constitutively produced) plasmid-mediated AmpC as opposed to acquired extended-spectrum β-lactamases in organisms such as E coli and Klebsiella.20
There are limited published data on the epidemiology of β-lactamase–mediated resistance specifically in pediatric patients. Jones et al21 collected over 12,000 strains from pediatric patients at 52 North American medical centers from 1998 to 2004. Among Enterobacter species, susceptibility to ceftriaxone, ceftazidime, and piperacillin-tazobactam ranged from 78% to 81%. Cefepime (99% susceptible) and imipenem (100% susceptible) were the most active agents. For P aeruginosa, ceftazidime susceptibility was 87.3%, piperacillin-tazobactam was 93.3%, cefepime was 90.8%, and imipenem susceptibility was 94.4%.
STUDIES OF THE RISK OF SELECTION FOR RESISTANT ISOLATES
Choosing appropriate β-lactam therapy for organisms with functional chromosomal ampC genes is complicated by the risk of selecting for stably de-repressed mutants. To quantify this risk in patients, we need to review studies that calculate the likelihood of a patient with an initially drug-susceptible isolate having a resistant organism of the same species isolated subsequent to antibacterial therapy. Ideally, such studies would perform molecular typing to determine whether paired of isolates were clonal, representing a change in susceptibility of the original infecting organism rather than acquisition of a new variant. Table 3 summarizes the proportion of isolates that developed resistance to various therapies across a number of studies. These studies typically included only adult patients or did not differentiate between isolates from adults or children. In the earliest study by Chow et al,22 of 31 patients with Enterobacter bacteremia initially susceptible to cephalosporins, 6 patients (19%) who received third-generation cephalosporins had another Enterobacter isolate with resistance to first- through third-generation cephalosporins isolated from a follow-up clinical culture (5 blood cultures, 1 intra-abdominal culture). Coadministration of aminoglycosides with cephalosporins did not appear to reduce the risk of emergence of resistance to the cephalosporin, as 4 of 6 patients in whom emergence of cephalosporin resistance occurred received a concomitant aminoglycoside. One of these 6 patients died (16.7%), similar to the mortality in patients without emergence of resistance (16.3%). Kaye et al24 performed a similar retrospective cohort study among 477 hospitalized patients who had cephalosporin-susceptible Enterobacter species isolated from clinical specimens. Emergence of resistance to cephalosporins was more common (19%) with extended-spectrum cephalosporins compared with penicillins (9%). On multivariate analysis, exposure to broad-spectrum cephalosporins, but not other drugs, was associated with an increased risk of emergence of resistance. This study controlled for the frequency of culturing but did not assess outcomes.
Table 3.
Clinical Studies of the Risk of Emergence of Resistance to Various Antibacterials
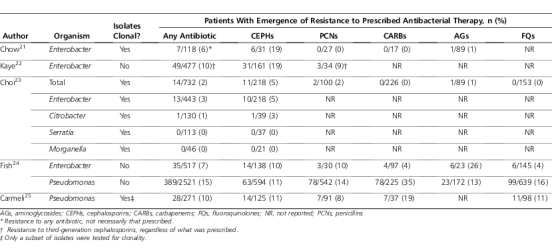
Selection for AmpC-overexpressing isolates can occur with organisms such as Serratia, Citrobacter, and Morganella in addition to Enterobacter. A study by Choi et al25 prospectively followed 732 patients with a clinical isolate of 1 of the previously mentioned organisms. Emergence of resistance was much more common when third-generation cephalosporins were used, compared with extended-spectrum penicillins, carbapenems, aminoglycosides, and fluoroquinolones. Emergence of resistance to cefepime did not occur (0/20 isolates). All but one case of emergence of resistance occurred in Enterobacter. The highest risk of emergence of resistance was seen among the subgroup of patients with Enterobacter bacteremia (4/30, 13.3%). Emergence of resistance overall was not lower with combination therapy compared with monotherapy (2.7% vs 1.8%) but somewhat lower in the subgroup of patients receiving broad-spectrum cephalosporin therapy (2.7% vs 5.5%).
Fish et al26 analyzed randomly selected clinical studies of antibacterials to examine development of resistance on therapy. Emergence of resistance to any antibacterial was significantly higher for Enterobacter (6.8%) and Pseudomonas (15.4%), compared with all organisms (4.0%, p<0.01 for both comparisons). Across all organisms, clinical success was significantly lower when emergence of resistance occurred (88.6% vs 83.2%, p<0.001).
Carmeli et al27 studied the emergence of resistance to ceftazidime, ciprofloxacin, imipenem, and piperacillin-tazobactam among 271 patients with Pseudomonas infection. Combination therapy with aminoglycosides did not have a statistically significant effect on the risk of emergence of resistance. Multivariate models adjusting for aminoglycoside use and frequency of culturing found that risk of emergence of resistance was highest with imipenem (hazard ratio [HR]=44, p=0.001), followed by ciprofloxacin (HR=9.2, p=0.04), piperacillin-tazobactam (HR=5.2, p=0.01), and ceftazidime (HR=0.8, p=0.7). Clinical outcomes were not reported. Georges et al28 prospectively followed 132 patients in the intensive care unit with isolates of Pseudomonas considered to represent colonization (55%) or infection (45%). Resistance to a β-lactam antibiotic developed in 42 patients (32%); there was an increased risk of development of resistance associated with imipenem (HR=6.0, p<0.001) and piperacillin-tazobactam (HR=3.8, p=0.02) but not with ceftazidime (HR=1.1, p=0.85). The proportion of treatment courses resulting in resistance for each drug was not presented. Mortality was 34% in the group without emergence of resistance compared with 26% among patients with emergence of resistance.
INTERPRETATION OF STUDIES AND RECOMMENDATIONS FOR THERAPY
Interpretation of these studies is limited by a number of factors. Most importantly, patients were not randomly assigned to the various therapies; thus, confounding factors might account for differences in the likelihood of emergence of resistance. Another key limitation is that dosage intensity is not reported. An increasing body of literature suggests that dosage regimens designed to optimize pharmacokinetic/pharmacodynamic (PK/PD) exposures (e.g. time>MIC) are associated with improved clinical and microbiologic outcomes and potentially the suppression of treatment-emergent resistance.29–32 Although the studies of emergence of resistance often reported on the use of combination therapy, the timing and duration of combination therapy is generally not provided. Finally, despite the fact that the isolation of a second resistant isolate generally represents microbiologic failure, emergence of resistance was not always associated with a higher rate of poorer clinical outcomes (e.g., mortality).
Resistance seems to be more likely to emerge among isolates causing more invasive infections, where the organism burden may be higher (e.g., bacteremia). Combination therapy (e.g., with aminoglycosides) does not appear to substantially reduce the emergence of resistance to third-generation cephalosporins. Empiric coverage for infections due to organisms that have the potential for overexpression of ampC should be tailored to local susceptibility data. Addition of an aminoglycoside for empiric coverage, especially when Pseudomonas is suspected, should be considered to ensure the regimen includes at least one active agent. Once final identification and susceptibility results are obtained, definitive therapy with a drug with confirmed in vitro activity should be initiated.
Among Enterobacter species, the risk of emergence of third-generation cephalosporin (presumably AmpC-mediated) resistance during therapy with third-generation cephalosporins appears to be between 5% and 20%. Use of these agents should probably be avoided for the treatment of serious infections (such as pneumonia or bacteremia), especially in severely ill patients. The risk of selection for resistant isolates appears to be lower, but still present, with extended-spectrum penicillins such as piperacillin and tazobactam. Data from a limited number of patients suggest cefepime may have a low risk of selecting AmpC-hyperproducing isolates, as might be expected by its greater stability to AmpC (and supported by some in vitro data).33 Thus, piperacillin-tazobactam and especially cefepime appear to convey a lower risk and should be considered for less serious infections (e.g., urinary tract infections) or for severe infections in clinically stable patients with careful monitoring. Non–β-lactams such as fluoroquinolones and aminoglycosides (and likely trimethoprim-sulfamethoxazole, although data are lacking) have comparatively lower risks of emergence of resistance to themselves. These would be reasonable choices for definitive therapy, although their toxicity risks are generally greater than β-lactams. The excellent oral bioavailability of fluoroquinolones and trimethoprim-sulfamethoxazole make them well suited for management of mild to moderate infections. It should be noted that the activity of oral cephalosporins (e.g., cefpodoxime, cefdinir) cannot always be inferred from the susceptibility to intravenous cephalosporins and should be used with caution and direct susceptibility testing if available. Carbapenems appear to have the lowest risk of all and would represent a reliable choice for seriously ill patients with severe infections, at the cost of administration of extremely broad-spectrum treatment. Addition of an aminoglycoside is generally unwarranted.
For P aeruginosa, because of the high risk of emergence of resistance across all drug classes, selecting a preferred agent is more difficult. Emergence of resistance to penicillins and cephalosporins appears to be of a similar order to that seen with Enterobacter. However, the risk of emergence of resistance to carbapenems and non–β-lactams is higher compared with Enterobacter, complicating the selection of alternative therapies. For serious infections, a carbapenem (imipenem, meropenem, or doripenem), ceftazidime, cefepime, or piperacillin-tazobactam are reasonable selections. Of these agents, ceftazidime has the narrowest spectrum of activity, and some data suggest that selection for resistance may be lower. Although there is little clinical evidence for the benefit of combination therapy with aminoglycosides or fluoroquinolones beyond the empiric period of therapy, some clinicians would continue these agents if no contraindications exist. Optimizing drug exposure through increasing the dose and/or extending the infusion of β-lactam agents is probably key in providing the best likelihood of good outcomes. Although most studies and simulations of PK/PD-based dosing have been performed in adults, an increasing number of studies are exploring such dosing in children.34,35 For less-severe infections, narrower spectrum therapy with ceftazidime or a fluoroquinolone is likely reasonable. Limited data for other species (Serratia, Citrobacter, Morganella) suggest the risk of emergence of resistance in lower than in Enterobacter or Pseudomonas.
CONCLUSION
The presence of an ampC gene capable of overexpression is a resistance mechanism “lying in wait” that complicates the treatment of infections due to a number of Gram-negative organisms, particularly Enterobacter and Pseudomonas. Understanding this complex mechanism of resistance can help clinicians to weigh the risks of β-lactam therapy and select among different β-lactams to reduce the likelihood of emergence of resistant organisms and potential clinical failure.
The next installment in this series will discuss the detection and clinical management of extended-spectrum β-lactamases in Enterobacteriaceae.
Abbreviations
- AmpC
chromosomal β-lactamase
- AmpD
cell wall precursor recycling protein
- AmpR
protein repressing transcription of gene for AmpC
- HR
hazard ratio
- MIC
minimum inhibitor concentration
- PK/PD
pharmacokinetic/pharmacodynamic
Footnotes
DISCLOSURES The author declares no conflicts or financial interest in any product or service mentioned in the manuscript, including grants, equipment, medications, employment, gifts, and honoraria.
REFERENCES
- 1.Pakyz AL, Gurgle HE, Ibrahim OM, et al. Trends in antibacterial use in hospitalized pediatric patients in United States academic health centers. Infect Control Hosp Epidemiol. 2009;30(6):600–603. doi: 10.1086/597545. [DOI] [PubMed] [Google Scholar]
- 2.Bush K, Jacoby GA. Updated functional classification of beta-lactamases. Antimicrob Agents Chemother. 2010;54(3):969–976. doi: 10.1128/AAC.01009-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jacoby GA. AmpC beta-lactamases. Clin Microbiol Rev. 2009;22(1):161–182. doi: 10.1128/CMR.00036-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Livermore DM. Clinical significance of beta-lactamase induction and stable derepression in gram-negative rods. Eur J Clin Microbiol. 1987;6(4):439–445. doi: 10.1007/BF02013107. [DOI] [PubMed] [Google Scholar]
- 5.Honoré N, Nicolas MH, Cole ST. Inducible cephalosporinase production in clinical isolates of Enterobacter cloacae is controlled by a regulatory gene that has been deleted from Escherichia coli. EMBO J. 1986;5(13):3709–3714. doi: 10.1002/j.1460-2075.1986.tb04704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deshpande LM, Jones RN, Fritsche TR, Sader HS. Occurrence of plasmidic AmpC type beta-lactamase-mediated resistance in Escherichia coli: report from the SENTRY Antimicrobial Surveillance Program (North America, 2004) Int J Antimicrob Agents. 2006;28(6):578–581. doi: 10.1016/j.ijantimicag.2006.07.025. [DOI] [PubMed] [Google Scholar]
- 7.Giamarellou H, Antoniadou A, Kanellakopoulou K. Acinetobacter baumannii: a universal threat to public health? Int J Antimicrob Agents. 2008;32(2):106–119. doi: 10.1016/j.ijantimicag.2008.02.013. [DOI] [PubMed] [Google Scholar]
- 8.Bennett PM, Chopra I. Molecular basis of beta-lactamase induction in bacteria. Antimicrob Agents Chemother. 1993;37(2):153–158. doi: 10.1128/aac.37.2.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dietz H, Pfeifle D, Wiedemann B. The signal molecule for beta-lactamase induction in Enterobacter cloacae is the anhydromuramyl-pentapeptide. Antimicrob Agents Chemother. 1997;41(10):2113–2120. doi: 10.1128/aac.41.10.2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schmidtke AJ, Hanson ND. Model system to evaluate the effect of ampD mutations on AmpC-mediated beta-lactam resistance. Antimicrob Agents Chemother. 2006;50(6):2030–2037. doi: 10.1128/AAC.01458-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peter K, Korfmann G, Wiedemann B. Impact of the ampD gene and its product on beta-lactamase production in Enterobacter cloacae. Rev Infect Dis. 1988;10(4):800–805. doi: 10.1093/clinids/10.4.800. [DOI] [PubMed] [Google Scholar]
- 12.Sanders CC, Bradford PA, Ehrhardt AF, et al. Penicillin-binding proteins and induction of AmpC beta-lactamase. Antimicrob Agents Chemother. 1997;41(9):2013–2015. doi: 10.1128/aac.41.9.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mushtaq S, Ge Y, Livermore DM. Doripenem versus Pseudomonas aeruginosa in vitro: activity against characterized isolates, mutants, and transconjugants and resistance selection potential. Antimicrob Agents Chemother. 2004;48(8):3086–3092. doi: 10.1128/AAC.48.8.3086-3092.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moya B, Juan C, Alberti S, et al. Benefit of having multiple ampD genes for acquiring beta-lactam resistance without losing fitness and virulence in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2008;52(10):3694–3700. doi: 10.1128/AAC.00172-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Strateva T, Yordanov D. Pseudomonas aeruginosa—a phenomenon of bacterial resistance. J Med Microbiol. 2009;58(9):1133–1148. doi: 10.1099/jmm.0.009142-0. [DOI] [PubMed] [Google Scholar]
- 16.Jones RN, Kirby JT, Rhomberg PR. Comparative activity of meropenem in US medical centers (2007): initiating the 2nd decade of MYSTIC program surveillance. Diagn Microbiol Infect Dis. 2008;61(2):203–213. doi: 10.1016/j.diagmicrobio.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 17.Rodriguez-Martinez J, Poirel L, Nordmann P. Molecular epidemiology and mechanisms of carbapenem resistance in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2009;53(11):4783–4788. doi: 10.1128/AAC.00574-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hocquet D, Nordmann P, El Garch F, et al. Involvement of the MexXY-OprM efflux system in emergence of cefepime resistance in clinical strains of Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2006;50(8):1347–1351. doi: 10.1128/AAC.50.4.1347-1351.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Livermore DM, Brown DF. Detection of beta-lactamase-mediated resistance. J Antimicrob Chemother. 2001;48(suppl 1):59–64. doi: 10.1093/jac/48.suppl_1.59. [DOI] [PubMed] [Google Scholar]
- 20.Doi Y, Paterson DL. Detection of plasmid-mediated class C beta-lactamases. Int J Infect Dis. 2007;11(3):191–197. doi: 10.1016/j.ijid.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 21.Jones RN, Sader HS, Fritsche TR, Pottumarthy S. Comparisons of parenteral broad-spectrum cephalosporins tested against bacterial isolates from pediatric patients: report from the SENTRY Antimicrobial Surveillance Program (1998–2004) Diagn Microbiol Infect Dis. 2007;57(1):109–116. doi: 10.1016/j.diagmicrobio.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 22.Chow JW, Fine MJ, Shlaes DM, et al. Enterobacter bacteremia: clinical features and emergence of antibiotic resistance during therapy. Ann Intern Med. 1991;115(8):585–590. doi: 10.7326/0003-4819-115-8-585. [DOI] [PubMed] [Google Scholar]
- 24.Kaye KS, Cosgrove S, Harris A, et al. Risk factors for emergence of resistance to broad-spectrum cephalosporins among Enterobacter spp. Antimicrob Agents Chemother. 2001;45(9):2628–2630. doi: 10.1128/AAC.45.9.2628-2630.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi S, Lee JE, Park SJ, et al. Emergence of antibiotic resistance during therapy for infections caused by Enterobacteriaceae producing AmpC beta-lactamase: implications for antibiotic use. Antimicrob Agents Chemother. 2008;52(3):995–1000. doi: 10.1128/AAC.01083-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fish DN, Piscitelli SC, Danziger LH. Development of resistance during antimicrobial therapy: a review of antibiotic classes and patient characteristics in 173 studies. Pharmacotherapy. 1995;15(3):279–291. [PubMed] [Google Scholar]
- 27.Carmeli Y, Troillet N, Eliopoulos GM, Samore MH. Emergence of antibiotic-resistant Pseudomonas aeruginosa: comparison of risks associated with different antipseudomonal agents. Antimicrob Agents Chemother. 1999;43(6):1379–1382. doi: 10.1128/aac.43.6.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Georges B, Conil J, Dubouix A, et al. Risk of emergence of Pseudomonas aeruginosa resistance to beta-lactam antibiotics in intensive care units. Crit Care Med. 2006;34(6):1636–1641. doi: 10.1097/01.CCM.0000215517.51187.CA. [DOI] [PubMed] [Google Scholar]
- 29.Ambrose PG, Bhavnani SM, Rubino CM, et al. Pharmacokinetics-pharmacodynamics of antimicrobial therapy: it's not just for mice anymore. Clin Infect Dis. 2007;44(1):79–86. doi: 10.1086/510079. [DOI] [PubMed] [Google Scholar]
- 30.Tam VH, Ledesma KR, Vo G, et al. Pharmacodynamic modeling of aminoglycosides against Pseudomonas aeruginosa and Acinetobacter baumannii: identifying dosing regimens to suppress resistance development. Antimicrob Agents Chemother. 2008;52(11):3987–3993. doi: 10.1128/AAC.01468-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Drusano GL, Louie A, Deziel M, Gumbo T. The crisis of resistance: identifying drug exposures to suppress amplification of resistant mutant subpopulations. Clin Infect Dis. 2006;42(4):525–532. doi: 10.1086/499046. [DOI] [PubMed] [Google Scholar]
- 32.Filho LS, Eagye KJ, Kuti JL, Nicolau DP. Addressing resistance evolution in Pseudomonas aeruginosa using pharmacodynamic modelling: application to meropenem dosage and combination therapy. Clin Microbiol Infect. 2007;13(6):579–585. doi: 10.1111/j.1469-0691.2007.01693.x. [DOI] [PubMed] [Google Scholar]
- 33.Chan WC, Li RC, Ling JM, et al. Markedly different rates and resistance profiles exhibited by seven commonly used and newer beta-lactams on the selection of resistant variants of Enterobacter cloacae. J Antimicrob Chemother. 1999;43(1):55–60. doi: 10.1093/jac/43.1.55. [DOI] [PubMed] [Google Scholar]
- 34.Bradley JS, Sauberan JB, Ambrose PG, et al. Meropenem pharmacokinetics, pharmacodynamics, and Monte Carlo simulation in the neonate. Pediatr Infect Dis J. 2008;27(9):794–799. doi: 10.1097/INF.0b013e318170f8d2. [DOI] [PubMed] [Google Scholar]
- 35.Courter JD, Kuti JL, Girotto JE, Nicolau DP. Optimizing bactericidal exposure for beta-lactams using prolonged and continuous infusions in the pediatric population. Pediatr Blood Cancer. 2009;53(3):379–385. doi: 10.1002/pbc.22051. [DOI] [PubMed] [Google Scholar]