

Abstract
The need for critical, well-designed comprehensive clinical pharmacology research in pediatrics that encompasses the age continuum, from the most premature infant through adolescence, may be more important today than ever. New drug regimens often require greater adherence to specific dose guidelines to maximize efficacy and minimize toxic potential. The climate that allowed the propagation of the “therapeutic orphan” concept is now mostly of historical perspective. Nevertheless, the negative impact of this concept continues to linger due to continued propagation of many, now outdated myths surrounding the effective study of optimal drug dosing in pediatrics. Advances in clinical medicine combined with the advances in study design, sampling, and analysis has dramatically improved the paradigm for clinical pharmacology research in infants and children. Capitalizing upon and thoughtfully using these many advances while dispelling these myths will result in greater research focused on optimal drug therapy in pediatric practice.
Keywords: clinical research, drug dosage calculations, pediatrics, pharmacodynamics, pharmacokinetics
INTRODUCTION
For decades the pediatric patient has been referred to as the “therapeutic orphan,” a phrase coined by the late Harry Shirkey. Dr Shirkey, a practicing pharmacist who subsequently trained as a pediatrician, dedicated his illustrious career to focusing on optimal drug therapy in children and highlighting the vast discrepancies that existed for drug research in adults vs children. The first edition (1964) of Dr Shirkey's textbook entitled Pediatric Therapy was the first of its kind to have a focus on drug therapy in infants and children. Clinical pharmacy, clinical pharmacology, and most importantly the children we care for have all benefited tremendously from the political connotations attached to this simple catchy phrase, therapeutic orphan, but it is time to recognize that the science and practice of pediatric clinical pharmacy and clinical pharmacology, continuing advances in pharmaceutical research and regulatory science, and the US Food and Drug Administration (FDA) have all worked very hard to address the issues underlying the genesis of the therapeutic orphan dilemma. Unfortunately, the decades of progress in defining age- and disease-based drug dosing across the age continuum, i.e., that is, the prematurely born infant through adolescence, is being threatened by the disbeliefs, unsubstantiated biases, and myths surrounding the performance of comprehensive clinical pharmacology trials in infants and children. Critical well-designed and powered studies remain absolutely necessary to determining the optimal drug dose regimen across the age continuum in pediatrics. We are the skilled professionals to incorporate contemporary methods to obtaining needed data and dispelling the old biases that have been used to exclude the pediatric patient from clinical pharmacology trials.
TO STUDY OR NOT TO STUDY SHOULD NO LONGER BE THE QUESTION
Myth: The phrase “Therapeutic Orphan” accurately describes the state of pediatric pharmacotherapeutics in the 21st century.
Myth: Severe limitations exist in pediatrics in the number of biologic fluid samples that can be obtained to conduct critical clinical pharmacology research.
Multiple reasons continue to be posed in support of excluding varying age groups from participating in premarket clinical pharmacology drug trials.1 Many times the exclusion of pediatric subjects from new drug research is premeditated, aggressive, and strong. Common reasons posed continue to include innovator concerns for return on investment (ROI) based on reduced consumption and small market size, the fact that most “blockbuster drugs” target adult indications, and a public that at many times is misinformed about the necessity for drug research in children. Furthermore, less substantiated concerns about perceived ethical hurtles and the myths suggesting children are at far greater risk for adverse drug effects and mishaps than their adult counterparts unfortunately remain. These issues are effectively nonissues in contemporary clinical pharmacology research and practice. However, members of our own broad community continue to propagate these myths to rationalize the lack of critical study much to the disservice of our profession.
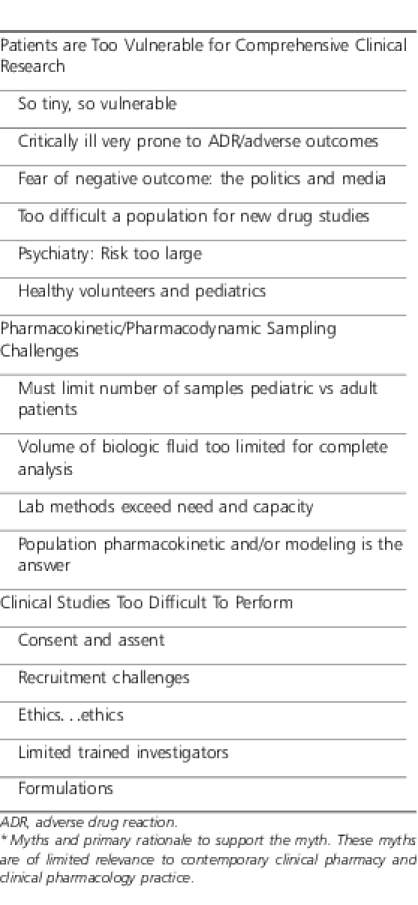
Advances in science, technology, and clinical pharmacy and clinical pharmacology practice have reversed and resolved many of the concerns outlined previously. These issues are highlighted in Table 1. In the 21st century, marketing issues and ROI are continuing to blur in favor of well-designed and controlled pediatric drug trials. For example, in the past a blockbuster drug like atorvastatin (e.g., Lipitor, Pfizer, Dublin, Ireland) would have limited use in pediatrics (e.g., familial hypercholesterolemia) and, thus, limited innovator interest in defining optimal dosing and safety in older children and adolescence leaving pediatric practitioners to estimate an optimal dose based on multiple characteristics. However, due to the scourge of childhood obesity (pre metabolic syndrome),2,3 the use of this drug in children has been increasing with innovator ROI. The performance of high-quality controlled clinical trials is really no more difficult in pediatrics than in adults. With certain exceptions a child's physiology is superior to an adult's just based on the lack of underlying disease(s) and long-standing pathologic insults. A child's major organ reserve or responsiveness to insult may be greater than that of an adult, limiting the magnitude of sequelae from an adverse study event. To underscore the negative impact of these myths is the continued, misplaced concerns regarding the number of biologic fluid samples and sample volume limitations inherent in a study of premature and newborn infants. These technical challenges are real but largely overcome by advances in quantitative pharmacology using highly sensitive and accurate analytic methodologies (e.g., HPLC-MS-MS) that accommodate very small sample volumes from highly instrumented infants. The bedside skill set involved in obtaining and processing these very small volumes from clinically used indwelling devices without compromising sterility circulatory access, or inducing any patient harm, has been available for decades.
Table 1.
Myths Obstructing Age- and Disease-Based Drug Research in Pediatrics*
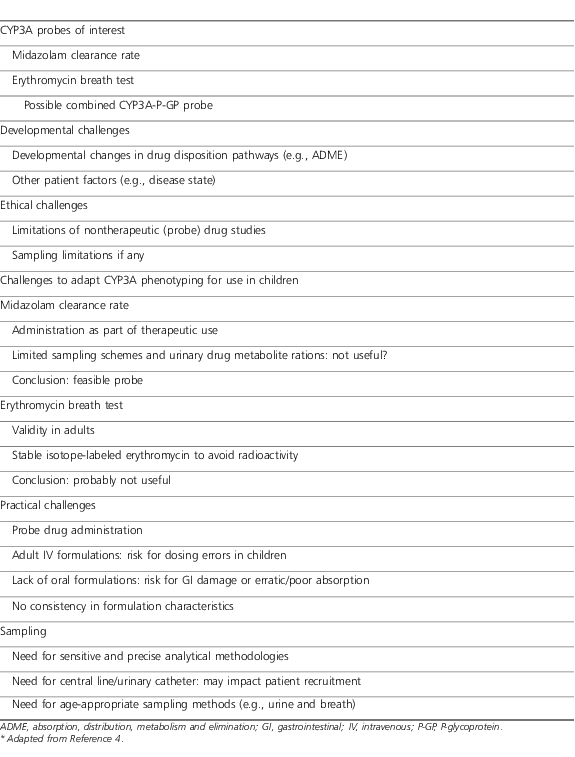
The pharmacogenomic (PG) era has fostered its own set of challenges to universal application in pediatric clinical research and practice. An example of pertinent issues relative to CYP 3A4 phenotyping for pediatric drug trials is outlined in Table 2.4 Like the fundamental challenges inherent to pharmacokinetic (PK) and pharmacodynamic (PD) studies, these challenges are largely addressed with current knowledge, capabilities, and capacity. The questions that remain lie in defining the true applicability of PG across the age spectrum. A recent description of a pharmacist-managed PG service within a cancer treatment facility5 demonstrates the evolving nature of incorporating PG into routine practice. This is an important caveat in pediatric practice as, with the exception of select cancer chemotherapies,6 PG has very limited utility in pediatric practice today.7
Table 2.
Challenges of Pediatric Drug Trials With an Emphasis on CYP3A Phenotyping*
What positions can we and should we take relative to dispelling these past myths and moving forward? First is to recognize that these previously legitimate barriers and challenges to comprehensive clinical research in pediatrics have been largely overcome and are rarely relevant today. Highly skilled pediatric trained investigators and programs of excellence exist for the performance of the most intricate clinical research in the most fragile of pediatric patients. There are no longer legitimate excuses to negating the performance of knowledge-targeted ontogenic research trials. Our posture should be there are very few pediatric patients across the entire age spectrum, including the most premature infant or critically ill child, who should be excluded from a well-designed, controlled clinical pharmacology trial when directed by trained pediatric researchers and practitioners. Today we should be asking ourselves why a specific pediatric patient is not eligible for enrollment into a clinical trial rather than why he or she should be excluded. Only with such a posture and perspective can the adopted therapeutic orphan blossom into a fully fledged “citizen”! A caveat: An important issue usually not addressed in dose finding, FDA label determining clinical drug trails in pediatrics is the long-term effects ( i.e., 5, 10 or 30 years of daily and repeated drug administration in the developing child through various stages of adulthood). Does age at the time of initiating therapy matter? This is an important knowledge gap that must be addressed in defining the comprehensive paradigm of clinical drug research across the age continuum.
PHARMACOKINETIC STUDY DESIGN: ONE SIZE DOES NOT FIT ALL
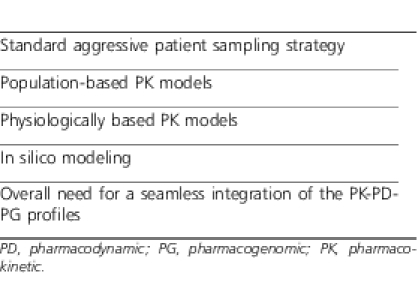
Debate has and continues to persist among clinical pharmacists, clinical pharmacologists, and clinical pharmacometricians8 regarding the optimal methodology to research protocol construct and data analysis for PK studies in children.8–11 Unfortunately, the emotion instilled into the argument for the need for limited sampling strategies12,13 with its inherent limitations as an excuse for the performance of less rigorous pediatric PK trials is unsubstantiated. Such a platform is ill conceived and probably irrelevant today when most patients are effectively instrumented and analytical chemistry methodologies accommodate very small biologic fluid volumes allowing multiple samples in even the smallest of patients. I do not desire to trivialize the complexity of sample collection, sterile technique, logistics, and expense but to simply underscore the need to address real limitations and move on from previous obstacles that are now largely solved. Numerous approaches to patient sampling to derive the most accurate age-appropriate PK data exist, some of which are outlined in Table 3. It would appear that each of these strategies has their advantages and disadvantages and the optimal approach depends on the research question and specific patient population. It would also seem that the best study design may incorporate the fundamental tenets of each of the strategies outlined in Table 3.
Table 3.
Different Methodologies for Pharmacokinetic Study Sampling and Analysis
Traditional PK studies involve multiple repeated sampling usually over a dosing interval providing the greatest confidence of describing the drug's PK profile in a specific patient at the time of study. The applicability of such individual data to the larger patient population has always been a source of debate. Examples of such PK study design is easily found for many drugs in children, particularly where investigators attempt to determine the PK-defined drug dose in a defined age group expecting to extrapolate the data to other children of similar age and disease severity. Many of my own initial research initiatives incorporated such a sample-rich approach to determine drug PK, specifically for drugs such as moxalactam,14 ranitidine,15 piperacillin,16 vancomycin,17 imipenem and cilistatin,18 ceftizoxime,19 meropenem,20 teicoplanin,21 azithromycin,22 cefepime,23 metoclopramide,24 and select antidepressants.25,26 These studies involved single dose but more often combined sample-rich first dose and steady-state PK evaluations to better determine parameter stability and variability with age (e.g., day of life influence in the case of very premature infants) and disease progression. Furthermore such paired first and multidose evaluations confirmed the linear or nonlinear character of the disposition characteristics for specific age groups. Despite the inherent limitations of relatively small sample sizes for specific age groups, the value of these data were important in describing an individual drug's PK profile and the variability inherent in each PK parameter. The ultimate precision of the PK data were “close enough for horse shoes!!” but, most importantly, were bedside applicable with real clinical results.27–33 The value of these initial, sample-rich PK studies served as the foundation for subsequent dose and dose regimen refinement using alternate population-based methodologies allowing for the construction of more creative and possibly more accurate dosing regimens in expanded age groups of varying disease severity.
Lastly, an important area often overlooked in pediatric PK evaluations is the intramuscular (IM) route of drug administration. There is no question that the intravenous (IV) route is preferred both for assurance of adequate bioavailability as well as administration control and patient comfort. Nevertheless in select clinical scenarios including the emergent situation or the need to initiate prompt drug therapy to an infant or child in an adult health care facility unaccustomed to caring for the child, the IM route remains a viable alternative to no dose due to inability to obtain IV access. In the absence of substantial abnormalities in circulation, the IM route of drug administration can ensure sufficient drug dose for initial emergent drug administration. Our data with cefepime demonstrated similar PK disposition and exposure with IV and IM administration.23 When possible the IM disposition should be evaluated with and without the coadministration of a local anesthetic (e.g., lidocaine). The use of lidocaine is of particular importance for drugs known to be particularly painful (from drug irritation and/or volume) after IM injection. The elegant study assessing ceftriaxone IM disposition when administered with and without lidocaine is a model for such an assessment.34
FIXED DOSE COMBINATIONS
Fixed dose drug combinations have a certain appeal to many in the health care field. A single drug preparation is easier to administer negating multiple administrations, may improve patient compliance, save time associated with drug administration, and depending upon market forces may be less expensive than the individual components. Some of these issues have greater relevance to pediatrics than others (e.g., simplicity of administration, compliance), but most combination drugs used in pediatrics reflect specific drug requirements. The most popular include the antibiotics ticarcillin and clavulanic acid, piperacillin and tazobactam, and amoxicillin and clavulanic acid, which incorporate a β-lactamase inhibitor to protect the accompanying antibiotic from destruction by the primary mechanism of pathogen resistance.35,36 Similarly, the antibiotic imipenem is cleared primarily via the kidney but is also destroyed within the kidney by renal dihydropeptidase. To prevent imipenem renal inactivation, cilistatin, a dihydropeptidase inhibitor, is coadministered to reduce the degree of renal imipenem destruction preventing a renal nidus for continued systemic infection and maintain the drug's efficacy for the treatment of renal bacterial infections.18 All of these agents and others37 are manufactured in a defined ratio of the amounts of the 2 components targeting specific ratios of 1 drug to the other within the body. Unfortunately, these combination drugs are usually manufactured at dose ratios defined for adults and not children. The best example of this is our data evaluating the first dose and linked multidose PK characteristics of imipenem and cilistatin in premature infants during the first week of life.18 The cilistatin concentrations determined in these infants far exceeded those observed in adults with similar weight adjusted dosing as well as the concentrations necessary to inhibit renal dihydropeptidase. Nevertheless, the dose ratio remained constant targeting the perceived in vivo optimal ratio for adults. A similar disparate finding in the ratio for a 2-drug combination was observed for ticarcillin and clavulanate32,35 and piperacillin and tazobactam.36 The clinical relevance for these in vivo concentration disparities is limited due to all the drugs' wide safety margins. Nevertheless, this fortunate circumstance of a wide safety margin does not negate the need for age-appropriate dose combinations for these agents. A classic example where a fixed dose ratio can lead to deleterious effects is our experience with the orally administered combination drug amoxicillin-clavulanate. The initial amoxicillin-clavulanate (Augmentin, GlaxoSmithKline, Triangle Park, NC) formulation incorporated a clavulanate dose much higher than needed and was associated with increased intestinal intolerance to the extent the product was reformulated to include a lower yet still just as effective clavulanate dose. Again, we were fortunate that the side effects associated with age-inappropriate dose ratios were limited—it does not negate our responsibility to continue to strive for age-appropriate dose ratios for combination products or simply have the agents available as individual agents for individualized pediatric dosing.
CAPITALIZING ON THE OPPORTUNITY TO DETERMINE DRUG DISTRIBUTION CHARACTERISTICS: THE PHARMACODYNAMIC CORRELATE
Pharmacokinetics as an applied discipline in pediatrics and adults is well established within the research and clinical realms. The PK characteristics and overall disposition profiles for most drugs have been well characterized for many of the routinely used drugs administered to infants and children of all ages. Less developed is our understanding of the PD profiles and how an agent's PK and PD profiles seamlessly and optimally integrate. Numerous factors underlie the difficulty in determining PD data in humans including lack of predictable biomarkers, ability to access the intact cellular mechanism, volume of biologic matrix, and others. Another important reason for this lack of PD sophistication is the common need for invasive techniques to determine drug distribution to the presumed target receptor site, if such anatomic sites are accessible. Considering that 60% to 70% of routine drug therapy in pediatrics involves infectious diseases treated by antimicrobial drugs, a great opportunity exists to assess drug distribution characteristics relative to the target receptor, the pathogen. Classic examples in pediatrics involve assessment of drug penetration into the central nervous system (e.g., meningitis, encephalitis), the lung (pneumonia—cystic fibrosis), and other so-called sanctuary sites for select chemotherapy drugs. Critical assessment of antimicrobial drug penetration into these anatomic sites not only allows assessment of specific drug PK-PD and disease interactions but provides a confirmatory pathway for assessment of drug physiochemical characteristics and body disposition. The best example form our work has been antibiotic concentrations within the cerebrospinal fluid27,38 and sputum39 as a surrogate for lung fluid drug concentrations in patients with cystic fibrosis. When possible these special biologic fluids should be repeatedly sampled over a specific dosing interval, preferably under steady-state conditions and compared with simultaneous assessment of classic systemic disposition characteristics. Such evaluations permit an assessment of not only total (and free) absolute concentration within the target biologic compartment but also the time course for penetration and elimination. Capitalizing upon the opportunity to obtain multiple cerebrospinal fluid concentrations in children undergoing ventriculoperitoneal shunt revisions we were able to assess the disposition characteristics of ceftriaxone central nervous system disposition.27
OPTIMAL DOSE RECOMMENDATIONS FOR CHILDREN: KEEPING THE QUESTIONS ALIVE
Myth: Drugs not FDA approved for use in children should not and cannot be used in children and their therapy costs should not be reimbursed.
As accurate drug disposition data for a large spectrum of drugs comprising many different drug classes including their physicochemical and metabolic characteristics continues to increase, the actual amount and sophistication of dosing data relative to specific age groupings continues to be suboptimal. Pharmaceutical companies who remain the primary innovators of new drugs and providers of grant funding for PK and PD research in children still, with few exceptions, target their drug development plan to complete necessary adult data for FDA approval before completing or, at times, even initiating pediatric trials. This data gap has and continues to pose challenges for the pediatric practitioner who wants to use the newest advanced therapy available for adults in their pediatric patients but is frustrated by the limited dosing data one can rely upon. This dilemma, though real and limits optimal drug dosing in varying age groups, is often used to deny, even today, drug use in children by the age-old saying “this drug cannot be used in children because it is not FDA approved for use in children.” Although the use of this false and inappropriately applied shield is decreasing, we as a profession must cease its use and bury it for one final time. The reality is the drug may not be labeled by the FDA for use in pediatrics but any licensed professional within the scope of his or her practice can use any medication deemed appropriate for a particular patient consistent with the standards of medical care in his or her community. Our insight and training often allows us to define projected optimal doses and most importantly the dynamic monitoring strategy for efficacy and tolerability with the limited data available for the pediatric patient. This concept may be very obvious to the readers of this journal, but it sadly remains a source of denying drug therapy for children and we must do all we can to eliminate this misinterpretation of current regulations.
Myth: FDA-labeled doses reflect the optimal dose and dose regimen—the antidote experience.
Examples of scenarios outlined previously where our training and experience afford us the ability to either define the pediatric drug development plan or question and project optimal pediatric doses are zolpidem for primary sleeping disorders in children,40,41 N-acetylcysteine (NAC) for acetaminophen overdose as noted by Kociancic and Reed42 and Blackford and colleagues (unpublished data), and fomepizole for toxic alcohol intoxication.43 Zolpidem, the most commonly prescribed soporific in the world with many appealing pharmacologic characteristics, appeared to have similar desirable characteristics for children with sleeping disorders. Unfortunately, no specific data were available to support its safe and effective use in children. To address this deficiency we developed a comprehensive drug development plan to define the optimal dose within the target age groupings 8 years and older. To our dismay, our zolpidem development plan was paused by concerns that “children do not have primary sleep disorders” and that sleep medications are uncommonly prescribed. More ridiculous was the notion that parents would only give sleep medications to their children to minimize their need for parenting! So prior to initiating the zolpidem development plan the many diverse drugs used as sleeping medications were reviewed and included in a Clinical Therapeutics Research supplement addressing the state of the art of sleep disorders in children.44 Once this obstacle was overcome, the zolpidem development studies were performed revealing the very interesting findings of failure to reduce latency to sleep using objective, validated criteria in children 6 to 17 years of age and, even more unexpected, differing dose tolerability profile between age groups. Simply proving the centuries old adage: Do the controlled trial!
Another therapeutic area for which limited drug dosing data exist is that for poisoning antidotes. This is very interesting when one considers most accidental poisonings addressed by the national poison control network involve children 5 years or younger. In practice however, antidote dosing information is often limited for individuals of all ages and simply reflective of the very complicated nature of antidote research in humans during an emergent and possibly life-threatening episode. Such studies are understandably very difficult regardless of subject age. As a result, antidote dose selection may not be determined by the performance of robust, critical dose response studies and are further hampered by the potential for animal to human discordance in disposition and receptor characteristics. Sometimes this limitation is simply overcome by empirically increasing the dose and/or dose interval as is the case with oral NAC for the treatment of acute acetaminophen poisoning.42 Understanding the metabolic disposition characteristics of acetaminophen relative to time and subject age linked to the molecular basis for toxicity raises serious questions with current antidote dose and a therapy duration that is long beyond the time when no toxic acetaminophen metabolite exists to bind to glutathione-deficient hepatocytes. These PK-PD facts continue to challenge current oral NAC dose recommendations and are further supported by the much shorter IV regimen.42 A similar situation exists for fomepizole, a highly effective competitive antagonist of alcohol dehydrogenase (ADH) used for the treatment of toxic alcohol ingestions, for example, methanol (windshield washer fluid) and ethylene glycol (automobile antifreeze). The affinity of fomepizole for ADH is more than 8000 times that of the natural substrate ethanol, underscoring the potency of the compound for ADH and its ability to block ADH conversion of methanol and ethylene glycol to their toxic metabolites, formic acid and glycolic acids, respectively. Current dose guidelines appear to incorporate apparent confusion from disparate PK and PD data from animals to humans, which may have also been exacerbated by misapplication of early toxicity profiles with the more toxic 4-methylpyrazole analogues all unrelated to fomepizole. Critical analysis of the available animal and human data43 does not support current dose recommendations and appears to clarify confusion regarding CYP 2E1 metabolism (enzyme induction vs stabilization), clear zero-order PK profile across a dose range, and serious questions regarding the need to lower the dose for 2 days after an initial loading dose to then increase the dose again. This unnecessary dosing strategy in an increasingly downsized medical team approach is a formula for dose errors. Fortunately, fomepizole has a wide safety margin; other than its high cost, lower doses equal lower costs. These experiences underscore the importance of our professions to continue to question the precision of recommended dosing regimens across the age spectrum using all basic and clinical pharmacology data available to better refine the PK-PD and soon to be integrated PK-PD-PG profile for optimal dosing. Furthermore, these experiences also underscore the need to continually, critically evaluate available data for its application to children of all ages and the value of incorporating in vitro, in vivo, animal to human data by applying confirmed age-dependent clinical pharmacology principles.
CONCLUSION
Many old myths surrounding the performance of highly sophisticated, critical, clinical pharmacology research in infants and children encompassing the spectrum of disease severity continues to hamper moving forward such research in a robust manner. The “Therapeutic Orphan” has been adopted, matured, probably graduated college, and is out having fun! Let's move beyond this mental obstacle, embracing the tool box and skill sets our training and experience have provided us to design and undertake effective clinical pharmacology research for drug use in pediatrics. Let's leave the past and aggressively address the challenges before us today and tomorrow. The availability of child-appropriate, palatable drug formulations (e.g., liquid, chewable, wafers) that are chemically stable under the most challenging of storage conditions (e.g., without clean water and refrigeration) can have such a dramatic impact on quality of life and even survival for an enormous number of children worldwide. Improved compliance by not only the child but the linked caregiver through medication education performed right in the pharmacy at the time of dispensing and counseling will also produce measurable results. The next frontier of applied PG, “personalized medicine” in pediatric practice is and will be a major challenge in defining appropriate therapeutic guidelines but most importantly ensuring the application of what PG has to offer into a proper perspective. PG is not the final answer; it is just another tool when combined with other important drug facts. The PK-PD-PG profiles allow us to better define an optimal dose regimen. Unfortunately, the hype for PG far exceeds its present utility for most pharmacotherapeutic interventions in pediatric practice.7 We as the clinical pharmacology professionals need to ensure proper utilization and application of PG just as we had for PK and PD assessments. When we do, the major beneficiaries are the children we have the privilege to serve.
Acknowledgments
The research described in this presentation would not have been possible without the trust, support, bravery, and constant encouragement of so many families, parents, grandparents, and sick children, all selfless, who simply wanted to make things better for the next sick child. I have also had the great fortune to work with so many brilliant colleagues, near and far, from around the world who taught me so much about applied clinical pharmacology and about life, who possess an unwavering dedication to the care of all children, and who have extended to me unconditional friendship with a measure of extreme tolerance for my many fallacies. I have been so privileged to interface with them and work with them and I continue to learn from them. I want to thank the PPAG for bestowing upon me this tremendous honor and to my family who have selflessly sacrificed much in their unwavering support.
Abbreviations
- ADH
alcohol dehydrogenase
- FDA
US Food and Drug Administration
- IM
intramuscular
- IV
intravenous
- NAC
N-acetylcysteine
- PD
pharmacodynamic
- PG
pharmacogenomic
- PK
pharmacokinetic
- ROI
for return on investment
Footnotes
DISCLOSURE The author declares no conflicts or financial interests, equipment, employment, or gifts in any product or service mentioned in the manuscript. The research described in this manuscript was supported by grants provided by the drugs innovator(s), select public grants (e.g., NICHD Pediatric Pharmacology Research Unit network), and institutional support. Specific funding sources for individual agents described are detailed in the original publication(s) cited within the manuscript and listed in references.
REFERENCES
- 1.Koren G, Kearns GL, Reed M, et al. Use of healthy children as volunteers in drug studies: the ethical debate. Clin Pharmacol Ther. 2003;73(3):147–152. doi: 10.1067/mcp.2003.16. [DOI] [PubMed] [Google Scholar]
- 2.Lane JR, Ben-Shachar G. Myocardial infarction in healthy adolescents. Pediatrics. 2007;120(4):e938–e943. doi: 10.1542/peds.2006-3123. [DOI] [PubMed] [Google Scholar]
- 3.Rotteveel J, Felius A, van Weissenbruch MM, et al. Insulin resistance and metabolic syndrome in obese children referred to an obesity center. J Pediatr Endocrinol Metab. 2010;23(9):943–951. doi: 10.1515/jpem.2010.151. [DOI] [PubMed] [Google Scholar]
- 4.deWildt SN, Shinya I, Koren G. Challenges for drug studies in children: CYP3A phenotyping as example. Drug Discov Today. 2009;14(1–2):6–15. doi: 10.1016/j.drudis.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 5.Crews KR, Cross SJ, McCormick JN, et al. Development and implementation of a pharmacist-managed clinical pharmacogenetics service. Am J Health Syst Pharm. 2011;68(2):143–150. doi: 10.2146/ajhp100113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Badquemont L, Alfirevic A, Amstutz U, et al. Practical recommendations for pharmacogenomics-based prescription: 2010 ESF-UB Conference on pharmacogenetics and pharmacogenomics. Pharmacogenomics. 2011;12(1):113–124. doi: 10.2217/pgs.10.147. [DOI] [PubMed] [Google Scholar]
- 7.Reed MD, Bestic M. Pharmacogenomics in 2011: navigating the maze to the bedside. eJournal Am Coll Osteopath Pediatr. 2011;(Jan) [Google Scholar]
- 8.Neely M, Jelliffe R. Practical, individualized dosing: 21st century therapeutics and the clinical pharmacometrician. J Clin Pharmacol. 2010;50(7):842–847. doi: 10.1177/0091270009356572. [DOI] [PubMed] [Google Scholar]
- 9.Anderson BJ, Allegaert K, Holford NHG. Population clinical pharmacology of children: general principles. Eur J Pediatr. 2006;165(11):741–746. doi: 10.1007/s00431-006-0188-y. [DOI] [PubMed] [Google Scholar]
- 10.Anderson BJ, Allegaert K, Holford NHG. Population clinical pharmacology of children: modeling covariate effects. Eur J Pediatr. 2006;165(12):819–829. doi: 10.1007/s00431-006-0189-x. [DOI] [PubMed] [Google Scholar]
- 11.Bouzom F, Walther B. Pharmacokinetic predictions in children by using the physiologically based pharmacokinetic modeling. Fundam Clin Pharmacol. 2008;22(6):579–587. doi: 10.1111/j.1472-8206.2008.00648.x. [DOI] [PubMed] [Google Scholar]
- 12.Panetta JC, Iacono LS, Adamson PC, et al. The importance of pharmacokinetic limited sampling models for childhood cancer drug development. Clin Cancer Res. 2003;9(14):5068–5077. [PubMed] [Google Scholar]
- 13.Drusano GL, Forrest A, Snyder MJ, et al. An evaluation of optimal sampling strategy and adaptive study design. Clin Pharmacol Ther. 1988;44(2):232–238. doi: 10.1038/clpt.1988.142. [DOI] [PubMed] [Google Scholar]
- 14.Reed MD, Aronoff SC, Myers CM, et al. Developmental pharmacokinetics of moxalactam. Antimicrob Agents Chemother. 1983;24(3):383–387. doi: 10.1128/aac.24.3.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blumer JL, Rothstein FC, Kaplan BS, et al. Pharmacokinetic determination of ranitidine pharmacodynamics in pediatric ulcer disease. J Pediatr. 1985;107(2):301–306. doi: 10.1016/s0022-3476(85)80156-6. [DOI] [PubMed] [Google Scholar]
- 16.Reed MD, Myers CM, Yamashita TS, et al. Developmental pharmacology and therapeutics of piperacillin in gram-negative infections. Dev Pharmacol Ther. 1986;9(2):102–114. doi: 10.1159/000457082. [DOI] [PubMed] [Google Scholar]
- 17.Reed MD, Kliegman RM, Weiner JS, et al. The clinical pharmacology of vancomycin in seriously ill preterm infants. Pediatr Res. 1987;22(3):360–363. doi: 10.1203/00006450-198709000-00024. [DOI] [PubMed] [Google Scholar]
- 18.Reed MD, Kliegman RM, Yamashita TS, et al. The clinical pharmacology of imipenem and cilastatin in premature infants during the first week of life. Antimicrob Agents Chemother. 1990;34(6):1172–1177. doi: 10.1128/aac.34.6.1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reed MD, Gooch WM, Minton S, et al. Ceftizoxime disposition in neonates and infants during the first six months of life. DICP Ann Pharmacother. 1991;25(4):344–347. doi: 10.1177/106002809102500401. [DOI] [PubMed] [Google Scholar]
- 20.Blumer JL, Reed MD, Kearns GL, et al. Sequential single-dose pharmacokinetic evaluation of meropenem in hospitalized infants and children. Antimicrob Agents Chemother. 1995;39(8):1721–1725. doi: 10.1128/aac.39.8.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reed MD, Yamashita TS, Myers CM, et al. The pharmacokinetics of teicoplanin in children. J Antimicrob Chemother. 1997;9(6):789–796. doi: 10.1093/jac/39.6.789. [DOI] [PubMed] [Google Scholar]
- 22.Stevens RC, Reed MD, Shenep JL, et al. Pharmacokinetics of azithromycin after single- and multiple-doses in children. Pharmacotherapy. 1997;17(5):874–880. [PubMed] [Google Scholar]
- 23.Reed MD, Yamashita TS, Knupp CK, et al. The pharmacokinetics of intravenously and intramuscularly administered cefepime in infants and children. Antimicrob Agents Chemother. 1997;41(8):1783–1787. doi: 10.1128/aac.41.8.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kearns GL, van den Anker J, Reed MD, et al. Pharmacokinetics of metoclopramide in neonates. J Clin Pharmacol. 1998;38(2):122–128. doi: 10.1002/j.1552-4604.1998.tb04400.x. [DOI] [PubMed] [Google Scholar]
- 25.Findling RL, Reed MD, Myers C, et al. Paroxetine pharmacokinetics in depressed children and adolescents. J Am Acad Child Adolesc Psychiatry. 1999;38(8):952–959. doi: 10.1097/00004583-199908000-00010. [DOI] [PubMed] [Google Scholar]
- 26.Findling RL, Preskorn SH, Marcus RN, et al. Nefazodone pharmacokinetics in depressed children and adolescents. J Am Acad Child Adolesc Psychiatry. 2000;39(8):1008–1016. doi: 10.1097/00004583-200008000-00016. [DOI] [PubMed] [Google Scholar]
- 27.Reed MD, Rekate HL, Aronoff SC, et al. Single dose plasma and cerebrospinal fluid pharmacokinetics of ceftriaxone in infants and children. Clin Pharm. 1983;2(6):558–563. [PubMed] [Google Scholar]
- 28.Aronoff SC, Murdell D, O'Brien CA, et al. Efficacy and safety of ceftriaxone in serious pediatric infections. Antimicrob Agents Chemother. 1983;24(5):663–666. doi: 10.1128/aac.24.5.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aronoff SC, Reed MD, O'Brien CA, et al. Comparison of the efficacy and safety of ceftriaxone to ampicillin/chloramphenicol in the treatment of childhood meningitis. J Antimicrob Chemother. 1984;13(2):143–151. doi: 10.1093/jac/13.2.143. [DOI] [PubMed] [Google Scholar]
- 30.Aronoff SC, Soles PV, Reed MD, et al. Evaluation of latamoxef as initial therapy of bone and joint infections in childhood. Chemotherapy. 1984;30(5):337–344. doi: 10.1159/000238290. [DOI] [PubMed] [Google Scholar]
- 31.Reed MD, Stern RC, O'Brien CA, et al. A randomized double-blind evaluation of ceftazidime dose ranging in hospitalized patients with cystic fibrosis. Antimicrob Agents Chemother. 1987;31(5):698–702. doi: 10.1128/aac.31.5.698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reed MD, Yamashita TS, Blumer JL. Pharmacokinetic-based ticarcillin/clavulanic acid dose recommendations for infants and children. J Clin Pharmacol. 1995;35(7):658–665. doi: 10.1002/j.1552-4604.1995.tb04105.x. [DOI] [PubMed] [Google Scholar]
- 33.Reed MD, O'Brien CA, Aronoff SC, et al. Ceftazidime as initial therapy for suspected bacterial infections in hospitalized pediatric patients. Antimicrob Agents Chemother. 1984;26(3):318–321. doi: 10.1128/aac.26.3.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patel IH, Weinfeld RE, Konikoff J, et al. Pharmacokinetics and tolerance of ceftriaxone in humans after single-dose intramuscular administration in water and lidocaine diluents. Antimicrob Agents Chemother. 1982;21(6):957–962. doi: 10.1128/aac.21.6.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reed MD. Rational prescribing of extended-spectrum penicillin beta-lactamase inhibitor combinations: focus on ticarcillin/clavulanic acid. Ann Pharmacother. 1998;32(1):S17–S21. doi: 10.1177/106002809803200105. [DOI] [PubMed] [Google Scholar]
- 36.Reed MD, Goldfarb J, Yamashita TS, et al. Single-dose pharmacokinetics of piperacillin and tazobactam in infants and children. Antimicrob Agents Chemother. 1994;38(12):2817–2826. doi: 10.1128/aac.38.12.2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reed MD, Stern RC, Bertino JS, Jr, et al. Dosing implications of rapid elimination of trimethoprim-sulfamethoxazole in patients with cystic fibrosis. J Pediatr. 1984;104(2):303–307. doi: 10.1016/s0022-3476(84)81019-7. [DOI] [PubMed] [Google Scholar]
- 38.Blumer JL, Aronoff SC, Myers CM, et al. Pharmacokinetics and cerebrospinal fluid penetration of ceftazidime in children with meningitis. Dev Pharmacol Ther. 1985;8(4):219–231. doi: 10.1159/000457041. [DOI] [PubMed] [Google Scholar]
- 39.Reed MD, Stern RC, Myers CM, et al. Lack of unique ciprofloxacin pharmacokinetic characteristics in patients with cystic fibrosis. J Clin Pharmacol. 1988;28(8):691–699. doi: 10.1002/j.1552-4604.1988.tb03202.x. [DOI] [PubMed] [Google Scholar]
- 40.Blumer J, Reed MD, Steinberg F, et al. Potential pharmacokinetic basis for zolpidem dosing in children with sleep difficulties. Clin Pharmacol Ther. 2008;83(4):551–558. doi: 10.1038/sj.clpt.6100380. [DOI] [PubMed] [Google Scholar]
- 41.Blumer JL, Findling RL, Shih WJ, et al. Controlled clinical trial of zolpidem in the treatment of insomnia associated with attention deficit hyperactivity disorder in children 6 to 17 years. Pediatrics. 2009;123(5):e770–e776. doi: 10.1542/peds.2008-2945. [DOI] [PubMed] [Google Scholar]
- 42.Kociancic T, Reed MD. Acetaminophen intoxication and length of treatment: how long is long enough? Pharmacotherapy. 2003;23(8):1052–1059. doi: 10.1592/phco.23.8.1052.32884. [DOI] [PubMed] [Google Scholar]
- 43.Bestic M, Blackford M, Reed M. Fomepizole: a critical assessment of current dose recommendations. J Clin Pharmacol. 2009;49(2):130–137. doi: 10.1177/0091270008327142. [DOI] [PubMed] [Google Scholar]
- 44.Reed MD, Findling RL. Overview of current management of sleep disorders in children. Curr Ther Res. 2002;63(suppl 2):B18–B37. [Google Scholar]