

Abstract
Obesity and related disorders are thought to have their roots in metabolic “thriftiness” that evolved to combat periodic starvation. The association of low birth weight with obesity in later life caused a shift in the concept from thrifty gene to thrifty phenotype or anticipatory fetal programming. The assumption of thriftiness is implicit in obesity research. We examine here, with the help of a mathematical model, the conditions for evolution of thrifty genes or fetal programming for thriftiness. The model suggests that a thrifty gene cannot exist in a stable polymorphic state in a population. The conditions for evolution of thrifty fetal programming are restricted if the correlation between intrauterine and lifetime conditions is poor. Such a correlation is not observed in natural courses of famine. If there is fetal programming for thriftiness, it could have evolved in anticipation of social factors affecting nutrition that can result in a positive correlation.
1. Introduction
Obesity and related disorders are on the rise throughout the globe at an alarming rate, the causes of which are not yet completely understood. On the one hand, individuals differ substantially in their tendency to accumulate fat, and there are strong familial tendencies suggesting that genetic predisposition may play a major role. On the other hand, it is obvious that no alleles can increase in frequency in a population with a rate matching the rate of spread of the obesity epidemic, indicating that genetics alone does not explain the rise in obesity. The most common perception, therefore, is that of a interplay between genes and environment. According to the current paradigm, a gene or a set of genes predisposing to obesity presumably evolved owing to a selective advantage in ancestral “feast and famine” environment and remained in polymorphic state in the population which is turning pathological in the modern urban environment selectively affecting individuals with the gene(s).
This line of thinking was first stated explicitly by Neel [1] who suggested that a “thrifty” gene helped storage of fat under conditions of better availability of nutrients and allowed reutilization under starvation. The thrifty gene was under positive selection pressure in ancestral life when seasonal and climatic conditions resulted into fluctuating food availability. This concept soon became almost an axiom, although no such “thrifty” gene or set of genes have been convincingly demonstrated. Later, the observation that individuals born small for gestational age had a greater probability to become obese and type 2 diabetic in later life led to the concept of fetal programming [2–4]. This hypothesis states that if a fetus faces inadequate nutrition in intrauterine life, the body is programmed to be “thrifty” as an adaptation. There are two possible components of this adaptation. One relates to an immediate gain in terms of survival during fetal and early infant life. The other is a predictive adaptive response in anticipation of starvation in later life [5]. This distinction is important in understanding the evolution of fetal programming.
Both the concepts of thrifty gene and thrifty phenotype by fetal programming have recently faced serious criticism on several grounds [6–11]. The main objections of the critics are as follows.
The original suggestion of Neel [1] was that in individuals prone to obesity and type 2 diabetes (T2D), a “quick insulin trigger” ensures rapid glucose uptake which is then converted into fat. In the early 1960s, it was well known that the levels of insulin are higher in prediabetics, and Neel's original proposal looked sound. However, insulin resistance was discovered soon, and it became clear that the “quick insulin trigger” is unlikely to work as believed by Neel [1]. Fat cell-specific insulin receptor knock-outs fail to accumulate fat [12], raising the possibility that insulin resistance actually arrests lipogenesis. It is therefore not logical to call the diabetes-prone genotypes as “thrifty.”
If thriftiness is due to lower metabolic rate conserving more energy which gets stored in fat tissue, a lower metabolic rate should be observed in people having a predisposition to obesity. Consistent correlations between birth weight and metabolic rate are not found in empirical data to demonstrate the supposed thriftiness of low birth weight individuals [13, 14]. Studies using doubly labeled water have not consistently found lower metabolic rates in people with sedentary lifestyle in the modern urban societies [15, 16].
Impaired fat oxidation rather than lower metabolic rate appears to be the main contributor to obesity of developmentally stunted individuals [17, 18]. If inability to reutilize stored fat is the major cause of obesity, the stored fat is unlikely to help under “famine” conditions, and this is a major blow to the thriftiness hypotheses. The doubly labeled water studies also suggest that obesity is more a product of hyperphagia than metabolic thriftiness [15, 16]. The known genetic mechanisms of obesity also work by interfering with appetite control rather than through metabolic thriftiness [19].
Evidence that obese people have a significantly better chance of surviving famines is debatable [8]. Therefore, it is doubtful whether obesity actually offered sufficient advantage during famines to get selected in spite of the fact that obesity is associated with reduced fecundity [20].
Obesity and insulin resistance is associated with a number of changes in the different body systems and their functions as diverse as ovulation, spermatogenesis, innate immunity, wound healing, memory, and cognitive brain functions [10]. The thriftiness hypotheses focus on energy homeostasis alone and offer no explanation as to why these diverse changes are associated with it.
Realizing the limitations, inadequacies, and flaws of the thrifty gene and fetal programming hypotheses, a number of alternatives have been suggested.
(a) As alternatives to the thrifty gene hypothesis, (i) Speakman [8] postulated genetic drift rather than selection for obesity-related genes. An upper limit on obesity was set in hunter-gatherer life which was effectively lifted when humans became free of predation. Subsequently, the obesity-related genes started spreading by genetic drift. (ii) Corbett et al. [21] argued that today's obese, diabetic, and PCOS-prone genotype was the ancestral one that had better fertility in famine conditions. In the modern era of food security since 1800 AD, an insulin-sensitive genotype that has better fertility under conditions of food abundance started spreading. (iii) Moalem et al. [22] hypothesized that high plasma glucose lowers the freezing point of blood which prevents formation of ice crystals in cells through supercooling, and this has been suggested as an adaptation to the ice age.
(b) Alternative explanations for fetal programming have also been offered. (i) Hattersley and Tooke [23] argued that the association between low birth weight and insulin resistance arises out of a reverse causation, that is, babies with insulin resistance genotype are more likely to survive fetal undernourishment. (ii) Wells [11, 24] argued that there is maternal advantage in fetal programming in the form of optimizing maternal inputs per fetus or bet hedging, that is, distribution of risk among offspring.
(c) There are hypotheses that account for genetic as well as intrauterine effects. (i) Watve and Yajnik [10] argued that insulin resistance is a socioecological adaptation that mediates two phenotypic transitions, namely, (i) transition in reproductive strategy from “r” (large number of offspring with little investment in each) to “K” (smaller number of offspring with more investment in each) and (ii) transition from “soldier to diplomat,” that is, from a physically aggressive behavior to a socially manipulative one. According to this hypothesis, insulin resistance changes the differential budget allocation to tissues. Since all tissues are not dependent on insulin for nutrient uptake, when insulin resistance develops, the uptake of insulin-dependent tissues reduces, and more nutrients become available for insulin independent tissues. Muscles are insulin dependent, and brain is insulin independent, and, therefore, insulin resistance results in disinvestment from muscles and increased investment in brain. Insulin resistance is likely to have evolved as a switch in reproductive and sustenance strategies rather than an adaptation to feast and famine. (ii) Extending this logic further, Rashidi et al. [25] explained why pancreatic beta cells and those of females in particular are more susceptible to oxidative damage. Under stress conditions, the release of stress hormones produces insulin resistance and, owing to reactive oxygen species (ROS) preventing beta cells from secreting insulin at the level required to maintain homeostasis, diverts glucose to insulin-independent tissues, such as the brain and the fetus. They suggest that pancreatic beta cells lost part of their antioxidant defense in association with brain evolution, and lost even more in females when placental mammals evolved.
The emergence of alternative hypotheses having different implications for the genetics of obesity has made it even more critical that the traditional hypothesis is examined analytically. The concept of thriftiness has been mostly discussed descriptively and qualitatively and almost never subject to quantitative methods commonly used by evolutionary geneticists to test or support any argument. We use a simple mathematical model here to examine the conditions under which thrifty gene or fetal programming is likely to gain a selective advantage and evaluate how likely it is for human ancestors to have evolved constitutive or programmable thriftiness. There are no empirical estimates of the actual fitness contributions of the hypothetical genes or phenotypes, and, therefore, a quantitatively predictive model cannot be attempted at this stage. The limited objective of our model is to conceptually examine whether a thrifty gene or thrifty programming can evolve in principle, whether stable polymorphism in this character is possible, if yes, what are the necessary conditions for its evolutionary stability, and whether the thriftiness hypotheses adequately explain the current obesity epidemic.
2. The Model
To model the possible evolution of thriftiness, we consider 3 hypothetical genotypes, namely, a nonthrifty wild type having no mechanism for thriftiness (n), a thrifty genotype (tg) which is genetically programmed for thriftiness and a genotype with a capacity for fetal programming for thriftiness (tp). Taking a year as a natural time unit of seasonality, we assume a simple dichotomy of years with adequate food supply (feast) and those with inadequate food supply (famine). Famines are assumed to occur randomly with a probability pf.
Fitness of an individual with nonthrifty genotype in feast conditions (nf1) is assumed to be greater than that of individual with thrifty genotype (tf1) because there is cost associated with thriftiness. When there is a feast, nonthrifty individuals will do better as they do not pay the cost for being thrifty (nf1 > tf1). The cost of thriftiness is justifiable based on the reproductive effects of obesity. We assume that thrifty individuals are fast to become obese in feast conditions, and there are multiple mechanisms by which obesity causes reduction in fecundity. On the one hand, obesity and insulin resistance are associated with ovulation disorders and are major risk factors for polycystic ovary syndrome (PCOS) [26]. On the other, reduced fecundity is also seen in obese women with regular cycles [20, 27]. Moreover, obesity also affects spermatogenesis in men [29], and the effects of obesity on male and female fertility become additive if a couple is obese [30].
Fitness of an individual with nonthrifty genotype in famine conditions (nf0) is assumed to be less than that of individuals with thrifty genotype (tf0) (nf0 < tf0).
The lifetime fitness of an individual is given as follows.
For individuals with nonthrifty genotype,
| (1) |
For individuals with thrifty genotype,
| (2) |
For individuals with thrifty phenotype or the capacity for irreversible fetal programming, assuming no correlation between birth and lifetime conditions, the total fitness is calculated as the sum of all years, with the assumption that in the birth year, the phenotype is best suited for given conditions. For the rest of the lifetime (S), the fitness fluctuates according to randomly fluctuating environmental conditions. Therefore,
| (3) |
The first term in (3) denotes fitness contribution of birth year, and the second term sums the fitness contribution of the rest of the lifespan.
Solution: —
Considering the nonthrifty and thrifty genotypes alone, it can be seen that, at large pf, thrifty gene has an advantage over nonthrifty gene, and, at small pf, nonthrifty gene cannot be invaded by the thrifty one. The transition is at Ln = Ltg and this happens when
(4) It is important to note that the areas of advantage are decided not by the absolute fitness values but only by the ratios of the differences in fitness in feast and famine conditions.
Equation (4) implies that the evolution of thrifty gene needs a high frequency of famine. Famines with significant mortality occur with a frequency of once in 100–150 years [8]. If surviving famines is the major selective force for thrifty gene, with pf = 0.01, the advantage of thrifty over nonthrifty phenotype in famine conditions should be more than 100-times the relative loss suffered by thrifty gene in feast conditions. If the obesity-induced reduction in fecundity is sizable, the advantage of thriftiness in famines should be exceedingly large for thrifty genes to evolve. Such a large advantage should be highly evident and easily measurable, but obese people have not been shown to survive famines significantly better than lean individuals [8].
Considering competition between nonthrifty and fetal programming genotypes, it can be seen that at higher pf and lower S, Ltp > Ln. The transition line resides at Ln = Ltp.
For a given pf, this condition is met at
| (5) |
Figure 1(a) shows the transition line demarcating the areas of selective advantage of Ln and Ltp when only these two genotypes compete. At very high pf, however, an all-time thrifty genotype might have an advantage over fetal programming. Therefore, we need to examine the areas of advantage of Ltp and Ltg.
Figure 1.
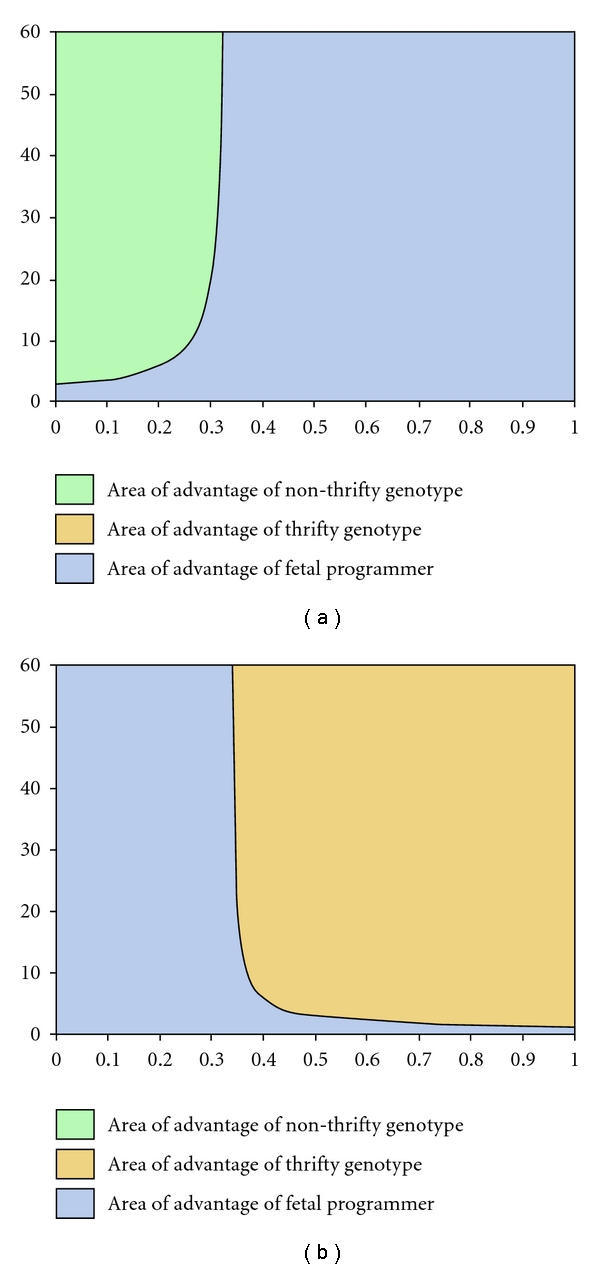
Parameter areas of advantage: (a) when only nonthrifty genotype and fetal programmer compete, (b) when only thrifty genotype and fetal programmer compete. For this and all other figures, (tf1 − nf1)/(nf0 − tf0) is maintained constant at 1.33.
Taking a similar approach as above, Ltg = Ltp when
| (6) |
A graph of pf versus S (Figure 1(b)) shows that, at greater probability of famine and greater longevity, thrifty gene(s) can evolve. The two results together (Figure 2) imply that at low probabilities of famine, a nonthrifty gene has a net advantage, and at high pf, thrifty gene would evolve leaving a very narrow area of advantage for fetal programming. The area is narrower for long-lived species whereas for short-lived ones, the birth year advantage is large enough as compared to lifetime, and, therefore, fetal programming has a much larger width of advantage.
Figure 2.

Parameter areas of advantage when nonthrifty genotype, thrifty genotype, and fetal programmer compete simultaneously. Fetal programming can evolve for species with short lifespan. If the life span is longer, fetal programming is unlikely to offer selective advantage over thrifty or nonthrifty genotypes except for a specific and very narrow range of pf.
The results so far are based on the assumption that seasonal or annual climatic variations are random with little or no predictability. We now consider the effects of such predictability. If there exists a correlation r between uterine conditions and lifetime conditions, then it follows that the lifetime probability of famine would be higher if birth was in a famine year. We can, therefore, write the expected probability of facing a famine if the birth year was a famine year as
| (7) |
The probability of facing a famine if the birth year was a feast year as
| (8) |
accordingly,
| (9) |
Now, the new transition between areas of advantage of non thrifty and fetal programming (when Ln = Ltp) is obtained at
| (10) |
Similarly, the transition between areas of advantage of thrifty and fetal programming is obtained at Ltg = Ltp. This condition is satisfied when
| (11) |
Figure 3 shows that as the birth-time and lifetime correlation increases, the area of advantage of fetal programmer widens. However, in long-lived species, since the advantage to fetal programmer in the absence of correlation is very small, even a small negative correlation drives the fetal programmer to extinction (Figure 3(c)). It is most important for the evolution of fetal programming that a positive correlation between birth-time and lifetime conditions exists. The correlation need not be very high. It can be seen that at r = 0.3, fetal programmer has an advantage over a wide range of pf.
Figure 3.
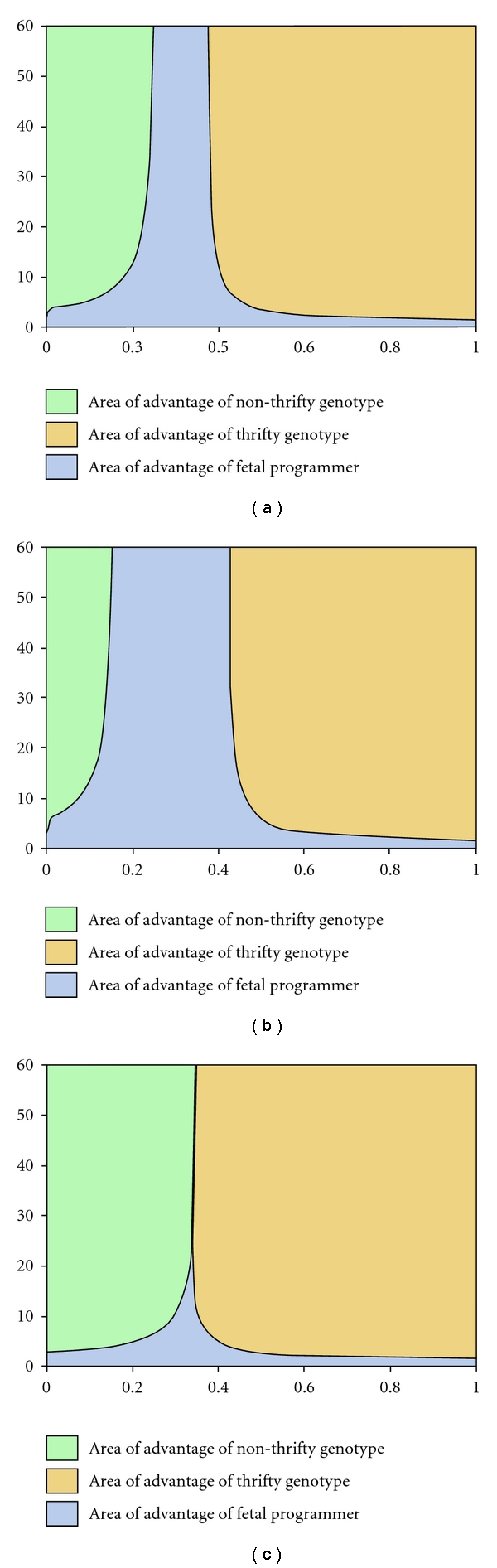
Parameter areas of advantage of the 3 genotypes when there is a correlation between birth-time and lifetime conditions: (a) r = 0.1, (b) r = 0.2, and (c) r = −0.05. With a small positive correlation, the advantage of fetal programmer increases substantially. However, even very weak negative correlation can drive fetal programmer to extinction when life expectancy is high. Selection for fetal programming, therefore, must be driven by factors that produce significant positive birth-time and lifetime correlations.
Climatic fluctuations from year to year are a complex phenomenon, and since prediction of important climatic features, such as rainfall, has important implications, there have been serious attempts to detect temporal patterns. However, temporal patterns are of little help in weather prediction since there are no consistent time lapse correlations in rainfall or other parameters. Since India has the largest population of diabetics and monsoon is the most important determinant of food availability in this region, it would be enlightening to see the patterns in the Indian monsoon. Based on the public-domain data on Indian monsoon by the Indian Institute of Tropical Meteorology (ftp://www.tropmet.res.in/pub/data/rain/iitm-subdivrf.txt), we investigated whether birth year and subsequent year's rainfall has any positive correlation in the short or long run for any of the 30 monsoon subdivisions in India. Table 1 shows that rainfall in a given year is not correlated with that of the subsequent year, subsequent 10 years, or 40 years cumulative. Over the 30 monsoon subdivisions, only 6 are statistically significant using individual α level of 0.05, out of which 4 are negative, contrary to the expectation. Using Bonferroni correction for significance level, applicable when a large number of tests are being done together, none of the correlations remain significant. As there is no detectable positive correlation between birth year and lifetime rainfall conditions, similarly no such correlational patterns in any other climatic variables are reported, fetal programming is unlikely to have evolved in anticipation of drought or famine.
Table 1.
Correlations of annual rainfall to that of the subsequent year, short-term (10 years), and long-term (40 years) cumulative: ∗ indicate significance of individual correlations at α = 0.05 level. Since a large number of statistical tests are being performed, a Bonferroni correction to the significance level is applicable. After the Bonferroni correction, none of the correlations are significant.
| S. No. | Subdivision | 1 year | 10 years cumulative | 40 years cumulative |
|---|---|---|---|---|
| 1 | Assam Meghalaya | 0.047 | 0.042 | −0.029 |
| 2 | Nagaland Manipur Mizoram and Tripura | 0.006 | 0.010 | −0.078 |
| 3 | Sub-Himalayan West Bengal | −0.061 | −0.103 | −0.023 |
| 4 | Gangetic West Bengal | −0.009 | 0.096 | −0.265* |
| 5 | Orissa | −0.111 | 0.135 | −0.096 |
| 6 | Jharkhand | −0.054 | −0.042 | −0.012 |
| 7 | Bihar | 0.070 | −0.158 | −0.048 |
| 8 | East Uttar Pradesh | 0.096 | −0.106 | −0.091 |
| 9 | West Uttar Pradesh Plains | −0.036 | 0.010 | −0.066 |
| 10 | Haryana | −0.010 | 0.050 | 0.041 |
| 11 | Punjab | −0.050 | 0.099 | 0.081 |
| 12 | Rajasthan | 0.048 | −0.200* | −0.094 |
| 13 | East Rajasthan | 0.047 | −0.034 | −0.017 |
| 14 | West Madhya Pradesh | 0.052 | 0.114 | 0.010 |
| 15 | East Madhya Pradesh | −0.076 | 0.040 | −0.034 |
| 16 | Gujarat | −0.007 | −0.121 | −0.110 |
| 17 | Saurashtra and Kutch | −0.037 | −0.118 | −0.152 |
| 18 | Konkan and Goa | 0.121 | 0.154 | 0.080 |
| 19 | Madhya Maharashtra | 0.227* | 0.010 | −0.163 |
| 20 | Marathwada | 0.171 | 0.043 | −0.101 |
| 21 | Vidarbha | −0.079 | 0.026 | −0.193* |
| 22 | Chhattisgarh | 0.039 | 0.278* | 0.010 |
| 23 | Coastal Andhra Pradesh | 0.082 | 0.023 | −0.053 |
| 24 | Telangana | 0.141 | 0.110 | 0.086 |
| 25 | Rayalaseema | −0.104 | −0.008 | −0.054 |
| 26 | Tamil Nadu | 0.049 | −0.139 | −0.169 |
| 27 | Coastal Karnataka | −0.035 | −0.042 | 0.079 |
| 28 | North Interior Karnataka | 0.132 | 0.086 | −0.020 |
| 29 | South Interior Karnataka | −0.025 | −0.087 | −0.202* |
| 30 | Kerala | 0.117 | 0.083 | 0.012 |
3. Discussion
The model suggests that a thrifty gene or a set of genes can evolve only if the frequency of famines is very high and under such conditions the thrifty allele(s) would not exist in a stable polymorphic state. Selection for the ability to program the fetus is likely in a very narrow range of frequency of famines, and at lower or higher frequency of famines, fetal programming would not evolve. The parameter space for the evolution of fetal programming becomes very narrow and highly specific for long-lived species owing to which evolution of fetal programming for thriftiness is highly unlikely.
The model results need to be interpreted carefully in the context of ancestral human ecology. One of the key questions is when in human history could selection for thriftiness, if any, have operated. We will examine three possible scenarios below.
(a) Selection during hunting-gathering stage: paleoarcheological data suggests that chronic starvation was uncommon during hunter-gatherer stage [31]. Today's hunter-gatherer societies do not seem to suffer starvation more frequently or more intensively than agricultural societies [9]. Therefore, the assumption that hunter-gatherer societies suffered frequent starvation is not well supported, but even if we assume hunter-gatherer societies to be prone to feast and famine selection, a number of other questions remain unanswered. Since hominids were hunter-gatherers for the most part of human evolutionary history, selection would have been prolonged, and we would expect alleles to have reached equilibrium frequencies. The model implies that selection cannot result in stable polymorphism of thrifty alleles. In the modern human society, there is considerable variation in the tendency to become obese or diabetic. Therefore, polymorphism with respect to genes predisposing to obesity and type 2 diabetes presumably exists. If there is no negative frequency dependence or heterozygote advantage, natural selection will be directional, resulting into the fixation of the advantageous genotype. In that case at no value of pf, the thrifty and nonthrifty genes can coexist stably. Theoretically, if a heterozygote of thrifty and nonthrifty alleles gets a dual advantage by expressing the right allele in the right environment, the two alleles can coexist, and the population at any given time will consist of thrifty, nonthrifty, and all time fit individuals. However, there is no empirical evidence of heterozygote advantage so far. Stable polymorphism is also possible if there is negative frequency-dependent selection. However, if fitness is decided by climatic conditions as assumed by the popular version of thrifty gene hypothesis, frequency dependence is unlikely. Therefore, selection during hunter-gatherer stage does not explain the prevalent polymorphism in predisposition to obesity.
(b) Selection after the beginning of agriculture: chronic starvation due to famines became more serious and common with the beginning of agriculture [7, 8]. Signs of chronic starvation on teeth, such as linear enamel hypoplasia, are more common in early agricultural societies than in hunter-gatherer societies [32]. This is owing to the fact that crops are highly seasonal in nature, and the failure of a single crop leads to long-term food scarcity. Such long-term food shortages are much less probable in hunter-gatherer life, particularly in biodiversity rich areas. Therefore, if selection for thriftiness started acting after the beginning of agriculture, there could be transient polymorphism. A testable prediction of the hypothesis would then be that ethnic groups that took to agriculture earlier should show higher tendencies to become obese and diabetic. This has not been rigorously tested with quantitative data. However, ethnic groups, such as the Australian aborigines, remained hunter gatherers until recently, and the recently urbanized individuals of this community developed a high prevalence of diabetes and hypertension [33]. It is difficult to argue, therefore, that thrifty genes evolved after the advent of agriculture.
(c) Selection in modern times: intensive agricultural and industrial societies are modern phenomena not more than a few hundred years old, and it is highly unlikely that this period could have brought about an evolutionary change. It can be seen from all the three possible scenarios that natural selection for the hypothetical thrifty gene(s) is unable to explain the polymorphism observed today.
Since different geographic regions of the world differ in the climatic conditions, seasonality, and food availability, ethnic groups evolved in different areas should show differential predisposition to obesity and related disorders. People evolved in arid or drought-prone areas could have suffered more frequent famines and, therefore, should have a greater tendency to develop obesity. Also, ethnic groups from harsh winter environments were unable to hunt, fish, or farm during the colder months and, thus, historically could have faced regular feast and famine conditions. Therefore, we may expect higher diabetic tendencies in them. Although substantial cross-ethnic differences are observed, the trends are not as expected by the thriftiness hypotheses. People from Caucasoid, Eskimo, and some of the Himalayan ethnic groups that have faced harsh winter environments have a considerably lower frequency of obesity and/or T2D [34–38] whereas almost all ethnic groups of warmer habitats have a high frequency on adopting a Western urban lifestyle [33, 39], contrary to the expectation.
The thrifty phenotype or fetal programming hypothesis suffers from a different set of problems. Fetal programming can offer two types of potential advantages. Short-term survival advantages in fetal and early infant life and long-term predictive adaptive responses. Our model incorporates both of the advantages separately. If the advantage is of a short duration, it is difficult to explain why a lifetime commitment to a particular metabolic state could have evolved. A number of developmental genes have age-specific expressions, and, therefore, any rigid lifelong programming for short-term advantage is a difficult proposition. If climatic fluctuations were the main selective force, it should evolve metabolic flexibility rather than lifelong rigid programming. Metabolic programming of a lifelong duration based on intrauterine conditions is unlikely to offer a fitness advantage except in two sets of conditions. As the model suggests, if a species has a very short life span, fetal programming for adapting to the birth year conditions can be beneficial, since the birth year itself is a substantial part of the total life span. Assuming one year to be the natural unit of seasonal cycles, species with a life span of less than 3–5 years can be expected to evolve lifelong fetal programming for thriftiness even though the adaptive advantage is of a short duration. For long-lived species, fetal programming is unlikely to evolve unless there is a significant positive correlation between birth conditions and lifelong conditions. Since such correlations are not seen in climatological data, there are problems in explaining evolution of lifelong thrifty programming.
Since the thriftiness hypotheses are inadequate or weak on several grounds, there is a need to rethink the paradigm and consider alternative hypotheses seriously. A detailed comparative analysis of all the alternative hypotheses is beyond the scope of this paper. Most of them are new, and all their implications have not yet come forward. We will only briefly evaluate the alternative hypotheses below to see whether they offer better explanations where the thriftiness hypotheses are weaker.
(1) Polymorphism: polymorphism in the predisposition to obesity can be potentially explained in three different ways. It is possible that the allelic composition in the population is close to equilibrium, and there is stable polymorphism. Out of all alternative hypotheses, only the Watve-Yajnik hypothesis is able to predict stable polymorphism since there is negative frequency-dependent selection in a Hawk and Dove like game [42]. An alternative view is that the human population today is not at a stable equilibrium proportion of alleles but is undergoing drift [8] or selection [21]. These alternative hypotheses try to explain polymorphism as a transient state. Speakman [7] also tries to quantify the drift dynamics; however, his calculation is based on the assumption that human ancestors became free from predators about 1.8 million years ago, and this estimate is debatable [41]. Moreover, if the drift is an ongoing process, it would take a different direction in different populations and the cross-ethnic differences are expected to be random. Instead, we see that almost all tropical ethnic groups have high tendency to develop obesity and T2D on urbanization, and such a generalizable pattern is not expected by the drift hypothesis. Also, as we know now, a large number of genes are associated with obesity, and if drift has to operate independently on all the genes, it is highly unlikely that it will result in a directional change.
Moalem et al. [22] presumed that selective pressures changed substantially by 1800 AD. It has not been critically questioned whether only about 200 years of selection is sufficient to generate the current levels of polymorphism. Any data to test the dynamics of both the hypotheses using a predictive model are currently absent.
The third possible explanation to the apparent polymorphism is that there is no real genetic polymorphism, but individuals are programmed differently depending upon environmental conditions faced in early life. Apart from the thrifty phenotype hypothesis, only the Watve and Yajnik [10] hypothesis accounts for lifetime programming in a way discussed below.
The cold adaptation hypothesis of Hattersley and Tooke [23] does not explain polymorphism at any of the above levels. If high blood glucose is adaptive in cold environments, then ethnic groups who evolved in cold climates should undergo directional selection leading to fixation. Polymorphism in that case can only arise by cross-breeding between ethnic groups coming from warmer and colder environments.
(2) Low birth weight effects: there is globally consistent and strong evidence that a small birth weight is associated with altered metabolic and endocrine states in adult life [3, 41, 42]. Different interpretations of the birth weight effects [23] have been offered, and the birth weight association itself cannot be considered a convincing evidence of fetal programming. However, rat experiments with maternal nutrient deficiencies have also given strong evidence in support of fetal programming [43]. Therefore, some fetal programming can be safely inferred. It can nevertheless be debated whether the programming is for thriftiness or for any other adaptation. Speakman's [7, 8] drifty gene hypothesis, Corbett et al. [21] hypothesis of reverse selection in modern times, and Moalem et al.'s [22] cold adaptation hypotheses are unable to explain the birth weight effect. Wells [44] and Watve and Yajnik [10] have different interpretations of the effect of maternal nutrient limitation, but both of them adequately account for birth weight effects. Wells' hypothesis [44] of maternal advantage does not have a predictive adaptation element and, therefore, does not explain lifetime rigidity in the programming. Watve and Yajnik [10] argue that fetal undernourishment affects muscle mass more than brain mass and, therefore, a brain-dependent “diplomat” lifestyle marked by insulin resistance is adaptive for low birth weight individuals. Since muscle cells do not replicate in adult life, this early developmental limitation persists throughout life, and, thus, lifetime programming could evolve. At the same time, low birth weight is not a necessary prerequisite for the Watve and Yajnik hypothesis to work. Any hypothesis exclusively based on fetal programming suffers from another problem in that although low birth weight individuals have a greater probability of developing obesity and T2D, a large proportion of adult type 2 diabetics are not born with low weights. Therefore, any hypothesis exclusively dependent upon fetal conditions are inherently inadequate.
(3) Birth time versus lifetime correlation: the model predicts that for lifetime fetal programming to evolve, a positive correlation between intrauterine and lifetime conditions is necessary. Although climatic variations are unlikely to cause such correlations, there can be other causes of food deprivation apart from climatic factors. A high population density can lead to increased competition, resulting into undernourishment. Although population density may oscillate, the oscillations typically span over several generations so that an individual born at a high population density is expected to see high population density through most of his life. Therefore, periodic food scarcity caused by population oscillations can result into positive correlations between birth year and lifetime nutritional conditions. In social species, the social hierarchy might also be related to differential access to food. An individual born smaller and weaker is more likely to face social subordination, and, therefore, an anticipatory fetal programming would be adaptive. These social causes can produce positive birth-lifetime correlations, and they are more likely candidates to select for fetal programming of lifelong duration. There exist a broad range of metabolic, endocrinological, behavioral, and cognitive adaptations that accompany social hierarchical positions. Therefore, if fetal programming is in response to social hierarchies, we expect that it need not be restricted to diet and energy homeostasis, but it affects a large number of systems of the body along with brain and behavior. This indeed, is the case, and type 2 diabetes involves almost all systems of the body [10, 45]. Social subordination is an important predisposing factor for type 2 diabetes [46], and the link between the two is elaborately explained by Belsare et al. [40].
We suggest that on exposure to intrauterine malnourishment, there is fetal programming in anticipation of physical weakness and social subordination or anticipation of high population density rather than anticipation of famine. In primate societies, it is known that social ranks of juveniles and subadults are influenced by the ranks of their mothers [47, 48]. If social rank influences preferential access to food resources, it is sufficient to produce the positive correlation between fetal and lifetime nutritional conditions needed for the evolution of fetal programming. It appears logical, therefore, that fetal programming could have evolved in anticipation of population- and social hierarchy-related factors than anticipation of famines or other climate-related factors.
(4) Multiple effects: obesity and T2D are not only about energy homeostasis but also about changes in innate immunity, sexual and reproductive function, vascular development and function, skin architecture, wound healing and tissue regeneration, memory, cognitive functions, behavior and mechanisms of decision making, social relations, and social signaling. Therefore, any hypothesis about the origins of obesity needs to account for all the network of changes adequately. Most hypotheses are too glucolipo-centric and, therefore, fall short of this criterion. The only possible exception is the Watve-Yajnik hypothesis which has an inherent expectation that a number of systems of the body would be simultaneously involved [10, 40].
Implications for genetics of obesity: a large body of research has now focused on the genetics of obesity. An increasing number of loci and mutants associated with obesity are being identified. However, there are certain internal paradoxes associated with these data. Studies prior to the genomic era that were based on familial, twin-pair, and adoption studies typically predicted a large heritable component in obesity (reviewed by [49]). The genome-wide association studies, on the other hand, have identified a large number of associations, but together they explain a very small fraction of variance in obesity parameters [50–54], leaving a large gap between the pregenomic and emerging genomic picture. We are yet to understand the reasons for this discrepancy, but a most likely implication goes against all hypotheses that assume a gene or a set of genes for obesity. These hypotheses include Neel's thrifty gene, Speakman's drifty gene, Corbett et al.'s reverse selection, and Moalem et al.'s cold adaptation. On the other hand, fetal programming has a promise for filling the gap between the familial studies and genome-wide association studies. The programming is likely to involve epigenetic mechanisms as well. Three of the above hypotheses involve fetal programming, namely, the classical thrifty phenotype hypothesis, maternal adaptation hypothesis by Wells [44], and behavioural switch hypothesis by Watve and Yajnik [10]. Since our model has indicated the inadequacies of the thrifty phenotype hypothesis, the other two need to be considered more seriously. We suggest, here, that more than one type of metabolic programming may be involved in obesity, and they may be induced in various stages of life. There are strong data for fetal programming, but behavioral programming is also likely to affect metabolism, since associations between neuroendocrine mechanisms of behavior and metabolic states are known [40].
The classical thriftiness family of hypotheses would expect genes associated with metabolic rates to be the obesity genes. The behavioural origins hypothesis, on the other hand, expects genes involved in sexual function, cognitive abilities, immunity, regulation of aggression, and other behavioral traits to be associated with obesity and related disorders. It would be useful to interpret the emerging data on genome-wide associations and, perhaps, near future studies on epigenetics of obesity in the light of the more promising hypotheses from the above.
We have shown with a simple theoretical model that the classical concepts of thrifty gene as well as fetal programming for thriftiness face a number of problems and are inadequate to account for the observable trends in the modern epidemic of obesity and related disorders. A number of alternative hypotheses have been suggested recently. It might be too early to discard any of the hypotheses right away, but a comparative analysis shows the strengths and weaknesses of each of them. Since search for obesity-associated genes has so far given limited success in terms of explaining population variance in obesity parameters, the hypotheses involving phenotypic programming look more promising. In understanding obesity and related disorders, the need for a combined genomic-phenomic approach is increasingly being felt [55, 56], and such an integration can be best achieved on the platform of an evolutionary insight into the phenomenon. Greater efforts are, therefore, needed to understand the evolutionary background of obesity and related pathophysiological conditions.
Acknowledgments
P. Belsare was supported by CSIR during the study. Discussions with Sulochana Gadgil (IISc) and Ashwini Kulkarni (IITM) were useful.
References
- 1.Neel JV. Diabetes mellitus: a “thrifty” genotype rendered detrimental by “progress”? American Journal of Human Genetics. 1962;14:353–362. [PMC free article] [PubMed] [Google Scholar]
- 2.Hales CN, Barker DJP. Type 2 (non-insulin-dependent) diabetes mellitus: the thrifty phenotype hypothesis. Diabetologia. 1992;35(7):595–601. doi: 10.1007/BF00400248. [DOI] [PubMed] [Google Scholar]
- 3.Barker DJP. In utero programming of chronic disease. Clinical Science. 1998;95(2):115–128. [PubMed] [Google Scholar]
- 4.Drake AJ, Walker BR. The intergenerational effects of fetal programming: non-genomic mechanisms for the inheritance of low birth weight and cardiovascular risk. Journal of Endocrinology. 2004;180(1):1–16. doi: 10.1677/joe.0.1800001. [DOI] [PubMed] [Google Scholar]
- 5.Gluckman PD, Hanson MA, Spencer HG. Predictive adaptive responses and human evolution. Trends in Ecology and Evolution. 2005;20(10):527–533. doi: 10.1016/j.tree.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 6.Speakman JR. Thrifty genes for obesity and the metabolic syndrome—time to call off the search? Diabetes and Vascular Disease Research. 2006;3(1):7–11. doi: 10.3132/dvdr.2006.010. [DOI] [PubMed] [Google Scholar]
- 7.Speakman JR. A non-adaptive scenario explaining the genetic predisposition to obesity: the “predation release” hypothesis. Cell Metabolism. 2007;6(1):5–12. doi: 10.1016/j.cmet.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 8.Speakman JR. Thrifty genes for obesity, an attractive but flawed idea, and an alternative perspective: the ‘drifty gene’ hypothesis. International Journal of Obesity. 2008;32(11):1611–1617. doi: 10.1038/ijo.2008.161. [DOI] [PubMed] [Google Scholar]
- 9.Benyshek DC, Watson JT. Exploring the thrifty genotype’s food-shortage assumptions: a cross-cultural comparison of ethnographic accounts of food security among foraging and agricultural societies. American Journal of Physical Anthropology. 2006;131(1):120–126. doi: 10.1002/ajpa.20334. [DOI] [PubMed] [Google Scholar]
- 10.Watve MG, Yajnik CS. Evolutionary origins of insulin resistance: a behavioral switch hypothesis. BMC Evolutionary Biology. 2007;7, article 61 doi: 10.1186/1471-2148-7-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wells JCK. Flaws in the theory of predictive adaptive responses. Trends in Endocrinology and Metabolism. 2007;18(9):331–337. doi: 10.1016/j.tem.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 12.Klöting N, Blüher M. Extended longevity and insulin signaling in adipose tissue. Experimental Gerontology. 2005;40(11):878–883. doi: 10.1016/j.exger.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 13.Eriksson J, Forsén T, Tuomilehto J, Osmond C, Barker D. Size at birth, fat-free mass and resting metabolic rate in adult life. Hormone and Metabolic Research. 2002;34(2):72–76. doi: 10.1055/s-2002-20518. [DOI] [PubMed] [Google Scholar]
- 14.Kensara OA, Wooton SA, Phillips DIW, et al. Substrate-energy metabolism and metabolic risk factors for cardiovascular disease in relation to fetal growth and adult body composition. American Journal of Physiology. 2006;291(2):E365–E371. doi: 10.1152/ajpendo.00599.2005. [DOI] [PubMed] [Google Scholar]
- 15.Westerterp KR, Speakman JR. Physical activity energy expenditure has not declined since the 1980s and matches energy expenditures of wild mammals. International Journal of Obesity. 2008;32(8):1256–1263. doi: 10.1038/ijo.2008.74. [DOI] [PubMed] [Google Scholar]
- 16.Black AE, Coward WA, Cole TJ, Prentice AM. Human energy expenditure in affluent societies: an analysis of 574 doubly-labelled water measurements. European Journal of Clinical Nutrition. 1996;50(2):72–92. [PubMed] [Google Scholar]
- 17.Zurlo F, Lillioja S, Puente AED, et al. Low ratio of fat to carbohydrate oxidation as predictor of weight gain: study of 24-h RQ. American Journal of Physiology. 1990;259(5):E650–E657. doi: 10.1152/ajpendo.1990.259.5.E650. [DOI] [PubMed] [Google Scholar]
- 18.Hoffman DJ, Sawaya AL, Verreschi I, Tucker KL, Roberts SB. Why are nutritionally stunted children at increased risk of obesity? Studies of metabolic rate and fat oxidation in shantytown children from Sao Paulo, Brazil. American Journal of Clinical Nutrition. 2000;72(3):702–707. doi: 10.1093/ajcn/72.3.702. [DOI] [PubMed] [Google Scholar]
- 19.Farooqi IS, O’Rahilly S. Genetic factors in human obesity. Obesity Reviews. 2007;8(1):37–40. doi: 10.1111/j.1467-789X.2007.00315.x. [DOI] [PubMed] [Google Scholar]
- 20.Norman RJ, Clark AM. Obesity and reproductive disorders: a review. Reproduction, Fertility and Development. 1998;10(1):55–63. doi: 10.1071/r98010. [DOI] [PubMed] [Google Scholar]
- 21.Corbett SJ, McMichael AJ, Prentice AM. Type 2 diabetes, cardiovascular disease, and the evolutionary paradox of the polycystic ovary syndrome: a fertility first hypothesis. American Journal of Human Biology. 2009;21(5):587–598. doi: 10.1002/ajhb.20937. [DOI] [PubMed] [Google Scholar]
- 22.Moalem S, Storey KB, Percy ME, Peros MC, Perl DP. The sweet thing about type 1 diabetes: a cryoprotective evolutionary adaptation. Medical Hypotheses. 2005;65(1):8–16. doi: 10.1016/j.mehy.2004.12.025. [DOI] [PubMed] [Google Scholar]
- 23.Hattersley AT, Tooke JE. The fetal insulin hypothesis: an alternative explanation of the association of low birthweight with diabetes and vascular disease. The Lancet. 1999;353(9166):1789–1792. doi: 10.1016/S0140-6736(98)07546-1. [DOI] [PubMed] [Google Scholar]
- 24.Wells JCK. Thrift: a guide to thrifty genes, thrifty phenotypes and thrifty norms. International Journal of Obesity. 2009;33(12):1331–1338. doi: 10.1038/ijo.2009.175. [DOI] [PubMed] [Google Scholar]
- 25.Rashidi A, Kirkwood TBL, Shanley DP. Metabolic evolution suggests an explanation for the weakness of antioxidant defences in beta-cells. Mechanisms of Ageing and Development. 2009;130(4):216–221. doi: 10.1016/j.mad.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 26.Pasquali R, Gambineri A, Pagotto U. The impact of obesity on reproduction in women with polycystic ovary syndrome. BJOG. 2006;113(10):1148–1159. doi: 10.1111/j.1471-0528.2006.00990.x. [DOI] [PubMed] [Google Scholar]
- 27.Gesink Law DC, Maclehose RF, Longnecker MP. Obesity and time to pregnancy. Human Reproduction. 2007;22(2):414–420. doi: 10.1093/humrep/del400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yilmaz N, Kilic S, Kanat-Pektas M, Gulerman C, Mollamahmutoglu L. The relationship between obesity and fecundity. Journal of Women’s Health. 2009;18(5):633–636. doi: 10.1089/jwh.2008.1057. [DOI] [PubMed] [Google Scholar]
- 29.Sallmén M, Sandler DP, Hoppin JA, Blair A, Baird DD. Reduced fertility among overweight and obese men. Epidemiology. 2006;17(5):520–523. doi: 10.1097/01.ede.0000229953.76862.e5. [DOI] [PubMed] [Google Scholar]
- 30.Ramlau-Hansen CH, Thulstrup AM, Nohr EA, Bonde JP, Sørensen TIA, Olsen J. Subfecundity in overweight and obese couples. Human Reproduction. 2007;22(6):1634–1637. doi: 10.1093/humrep/dem035. [DOI] [PubMed] [Google Scholar]
- 31.Sahlins M. Stone Age Economics. London, UK: Tavistock Publications; 1974. [Google Scholar]
- 32.Lukacs JR, Walimbe SR. Physiological stress in prehistoric India: new data on localized hypoplasia of primary canines linked to climate and subsistence change. Journal of Archaeological Science. 1998;25(6):571–585. [Google Scholar]
- 33.O’Dea K. Westernisation, insulin resistance and diabetes in Australian Aborigines. Medical Journal of Australia. 1991;155(4):258–264. doi: 10.5694/j.1326-5377.1991.tb142236.x. [DOI] [PubMed] [Google Scholar]
- 34.Swinburn BA. The thrifty genotype hypothesis: how does it look after 30 years? Diabetic Medicine. 1996;13(8):695–699. doi: 10.1002/(SICI)1096-9136(199608)13:8<695::AID-DIA170>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 35.Beck-Nielsen H. General characteristics of the insulin resistance syndrome prevalence and heritability. Drugs. 1999;58(supplement 1):7–10. doi: 10.2165/00003495-199958001-00003. [DOI] [PubMed] [Google Scholar]
- 36.Vilbergsson S, Sigurdsson G, Sigvaldason H, Hreidarsson AB, Sigfusson N. Prevalence and incidence of NIDDM in Iceland: evidence for stable incidence among males and females 1967–1991—the Reykjavik Study. Diabetic Medicine. 1997;14(6):491–498. doi: 10.1002/(SICI)1096-9136(199706)14:6<491::AID-DIA365>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 37.Mouratoff GJ, Carroll NV, Scott EM. Diabetes mellitus in Eskimos. Journal of the American Medical Association. 1967;199(13):107–112. doi: 10.1001/jama.199.13.107. [DOI] [PubMed] [Google Scholar]
- 38.Vaz M, Ukyab TT, Padmavathi R, et al. Body fat topography in Indian and Tibetan males of low and normal body mass index. Indian Journal of Physiology and Pharmacology. 1999;43(2):179–185. [PubMed] [Google Scholar]
- 39.Yudkin JS. Non-insulin-dependent diabetes mellitus (NIDDM) in Asians in the UK. Diabetic Medicine. 1996;13(6):S16–S18. [PubMed] [Google Scholar]
- 40.Belsare PV, Watve MG, Ghaskadbi SS, Bhat DS, Yajnik CS, Jog M. Metabolic syndrome: aggression control mechanisms gone out of control. Medical Hypotheses. 2010;74(3):578–589. doi: 10.1016/j.mehy.2009.09.014. [DOI] [PubMed] [Google Scholar]
- 41.Fall CHD, Stein CE, Kumaran K, et al. Size at birth, maternal weight, and type 2 diabetes in South India. Diabetic Medicine. 1998;15(3):220–227. doi: 10.1002/(SICI)1096-9136(199803)15:3<220::AID-DIA544>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 42.Li C, Johnson MS, Goran MI. Effects of low birth weight on insulin resistance syndrome in Caucasian and African-American children. Diabetes Care. 2001;24(12):2035–2042. doi: 10.2337/diacare.24.12.2035. [DOI] [PubMed] [Google Scholar]
- 43.Martin JF, Johnston CS, Han CT, Benyshek DC. Nutritional origins of insulin resistance: a rat model for diabetes-prone human populations. Journal of Nutrition. 2000;130(4):741–744. doi: 10.1093/jn/130.4.741. [DOI] [PubMed] [Google Scholar]
- 44.Wells JCK. The thrifty phenotype as an adaptive maternal effect. Biological Reviews. 2007;82(1):143–172. doi: 10.1111/j.1469-185X.2006.00007.x. [DOI] [PubMed] [Google Scholar]
- 45.Koshiyama H, Ogawa Y, Tanaka K, Tanaka I. Diabetes mellitus as dysfunction of interactions among all organs: “Ominous Orchestra of Organs,”. Clinical Medicine: Endocrinology and Diabetes. 2008;1:1–6. [Google Scholar]
- 46.Agardh EE, Ahlbom A, Andersson T, et al. Work stress and low sense of coherence is associated with type 2 diabetes in middle-aged Swedish women. Diabetes Care. 2003;26(3):719–724. doi: 10.2337/diacare.26.3.719. [DOI] [PubMed] [Google Scholar]
- 47.Johnson JA. Dominance rank in juvenile olive baboons, Papio anubis: the influence of gender, size, maternal rank and orphaning. Animal Behaviour. 1987;35(6):1694–1708. [Google Scholar]
- 48.Holekamp KE, Smale L. Dominance acquisition during mammalian social development: the “inheritance” of maternal rank. American Zoologist. 1991;31(2):306–317. [Google Scholar]
- 49.Maes HHM, Neale MC, Eaves LJ. Genetic and environmental factors in relative body weight and human adiposity. Behavior Genetics. 1997;27(4):325–351. doi: 10.1023/a:1025635913927. [DOI] [PubMed] [Google Scholar]
- 50.Rankinen T, Zuberi A, Chagnon YC, et al. The human obesity gene map: the 2005 update. Obesity. 2006;14(4):529–644. doi: 10.1038/oby.2006.71. [DOI] [PubMed] [Google Scholar]
- 51.Li S, Zhao JH, Luan J, et al. Cumulative effects and predictive value of common obesity-susceptibility variants identified by genome-wide association studies. American Journal of Clinical Nutrition. 2010;91(1):184–190. doi: 10.3945/ajcn.2009.28403. [DOI] [PubMed] [Google Scholar]
- 52.Li S, Zhao JH, Luan J, et al. Physical activity attenuates the genetic predisposition to obesity in 20,000 men and women from EPIC-Norfolk prospective population study. PLoS Medicine. 2010;7(8) doi: 10.1371/journal.pmed.1000332. Article ID e1000332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thorleifsson G, Walters GB, Gudbjartsson DF, et al. Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nature Genetics. 2009;41(1):18–24. doi: 10.1038/ng.274. [DOI] [PubMed] [Google Scholar]
- 54.Mutch DM, Clément K. Unraveling the genetics of human obesity. PLoS Genetics. 2006;2(12):1956–1963. doi: 10.1371/journal.pgen.0020188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hegele RA, Pollex RL. Hypertriglyceridemia: phenomics and genomics. Molecular and Cellular Biochemistry. 2009;326(1-2):35–43. doi: 10.1007/s11010-008-0005-1. [DOI] [PubMed] [Google Scholar]
- 56.Houle D. Numbering the hairs on our heads: the shared challenge and promise of phenomics. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(1):1793–1799. doi: 10.1073/pnas.0906195106. [DOI] [PMC free article] [PubMed] [Google Scholar]