

Abstract
Inflammatory pain represents an important unmet clinical need with important socioeconomic implications. Ceramide, a potent proinflammatory sphingolipid, has been shown to elicit mechanical hyperalgesia, but the mechanisms remain largely unknown. We now demonstrate that, in addition to mechanical hyperalgesia, intraplantar injection of ceramide (10 μg) led to the development of thermal hyperalgesia that was dependent on induction of the inducible cyclooxygenase (COX-2) and subsequent increase of prostaglandin E2 (PGE2). The development of mechanical and thermal hyperalgesia and increased production of PGE2 was blocked by NS-398 (15–150 ng), a selective COX-2 inhibitor. The importance of the COX-2 to PGE2 pathway in ceramide signaling was underscored by the findings that intraplantar injection of a monoclonal PGE2 antibody (4 μg) blocked the development of hyperalgesia. Our results further revealed that COX-2 induction is regulated by NF-κB and p38 kinase activation, since intraplantar injection of SC-514 (0.1–1 μg) or SB 203580 (1–10 μg), well-characterized inhibitors of NF-κB and p38 kinase activation, respectively, blocked COX-2 induction and increased formation of PGE2 and thermal hyperalgesia in a dose-dependent manner. Moreover, activation of NF-κB was dependent on upstream activation of p38 MAPK, since SB 203580 (10 μg) blocked p65 phosphorylation, whereas p38 kinase phosphorylation was unaffected by NF-κB inhibition by SC-514 (1 μg). Our findings not only provide mechanistic insight into the signaling pathways engaged by ceramide in the development of hyperalgesia, but also provide a potential pharmacological basis for developing inhibitors targeting the ceramide metabolic-to-COX-2 pathway as novel analgesics.—Doyle, T., Chen, Z., Muscoli, C., Obeid, L. M., Salvemini, D. Intraplantar-injected ceramide in rats induces hyperalgesia through an NF-κB- and p38 kinase-dependent cyclooxygenase 2/prostaglandin E2 pathway.
Keywords: pain, sphingolipid, sphingosine-1-phosphate, COX inhibitor, NS-398
Ceramide, a potent proinflammatory sphingolipid (1), is generated from enzymatic hydrolysis of sphingomyelin by sphingomyelinases (sphingomyelin pathway) and from de novo synthesis by serine palmitoyltransferase and ceramide synthase (de novo pathway) (2). Besides its well-established role in inflammation, a potential role of ceramide in peripheral sensitization and mechanical hyperalgesia is documented by the observations that intradermal injection of ceramide in rats produces dose-dependent hyperalgesia (3) and that TNF-α-mediated peripheral sensitization and hyperalgesia is driven at least in part by ceramide (3). Furthermore, and as shown in in vitro studies, ceramide increases the excitability of small-diameter sensory neurons and is an important mediator in nerve growth factor (NGF)-induced sensitization of sensory neurons (4). The signaling pathways engaged by ceramide in the development of hypersensitivity remain largely undefined and were explored in this study by intraplantar injection of this sphingolipid in rats.
Cyclooxygenase (COX) catalyzes the conversion of arachidonic acid to PGH2, the initial step in the formation of prostaglandins (PGs) and thromboxane (5). In mammals, two main COX isoforms have been described. Constitutively expressed COX-1, also known as the “housekeeping” enzyme, makes prostaglandins that are important for maintaining physiological functions and an inducible COX-2 that releases large quantities of the proinflammatory and pronociceptive PGE2 (6). Evidence from several independent lines of investigation in fields unrelated to pain links ceramide to the COX-2 pathway. Indeed, exogenous application of ceramide or enzymes leading to its biosynthesis induces COX-2 and increases PGE2 synthesis in several cell lines (7–12). COX-2 induction is promoted by numerous inflammatory mediators, and its expression is tightly regulated, at least in part, by the redox-sensitive transcription factors NF-κB and the MAPK kinase, p38 kinase (13, 14). Ceramide is a potent activator of both NF-κB (15–18) and p38 kinase (19–21), and inhibitors of NF-κB (11, 22) or p38 kinase (11, 12) block COX-2 induction and increased PGE2 formation.
These observations in unrelated fields of research prompted us to consider and test in the present study whether the COX-to-PGE2 pathway contributes to ceramide-induced peripheral sensitization and ensuing hyperalgesia. Our results demonstrate that the development of hyperalgesia (mechanical and thermal) following the intraplantar injection of exogenous ceramide in rats is driven by increased in situ formation of PGE2 derived from a NF-κB/p38 kinase-dependent induction of COX-2. These findings provide a mechanistic link engaged by ceramide in the development of peripheral sensitization and ensuing hyperalgesia and suggest that targeting the ceramide metabolic pathway may provide a novel approach in pain management.
MATERIALS AND METHODS
Materials
C2-ceramide (d-erythro-sphingosine, N-acetyl) and dihydro-C2-ceramide (d-erythro-sphingosine, dihydro-, N-acetyl) were purchased from Calbiochem (La Jolla, CA, USA). NS-398, SB 203580, SC-514, and the anti-PGE2 antibody were purchased from Cayman Chemical (Ann Arbor, MI, USA). Rabbit polyclonal antibodies to total p38, rabbit monoclonal antibodies to phospho-p65 (p-p65) and total p65, and mouse monoclonal antibodies to phospho-p38 (p-p38) were from Cell Signaling Technology (Boston, MA, USA). The mouse monoclonal COX-1 antibody and the rabbit polyclonal COX-2 antibody were from Cayman Chemical. The mouse monoclonal α-tubulin antibody was from Sigma-Aldrich (St. Louis, MO, USA). The horseradish-peroxidase (HRPO)-conjugated goat anti-rabbit and anti-mouse secondary antibodies and the reagents for enhanced chemiluminescent detection and the bicinchoninic acid (BCA) assay were obtained from Thermo-Fisher (Rockford, IL, USA). The polyacrylamide gels for electrophoresis (10% and 4–20% Tris-glycine) were obtained from Bio-Rad (Hercules, CA, USA). Unless otherwise noted, all other chemicals and reagents were from Sigma-Aldrich.
Experimental animals
Male Sprague-Dawley rats (200–220 g) were purchased from Harlan (Indianapolis, IN, USA), housed 3 or 4 per cage, and maintained in a controlled environment (12-h light-dark cycles) with food and water available ad libitum. All experiments were performed in accordance with the International Association for the Study of Pain and the U.S. National Institutes of Health guidelines on laboratory animal welfare and the recommendations by Saint Louis University Institutional Animal Care and Use Committee.
Drug administration and induction of thermal and mechanical hyperalgesia
NS-398, SB 203580, SC-514 (or their respective vehicle, 50% DMSO in saline), or an anti-PGE2 monoclonal antibody or its vehicle (saline) were given by intraplantar injection into the hindpaw of rats 15 min before intraplantar injections of C2-ceramide or its vehicle, DMSO. DMSO was also used to dissolve dihydro-C2-ceramide. All drugs were injected in a 5-μl injection volume using a Hamilton gauge needle (3.5 inch; Hamilton, Reno, NV, USA) in lightly anesthetized rats (80% CO2/20% O2). Hyperalgesic responses to heat were determined by the Hargreaves method using a Basile plantar test (Ugo Basile, Comeria, Italy; ref. 23) with a cutoff latency of 20 s employed to prevent tissue damage. Rats were individually confined to Plexiglas chambers and allowed to habituate and acclimate for 15 min prior to behavioral testing. A mobile infrared generator was positioned to deliver a thermal stimulus directly to an individual hindpaw from beneath the chamber. The withdrawal latency period of injected paws was determined with an electronic clock circuit and thermocouple. Two readings were acquired for each paw to determine a mean latency for each animal. Thermal hyperalgesia results are the mean latency for each group and expressed as paw withdrawal latency (s). Mechanical hyperalgesia was assessed by the Randall and Sellitto paw pressure test (24), using a Ugo Basile analgesiometer, which applies a linearly increasing mechanical force to the dorsum of the rat's hindpaw. The nociceptive threshold was defined as the force (g) at which the rat withdrew its paw, with baseline threshold forces of ∼99–111 g and cutoff set at 250 g to avoid tissue damage. Two readings were acquired for each paw to determine a mean threshold for each animal. Mechanical hyperalgesia results are expressed as paw withdrawal threshold (g). To assess and account for intra-assay and interassay variability in behavioral testing, 2 animals from each treatment group were treated, tested, and sacrificed for tissue harvest on a given day. This was repeated with an additional pair of animals from each group on a subsequent day for an n value of 4 animals/group. All experiments were conducted with the experimenters masked to treatment conditions. Behavioral testing was done at baseline in all rats prior to drug or vehicle administration, at 15 min after drug or vehicle administration (defined as time 0), and subsequently at different time points after intraplantar injection of ceramide, dihydro-C2-ceramide, or vehicle.
Determinations of PGE2 release in ceramide-injected rat paws
Prostaglandin E2 released in the paw exudates was measured as described previously by our group (25–27). Briefly, at the required time points, rats in each group were sacrificed, and each paw was excised at the level of the calcaneus bone. Saline (200 μl) was injected into each paw, which was then gently centrifuged at 250 g for 20 min to recover a sample of the edematous fluid, and the volume of fluid recovered from each paw was measured. Prostaglandin E2 levels were measured by ELISA using commercially available kits (Cayman Chemical), and the results were normalized to the amount of exudates recovered from each paw and expressed as nanograms per paw tissue fluid. All determinations were performed in duplicate.
Immunoblotting
Plantar lysates (n=3–6) were obtained from flash-frozen plantar soft tissue, and the relative protein expression was assessed by immunoblot assays. Frozen tissues were pulverized with a liquid nitrogen-chilled mortar and pestle, then homogenized in 0.7–1.5 ml (4 vol) of homogenization buffer [20 mM Tris-Cl, pH 7.4; 150 mM NaCl; 16.3 mM CHAPS; 0.5% Triton X-100; 0.1% SDS; 2 mM EGTA; 5% glycerol; 50 mM sodium fluoride; 1 mM sodium orthovanadate; 1 mM sodium molybdinate; 2 mM PMSF; 1× phosphatase inhibitor cocktail (Sigma-Aldrich; containing cantharidin, bromotetramisole, and microcystin LR), and 1× protease inhibitor cocktail (Sigma-Aldrich; final concentration: 1 mM 4-(2-aminoethyl)benzenesulfonyl fluoride, 15 μM pepstatin A, 14 μM E-64, 40 μM bestatin, 20 μM leupeptin, and 850 nM aprotinin)]. Each lysate was incubated for 1 h, with rotation, at 4°C; sonicated for 10 min at 4°C; and clarified by centrifugation at 13,000 g, 4°C for 10 min. Each sample (200 μg as determined by BCA assay) was heat denatured in Laemmli buffer and stored at −20°C until assayed. Proteins (30–100 μg/well) were resolved by Tris-glycine-SDS electrophoresis with 10% or 4–20% Tris-glycine gel at 170 V, and then transferred to nitrocellulose membrane (Bio-Rad) for 90 min at 0.35 mA, 4°C in Towbin buffer. After blocking with 5% nonfat milk in PBS-T (1× PBS, pH 7.4; 0.05% Tween-20; and 0.1% thimerosol), the membranes were probed overnight at 4°C with rabbit anti-COX-2 antibody (1:500), mouse anti-COX-1 antibody (1:500), rabbit anti-p-p65 (S536; 1:1000), rabbit anti-p65 (1:1000), mouse anti-p-p38 (T180/Y182; 1:500), rabbit p38 (1:1000), or mouse anti-α-tubulin antibody (1:1000) in 2.5% nonfat milk in PBS-T. Each membrane was washed in PBS-T, and the bands were visualized with HRPO-conjugated goat anti-rabbit or anti-mouse secondary antibodies in 2.5% low-fat milk in PBS-T and enhanced chemiluminescence. Membranes were stripped 15 min at 50°C with 62.5 mM Tris-HCl (pH 6.8), 2% SDS, and 100 mM β-mercaptoethanol, washed, blocked, and reprobed.
Images were captured using Fujifilm LAS-3000 imaging system and Image Reader LAS-3000 2.2 software (Fujifilm, Tokyo Japan) to acquire high sensitivity, incremental exposures every 10–60 s up to 20 min. The relative density of each protein band was analyzed from the original captured image using Multi Gauge 3.0 (Fujifilm), background adjusted, and normalized to α-tubulin. All immunoblot data are expressed as percentage of band of interest vs. α-tubulin (e.g., %COX-2/tubulin). For image clarity only, the entire image was rotated and a multiply mask was applied before being cropped using Photoshop CS2 9.0 (Adobe Systems, San Jose, CA, USA).
Statistical analysis
All data are expressed as means ± se. The differences in paw withdrawal threshold (g) and paw withdrawal latency (s) were assessed by 2-way ANOVA with Bonferroni post hoc comparisons to ceramide-treated animals. Ceramide-mediated protein expression or PGE2 formation were analyzed, as noted, by unpaired Student's t test or 1-way ANOVA with Dunnett's post hoc comparisons to ceramide-treated animals or to t = 0. Significance was defined as P < 0.05.
RESULTS
Ceramide-induced hyperalgesia
Previous studies by Joseph and Levine (3) revealed that intraplantar injection of ceramide in rats led to the development of mechanical hyperalgesia. When compared to the vehicle group (DMSO), intraplantar injection of C2-ceramide (10 μg, n=4), given at a dose previously shown to elicit mechanical hyperalgesia (3), led to a time-dependent development of mechanical and thermal hyperalgesia that peaked by 1 h, plateaued by 2 h, and remained sustained up to the 5-h time point (Fig. 1). Intraplantar injection of dihydro-C2-ceramide, an inactive analog of ceramide lacking the double bond between carbons 4 and 5 of the sphingoid backbone, did not evoke mechanical or thermal hyperalgesia (Fig. 1). Our studies confirm previous observations that ceramide leads to the development of mechanical hyperalgesia and extend them to demonstrate that ceramide is also a potent mediator of thermal hyperalgesia. All mechanistic studies undertaken in the next series of experiments were carried out with ceramide at 2 h, the plateau phase.
Figure 1.

Ceramide-induced hyperalgesia. When compared to vehicle (Veh, ▵), intraplantar injection of 10 μg C2-ceramide (●), but not dihydro-C2-ceramide (◊), led to a time-dependent development of mechanical (A) and thermal hyperalgesia (B). Results are expressed as means ± se for 4 rats. BL, baseline. *P < 0.001 vs. Veh; 2-way ANOVA with Bonferroni post hoc tests.
The COX-2-to-PGE2 pathway plays a critical role in ceramide-mediated hyperalgesia
When compared to the vehicle group, the development of ceramide-induced hyperalgesia (mechanical and thermal) was associated with a significant increase in COX-2 (Fig. 2A), but not COX-1 protein (Fig. 2B), as measured by Western blot analysis, and an increase in the formation of PGE2 (Fig. 2C). When compared to the vehicle group (intraplantar injection of the vehicle used for NS-398 followed by intraplantar injection of ceramide), intraplantar injection of NS-398, a selective COX-2 inhibitor (28), attenuated ceramide-induced mechanical and thermal hyperalgesia in a dose-dependent fashion (15–150 ng, n=4; Fig. 3A, B), as well as the increased production of PGE2 (Fig. 3C); thus implicating the contribution of PGE2 in ceramide's effects. Furthermore, intraplantar injection of an anti-PGE2 antibody (4 μg, n=4; ref. 29), but not its vehicle (saline), blocked the development of ceramide-induced mechanical and thermal hyperalgesia (Fig. 3D, E); thus confirming the role of PGE2 in ceramide's effects. When tested alone and compared to rats that received an intraplantar injection of the vehicle used for ceramide, NS-398 (150 ng, n=4; Fig. 3A, B) or the anti-PGE2 antibody (4 μg, n=4) had no effect on baseline withdrawal latencies (Fig. 3D, E). These results establish that hyperalgesic responses to ceramide are mediated, at least in part, through peripheral COX-2 induction and that PGE2 is an important signaling eicosanoid involved in this response.
Figure 2.

Role of COX-2 and PGE2 in ceramide-induced hyperalgesia. A, B) When compared to baseline values at t = 0 h, intraplantar injection of C2-ceramide (10 μg) led to a significant increase in COX-2 (A), but not COX-1 (B), expression by 2 h, which was sustained through 5 h, as measured by Western blot analysis. Top panels: bands are representative replicates of separate animals (n=3 rats) taken from the same gel and exposure. Bottom panels: band densities were normalized to α-tubulin (%COX-1 or -2/tubulin). C) Increased COX-2 corresponded with a significant increase in production of PGE2 at 2 h, as measured in paw fluids (n=4 rats). Results are expressed as means ± se. *P < 0.05, **P < 0.01, ***P < 0.001 vs. t = 0 h; ANOVA with Dunnett's post hoc test (A, B) or unpaired Student's t test (C).
Figure 3.

Inhibition of ceramide-induced hyperalgesia by NS-398. A, B) When compared to rats administered intraplantar NS-398 vehicle and ceramide vehicle (Veh; ▵), an intraplantar injection of ceramide (Cer; 10 μg, ◊) led to a time-dependent development of mechanical (A) and thermal hyperalgesia (B) that was attenuated in a dose-dependent manner by intraplantar NS-398 given at 15 ng (●), 45 ng (■), or 150 ng (♦). Intraplantar administration of NS-398, when tested at the highest dose and compared to rats that received vehicle for ceramide, had no effect on baseline (BL) withdrawal latency (○). C) When tested at the highest dose, NS-398 (150 ng) attenuated the increased production of PGE2 in paw tissue fluids, as measured at t =2 h. D, E) When compared to rats administered ceramide vehicle (▵), an intraplantar injection of ceramide (10 μg; ◊) led to a time-dependent development of mechanical (D) and thermal hyperalgesia (E) that was attenuated by intraplantar injection of a monoclonal antibody to PGE2 (▾). Anti-PGE2 antibody alone had no effect on baseline withdrawal latency (▿). Results are expressed as means ± se (n=4 rats). *P < 0.001 vs. Veh; †P < 0.05, ††P < 0.01, †††P < 0.001 vs. Cer; 2-way ANOVA with Bonferroni post hoc test (A, B, D, E) or 1-way ANOVA with Dunnett's post hoc test (C).
Role for NF-κB in ceramide-induced COX-2 induction and development of hyperalgesia
In response to a variety of agonists, the inhibitory κB kinase (IKK), a multisubunit complex containing IKK-1 and 2 catalytic subunits and regulatory IKKγ subunit (13), relieves the inhibitory κB (IκB) cytosolic suppression of NF-κB translocation to the nucleus (30, 31). Gene-knockout studies revealed that IKK-2 and IKKγ are required for NF-κB activation by all known inflammatory stimuli (13). SC-514 is a selective inhibitor of IKK-2 and, consequently, inflammatory gene expression (32). By binding to the ATP-binding site of IKK-2, SC-514 is selective for IKK-2 over 30 other kinases, including those in the MAP kinase inflammatory pathways, such as p38, c-Jun N-terminal kinase, and ERK (32). When compared to the vehicle group (intraplantar injection of the vehicle used for SC-514 followed by intraplantar injection of ceramide), intraplantar injection of SC-514 blocked the development of ceramide-induced mechanical and thermal hyperalgesia in a dose-dependent manner (0.1–1 μg, n=4; Fig. 4A, B). When tested alone at the highest dose used and compared to rats that received an intraplantar injection of the vehicle used for ceramide, SC-514 (1 μg, n=4) had no effect on baseline withdrawal latencies (Fig. 4A, B). When tested at the highest dose, SC-514 (1 μg) blocked the induction of COX-2 (n=4; Fig. 4C) and the increased production of PGE2 (n=4; Fig. 4D).
Figure 4.

Role of NF-κB in ceramide-induced hyperalgesia. A, B) When compared to rats administered intraplantar SC-514 vehicle and ceramide vehicle (Veh; ▵), an intraplantar injection of ceramide (Cer; 10 μg, ◊) led to a time-dependent development of mechanical (A) and thermal (B) hyperalgesia that was attenuated in a dose-dependent manner by intraplantar SC-514 given at 0.1 μg (●), 0.3 μg (■), or 1 μg (♦). Intraplantar administration of SC-514, when tested at the highest dose and compared to rats that received vehicle for ceramide, had no effect on baseline (BL) withdrawal latency (○). C, D) When tested at the highest dose, SC-514 (1 μg) blocked the induction of COX-2 (C), and the increased production of PGE2 (D), as measured at t = 2 h. Results are expressed as means ± se (n=4 rats; A–C). Band densities (C, bottom panel) were normalized to α-tubulin (%COX-2/tubulin). *P < 0.001 vs. Veh; †P < 0.05, ††P < 0.01, †††P < 0.001 vs. Cer; 2-way ANOVA with Bonferroni post hoc test (A, B) or 1-way ANOVA with Dunnett's post hoc test (C, D).
Role for p38 kinase in ceramide-induced COX-2 induction and development of thermal hyperalgesia
When compared to the vehicle group (intraplantar injection of the vehicle used for SB 203580, followed by intraplantar injection of ceramide), intraplantar injection of SB 203580, a well-characterized pyridinyl imidazole inhibitor of p38 kinase that acts as an ATP substrate competitor, preventing p38 phosphorylation of downstream targets (33), blocked the development of ceramide-induced mechanical and thermal hyperalgesia in a dose-dependent manner (1–10 μg, n=4; Fig. 5A, B). When tested alone at the highest dose used and compared to rats that received an intraplantar injection of the vehicle used for ceramide, SB 203580 (10 μg, n=4) had no effect on baseline paw withdrawal latencies (Fig. 5A, B). At the highest dose, SB 203580 (10 μg) blocked COX-2 induction (n=4; Fig. 5C) and the increased production of PGE2 (n=4; Fig. 5D).
Figure 5.

Role of p38 in ceramide-induced hyperalgesia. A, B) When compared to rats administered intraplantar SB 203580 vehicle and ceramide vehicle (Veh; ▵), an intraplantar injection of ceramide (Cer; 10 μg, ◊) led to a time-dependent development of mechanical (A) and thermal hyperalgesia (B) that was attenuated in a dose-dependent manner by intraplantar SB 203580 given at 1 μg (●), 3 μg (■), or 10 μg (♦). Intraplantar administration of SB 203580, when tested at the highest dose and compared to rats that received vehicle for ceramide, had no effect on baseline withdrawal latency (○). C, D) When tested at the highest dose, SB 203580 (10 μg) blocked the induction of COX-2 (C) and the increased production of PGE2 (D) as measured at t = 2 h. Results are expressed as means ± se (n=4 rats). Gels (C, top panel) are replicates from separate animals representative of n = 4 rats. Band densities (C, bottom panel) were normalized to α-tubulin (%COX-2/tubulin). *P < 0.01, **P < 0.001 vs. Veh; †P < 0.05, ††P < 0.01, †††P < 0.001 vs. Cer; 2-way ANOVA with Bonferroni post hoc test (A, B) or 1-way ANOVA with Dunnett's post hoc test (C, D).
Role of p38 in regulating NF-κB in ceramide-induced COX-2 induction and development of hyperalgesia
Activation of NF-κB can be regulated by p38 MAPK (34–38), which can involve the phosphorylation of the serine 536 residue of p65 (36–38). Thus, we sought to determine whether p38 was involved in regulating ceramide-induced activation of NF-κB. Our results demonstrate that when compared to vehicle, intraplantar ceramide increases phosphorylation of p38 MAPK at threonine 180 and tyrosine 182 (n=5; Fig. 6A) and phosphorylation of p65 at serine residue 536 rat paws at 2 h (n=6; Fig. 6B). Blocking the activation of NF-κB signaling with SC-514 had no effect on p38 MAPK phosphorylation (n=5; Fig. 6A). On the other hand, inhibiting p38 MAPK signaling with SB 203580 blocked the increase in ceramide-induced p65 phosphorylation (n=6; Fig. 6B). No significant differences in total p38 (n=5, P=0.9) or in total p65 (n=6; P=0.1) expression were detected between groups. These results suggest that ceramide-induced activation of NF-κB depends at least in part by activation of p38 kinase; conversely, our results do not support potential regulation of p38 by NF-κB activation (Fig. 7).
Figure 6.

Ceramide-induced p65 phosphorylation is dependent on p38 MAPK activation. When compared with rats administered intraplantar vehicles for the inhibitors SB 203580 or SC-514 and for ceramide (Veh), an intraplantar injection of ceramide (Cer, 10 μg) increased the p-p38 (T180/Y182; A) and p-p65 (S536; B) at 2 h. When tested at the highest dose, SB 203580 (Cer+SB 203580; 10 μg) blocked ceramide-induced p65 phosphorylation (B). In contrast, SC-514 (Cer + SC-514; 1 μg) did not block ceramide-induced p38 phosphorylation (A). There were no significant differences in total p38 or p65 between groups at 2 h. Top panels: gels are replicates from n = 5 rats for p38 and n = 6 rats for p65. Bottom panels: band densities were normalized to α-tubulin (%p-p38/tubulin or %p-p65/tubulin) and expressed as means ± se. *P < 0.05, **P < 0.001 vs. Veh; †P < 0.05 vs. Cer; 1-way ANOVA with Dunnett's post hoc test.
Figure 7.
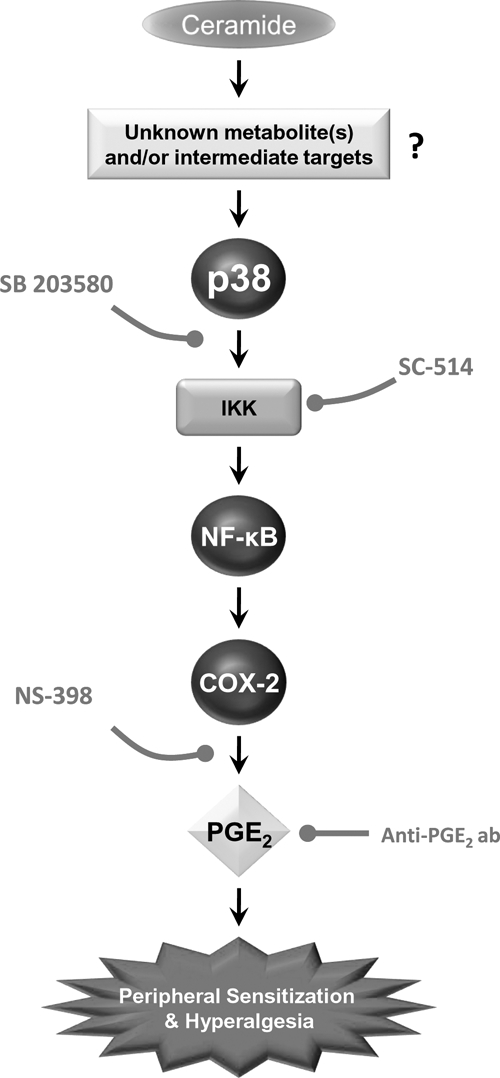
Cartoon summarizing the key findings of this study. Intraplantar injection of ceramide is a potent hyperalgesic sphingolipid acting, at least in part, via COX-2 induction through activation of a p38 MAPK-dependent NF-κB pathway. Whether ceramide acts alone or in concert with one of its active metabolites (such as sphingosine-1-phosphate) is certainly a possibility, but not a requirement, since it is well established that ceramide does not need to be metabolized in order to exert its biological responses (1). Inhibiting this pathway with p38 MAPK inhibitor, SB 203580, or the IKK-2 inhibitor, SC-514, prevents the increased expression of COX-2 and the subsequent formation of PGE2, which are essential to the development of ceramide-induced hyperalgesia. Dual inhibition of ceramide formation and COX inhibitors may provide a novel therapeutic approach to the management of pain: effective analgesia with reduced side effects typically associated with the use of COX inhibitors.
DISCUSSION
There are 50 million annual cases of inflammatory pain (i.e., arthritis and fibromyalgia) in the United States (39), with a projected 67 million cases by 2030 (40). Although nonsteroidal anti-inflammatory drugs are the most commonly used class of analgesics for moderate/severe inflammatory pain with more than 60 million prescribed annually in the United States (41), the well-documented side effects associated with prolonged use of nonselective COX-1/COX-2 inhibitors or more selective COX-2 inhibitors (42–44) necessitate novel approaches to the management of inflammatory pain. Emerging evidence points to the ceramide metabolic pathway as a potential therapeutic target in pain (3, 4, 45–52) with signaling pathways involved being currently explored. Results of our studies summarized in the schematic shown in Fig. 7 contribute to the mechanistic understanding of the signaling pathways engaged by ceramide in the development of peripheral sensitization and hyperalgesia. To this end, we demonstrate that ceramide-induced hyperalgesia is mediated, at least in part, by a COX-2-to-PGE2 pathway, since the development of hyperalgesia was associated with induction of COX-2, but not COX-1, in paw tissues, and increased production of PGE2 that was blocked by the well-characterized COX-2 inhibitor, NS-398 (28), and an anti-PGE2 antibody. PGE2 is one of the most potent and best-studied pronociceptive prostaglandins. Indeed, and when administered exogenously, PGE2 causes hyperalgesia in humans and experimental animals (53–57). In addition, PGE2 is well known to sensitize primary afferent nociceptors (58–61). These pronociceptive effects of PGE2 are mediated via adenyl cyclase-cAMP-protein kinase A, since agents that inhibit this kinase and those that inhibit adenyl cyclase attenuate PGE2-induced hyperalgesia (62–64). Besides COX-2, arachidonic acid production by cPLA2 is also rate limiting in the production of PGE2. Interestingly, ceramide-1-phosphate generated from ceramide by ceramide kinase can increase PGE2 following translocation and activation of cPLA2 to the membrane (65). It is, therefore, possible that ceramide, through the formation of ceramide-1-phosphate, provides a stimulus of arachidonic acid production that can in turn be metabolized by COX-2 to further increase PGE2 formation as observed in our studies. This interesting possibility will need to be examined in future studies.
Our results further demonstrate that COX-2 induction in response to ceramide relies on p38 kinase and NF-κB, which are known to modulate the expression of a variety of genes, including those encoding COX-2 (13, 66), since their respective inhibition blocked hyperalgesia, the associated induction of COX-2, and the increased formation of PGE2 in paw tissues. Our data also suggest that p38 MAPK following ceramide treatment regulates p65 phosphorylation. Activation of NF-κB by p38 MAPK signaling is well documented (34–38, 67), and phosphorylation of p65 at S536 can be blocked by SB 203580 treatment (36–38). Whether p38 MAPK regulates phosphorylation of p65 at S536 (36–38) or S276 (68, 69) or through acetylation and recruitment to its promoter sequence (70, 71) or some combination of these processes remains controversial. However, our p-p65 (S536) data provide evidence that exogenous administration of ceramide induces phosphorylation of p65 at serine 536 through upstream activation of p38 MAPK. Although the mechanisms by which exogenously administered ceramide activates the p38/NF-κB pathway in the rat paw are not known, ceramide has been shown to activate p38 through protein kinase C (PKC; ref. 12) and Rac1 (20) signaling and to activate NF-κB through PKC-ζ (72) or mitochondrial-derived superoxide formation (73, 74). Ceramide-induced p38 and NF-κB activity may also result from activation of a broader unfolded protein response (UPR). Conditions eliciting UPR are often associated with increased ceramide and sphingomyelinase production (75, 76), enhanced plasma membrane translocation of sphingomyelinases (75), and increased COX-2 expression (77, 78). Exogenous short-chain ceramides rapidly localize to the endoplasmic reticulum on administration (79), where they are converted to long-chain ceramides (80) that can trigger UPR (81, 82). Moreover, activation of p38 MAPK can also trigger UPR (83) by activating inositol-requiring enzyme-1 (IRE-1). Once initiated, UPR can activate NF-κB through IRE-1 activity (84) and p38 through apoptosis signal-regulating kinase 1 (22), a member of the MAPK kinase kinase (MKK) family that acts upstream of p38 MAPK through MKK3/6 (85). Therefore, it is plausible that ceramide could mediate COX-2 production and inflammatory pain through p38 and NF-κB signaling associated with the UPR. Each of these potential mechanisms of ceramide signaling in peripheral sensitization and hyperalgesia is a target for continued lines of study.
Activation of NF-κB and p38 kinase leads to the formation of various cytokines, such as TNF-α, IL-1β, and IL-6. Thus, we cannot exclude the likely possibility that, in addition to PGE2, ceramide contributes to the development of peripheral sensitization and hyperalgesia by favoring the in situ production of such cytokines that, in addition to their proinflammatory effects, are also known to sensitize peripheral nociceptors (86–88). Besides the role of ceramide in peripheral sensitization, ceramide is also emerging as an important signaling molecule in the development of central sensitization. Indeed, ceramide has been implicated in the development of central sensitization observed in a model of carrageenan-induced orofacial nociception (52) and as recently reported by our group in the development of central sensitization associated with the induction of morphine-induced hyperalgesia and antinociceptive tolerance (46–48).
Collectively, and more broadly, our results clearly demonstrated that the COX-2 to PGE2 pathway is important in events leading to ceramide-induced hyperalgesia, thus supporting the concept that therapeutic manipulations of ceramide are mechanistically grounded targets for the development of novel nonnarcotic analgesics.
Acknowledgments
The authors thank Amanda Finley (Department of Pharmacological and Physiological Science, St. Louis University School of Medicine) for her help in performing p38 Western blot analysis.
This study was supported by U.S. National Institutes of Health (NIH) grants R01 DA024074 and R21 DA023056 to D.S. and a U.S. Department of Veterans Affairs Merit Award and NIH grant RO1 GM062887 to L.O. The authors declare no conflicts of interest.
REFERENCES
- 1. Hannun Y. A., Obeid L. M. (2008) Principles of bioactive lipid signalling: lessons from sphingolipids. Nat. Rev. Mol. Cell. Biol. 9, 139–150 [DOI] [PubMed] [Google Scholar]
- 2. Delgado A., Casas J., Llebaria A., Abad J. L., Fabrias G. (2006) Inhibitors of sphingolipid metabolism enzymes. Biochim. Biophys. Acta 1758, 1957–1977 [DOI] [PubMed] [Google Scholar]
- 3. Joseph E. K., Levine J. D. (2004) Caspase signalling in neuropathic and inflammatory pain in the rat. Eur. J. Neurosci. 20, 2896–2902 [DOI] [PubMed] [Google Scholar]
- 4. Zhang Y. H., Vasko M. R., Nicol G. D. (2002) Ceramide, a putative second messenger for nerve growth factor, modulates the TTX-resistant Na+ current and delayed rectifier K+ current in rat sensory neurons. J. Physiol. 544, 385–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vane J. R. (1971) Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat. New Biol. 231, 232–235 [DOI] [PubMed] [Google Scholar]
- 6. Simmons D. L., Botting R. M., Hla T. (2004) Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol. Rev. 56, 387–437 [DOI] [PubMed] [Google Scholar]
- 7. Candela M., Barker S. C., Ballou L. R. (1991) Sphingosine synergistically stimulates tumor necrosis factor alpha-induced prostaglandin E2 production in human fibroblasts. J. Exp. Med. 174, 1363–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ballou L. R., Chao C. P., Holness M. A., Barker S. C., Raghow R. (1992) Interleukin-1-mediated PGE2 production and sphingomyelin metabolism. Evidence for the regulation of cyclooxygenase gene expression by sphingosine and ceramide. J. Biol. Chem. 267, 20044–20050 [PubMed] [Google Scholar]
- 9. Hayakawa M., Jayadev S., Tsujimoto M., Hannun Y. A., Ito F. (1996) Role of ceramide in stimulation of the transcription of cytosolic phospholipase A2 and cyclooxygenase 2. Biochem. Biophys. Res. Commun. 220, 681–686 [DOI] [PubMed] [Google Scholar]
- 10. Newton R., Hart L., Chung K. F., Barnes P. J. (2000) Ceramide induction of COX-2 and PGE(2) in pulmonary A549 cells does not involve activation of NF-κB. Biochem. Biophys. Res. Commun. 277, 675–679 [DOI] [PubMed] [Google Scholar]
- 11. Chen C. C., Sun Y. T., Chen J. J., Chang Y. J. (2001) Tumor necrosis factor-alpha-induced cyclooxygenase-2 expression via sequential activation of ceramide-dependent mitogen-activated protein kinases, and IκB kinase 1/2 in human alveolar epithelial cells. Mol. Pharmacol. 59, 493–500 [DOI] [PubMed] [Google Scholar]
- 12. Subbaramaiah K., Chung W. J., Dannenberg A. J. (1998) Ceramide regulates the transcription of cyclooxygenase-2. Evidence for involvement of extracellular signal-regulated kinase/c-Jun N-terminal kinase and p38 mitogen-activated protein kinase pathways. J. Biol. Chem. 273, 32943–32949 [DOI] [PubMed] [Google Scholar]
- 13. Li N., Karin M. (2000) Signaling pathways leading to nuclear factor-κB activation. Methods Enzymol. 319, 273–279 [DOI] [PubMed] [Google Scholar]
- 14. Cohen S., Fleischmann R. (2010) Kinase inhibitors: a new approach to rheumatoid arthritis treatment. Curr. Opin. Rheumatol. 22, 330–335 [DOI] [PubMed] [Google Scholar]
- 15. Machleidt T., Wiegmann K., Henkel T., Schutze S., Baeuerle P., Kronke M. (1994) Sphingomyelinase activates proteolytic IκB-α degradation in a cell-free system. J. Biol. Chem. 269, 13760–13765 [PubMed] [Google Scholar]
- 16. Lozano J., Berra E., Municio M. M., Diaz-Meco M. T., Dominguez I., Sanz L., Moscat J. (1994) Protein kinase C zeta isoform is critical for κB-dependent promoter activation by sphingomyelinase. J. Biol. Chem. 269, 19200–19202 [PubMed] [Google Scholar]
- 17. Majumdar S., Lamothe B., Aggarwal B. B. (2002) Thalidomide suppresses NF-κB activation induced by TNF and H2O2, but not that activated by ceramide, lipopolysaccharides, or phorbol ester. J. Immunol. 168, 2644–2651 [DOI] [PubMed] [Google Scholar]
- 18. Schutze S., Potthoff K., Machleidt T., Berkovic D., Wiegmann K., Kronke M. (1992) TNF activates NF-κB by phosphatidylcholine-specific phospholipase C-induced “acidic” sphingomyelin breakdown. Cell 71, 765–776 [DOI] [PubMed] [Google Scholar]
- 19. Jarvis W. D., Fornari F. A., Jr., Auer K. L., Freemerman A. J., Szabo E., Birrer M. J., Johnson C. R., Barbour S. E., Dent P., Grant S. (1997) Coordinate regulation of stress- and mitogen-activated protein kinases in the apoptotic actions of ceramide and sphingosine. Mol. Pharmacol. 52, 935–947 [DOI] [PubMed] [Google Scholar]
- 20. Brenner B., Koppenhoefer U., Weinstock C., Linderkamp O., Lang F., Gulbins E. (1997) Fas- or ceramide-induced apoptosis is mediated by a Rac1-regulated activation of Jun N-terminal kinase/p38 kinases and GADD153. J. Biol. Chem. 272, 22173–22181 [DOI] [PubMed] [Google Scholar]
- 21. Reunanen N., Westermarck J., Hakkinen L., Holmstrom T. H., Elo I., Eriksson J. E., Kahari V. M. (1998) Enhancement of fibroblast collagenase (matrix metalloproteinase-1) gene expression by ceramide is mediated by extracellular signal-regulated and stress-activated protein kinase pathways. J. Biol. Chem. 273, 5137–5145 [DOI] [PubMed] [Google Scholar]
- 22. Chen C. L., Lin C. F., Chang W. T., Huang W. C., Teng C. F., Lin Y. S. (2008) Ceramide induces p38 MAPK and JNK activation through a mechanism involving a thioredoxin-interacting protein-mediated pathway. Blood 111, 4365–4374 [DOI] [PubMed] [Google Scholar]
- 23. Hargreaves K., Dubner R., Brown F., Flores C., Joris J. (1988) A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 32, 77–88 [DOI] [PubMed] [Google Scholar]
- 24. Randall L. O., Selitto J. J. (1957) A method for measurement of analgesic activity on inflamed tissue. Arch. Int. Pharmacodyn. Ther. 111, 409–419 [PubMed] [Google Scholar]
- 25. Wang Z. Q., Porreca F., Cuzzocrea S., Galen K., Lightfoot R., Masini E., Muscoli C., Mollace V., Ndengele M., Ischiropoulos H., Salvemini D. (2004) A newly identified role for superoxide in inflammatory pain. J. Pharmacol. Exp. Ther. 309, 869–878 [DOI] [PubMed] [Google Scholar]
- 26. Salvemini D., Wang Z. Q., Wyatt P. S., Bourdon D. M., Marino M. H., Manning P. T., Currie M. G. (1996) Nitric oxide: a key mediator in the early and late phase of carrageenan-induced rat paw inflammation. Br. J. Pharmacol. 118, 829–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ndengele M. M., Cuzzocrea S., Esposito E., Mazzon E., Di Paola R., Matuschak G. M., Salvemini D. (2008) Cyclooxygenases 1 and 2 contribute to peroxynitrite-mediated inflammatory pain hypersensitivity. FASEB J. 22, 3154–3164 [DOI] [PubMed] [Google Scholar]
- 28. Futaki N., Yoshikawa K., Hamasaka Y., Arai I., Higuchi S., Iizuka H., Otomo S. (1993) NS-398, a novel non-steroidal anti-inflammatory drug with potent analgesic and antipyretic effects, which causes minimal stomach lesions. Gen. Pharmacol. 24, 105–110 [DOI] [PubMed] [Google Scholar]
- 29. Oliveira M. S., Furian A. F., Royes L. F., Fighera M. R., Fiorenza N. G., Castelli M., Machado P., Bohrer D., Veiga M., Ferreira J., Cavalheiro E. A., Mello C. F. (2008) Cyclooxygenase-2/PGE2 pathway facilitates pentylenetetrazol-induced seizures. Epilepsy Res. 79, 14–21 [DOI] [PubMed] [Google Scholar]
- 30. Brown K., Gerstberger S., Carlson L., Franzoso G., Siebenlist U. (1995) Control of IκB-alpha proteolysis by site-specific, signal-induced phosphorylation. Science 267, 1485–1488 [DOI] [PubMed] [Google Scholar]
- 31. DiDonato J. A., Mercurio F., Karin M. (1995) Phosphorylation of IκBα precedes but is not sufficient for its dissociation from NF-κB. Mol. Cell. Biol. 15, 1302–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kishore N., Sommers C., Mathialagan S., Guzova J., Yao M., Hauser S., Huynh K., Bonar S., Mielke C., Albee L., Weier R., Graneto M., Hanau C., Perry T., Tripp C. S. (2003) A selective IKK-2 inhibitor blocks NF-κB-dependent gene expression in interleukin-1 β-stimulated synovial fibroblasts. J. Biol. Chem. 278, 32861–32871 [DOI] [PubMed] [Google Scholar]
- 33. Cuenda A., Rouse J., Doza Y. N., Meier R., Cohen P., Gallagher T. F., Young P. R., Lee J. C. (1995) SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett. 364, 229–233 [DOI] [PubMed] [Google Scholar]
- 34. Arthur J. S. (2008) MSK activation and physiological roles. Front. Biosci. 13, 5866–5879 [DOI] [PubMed] [Google Scholar]
- 35. Kim H. J., Lee H. S., Chong Y. H., Kang J. L. (2006) p38 Mitogen-activated protein kinase up-regulates LPS-induced NF-κB activation in the development of lung injury and RAW 264.7 macrophages. Toxicology 225, 36–47 [DOI] [PubMed] [Google Scholar]
- 36. Huang C. Y., Lee C. Y., Chen M. Y., Tsai H. C., Hsu H. C., Tang C. H. (2010) Adiponectin increases BMP-2 expression in osteoblasts via AdipoR receptor signaling pathway. J. Cell. Physiol. 224, 475–483 [DOI] [PubMed] [Google Scholar]
- 37. Huang H., Ryu J., Ha J., Chang E. J., Kim H. J., Kim H. M., Kitamura T., Lee Z. H., Kim H. H. (2006) Osteoclast differentiation requires TAK1 and MKK6 for NFATc1 induction and NF-κB transactivation by RANKL. Cell Death Differ. 13, 1879–1891 [DOI] [PubMed] [Google Scholar]
- 38. Vanden Berghe W., Plaisance S., Boone E., De Bosscher K., Schmitz M. L., Fiers W., Haegeman G. (1998) p38 and extracellular signal-regulated kinase mitogen-activated protein kinase pathways are required for nuclear factor-κB p65 transactivation mediated by tumor necrosis factor. J. Biol. Chem. 273, 3285–3290 [DOI] [PubMed] [Google Scholar]
- 39. Cheng Y. J., Hootman J. M., Murphy L. B., Langmaid G. A., Helmick C. G. (2010) Prevelence of doctor-diagnosed arthritis and arthritis-attributable activity limitation-United States, 2007–2009. MMWR Morb. Mortal. Wkly. Rep. 59, 1261–1265 [PubMed] [Google Scholar]
- 40. Hootman J. M., Helmick C. G. (2006) Projections of US prevalence of arthritis and associated activity limitations. Arthritis Rheum. 54, 226–229 [DOI] [PubMed] [Google Scholar]
- 41. Wilcox C. M., Allison J., Benzuly K., Borum M., Cryer B., Grosser T., Hunt R., Ladabaum U., Lanas A., Paulus H., Regueiro C., Sandler R. S., Simon L. (2006) Consensus development conference on the use of nonsteroidal anti-inflammatory agents, including cyclooxygenase-2 enzyme inhibitors and aspirin. Clin. Gastroenterol. Hepatol. 4, 1082–1089 [DOI] [PubMed] [Google Scholar]
- 42. Grosser T., Fries S., FitzGerald G. A. (2006) Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J. Clin. Invest. 116, 4–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Warner T. D., Mitchell J. A. (2004) Cyclooxygenases: new forms, new inhibitors, and lessons from the clinic. FASEB J. 18, 790–804 [DOI] [PubMed] [Google Scholar]
- 44. Mitchell J. A., Warner T. D. (2006) COX isoforms in the cardiovascular system: understanding the activities of non-steroidal anti-inflammatory drugs. Nat. Rev. Drug Discov. 5, 75–86 [DOI] [PubMed] [Google Scholar]
- 45. Chi X. X., Nicol G. D. (2010) The sphingosine 1-phosphate receptor, S1PR1, plays a prominent but not exclusive role in enhancing the excitability of sensory neurons. J Neurophysiol. 104, 2741–2748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ndengele M. M., Cuzzocrea S., Masini E., Vinci M. C., Esposito E., Muscoli C., Petrusca D. N., Mollace V., Mazzon E., Li D., Petrache I., Matuschak G. M., Salvemini D. (2009) Spinal ceramide modulates the development of morphine antinociceptive tolerance via peroxynitrite-mediated nitroxidative stress and neuroimmune activation. J. Pharmacol. Exp. Ther. 329, 64–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bryant L., Doyle T., Chen Z., Cuzzocrea S., Masini E., Vinci M. C., Esposito E., Mazzon E., Petrusca D. N., Petrache I., Salvemini D. (2009) Spinal ceramide and neuronal apoptosis in morphine antinociceptive tolerance. Neurosci. Lett. 463, 49–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Muscoli C., Doyle T., Dagostino C., Bryant L., Chen Z., Watkins L. R., Ryerse J., Bieberich E., Neumman W., Salvemini D. (2010) Counter-regulation of opioid analgesia by glial-derived bioactive sphingolipids. J. Neurosci. 30, 15400–15408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang Y. H., Fehrenbacher J. C., Vasko M. R., Nicol G. D. (2006) Sphingosine-1-phosphate via activation of a G-protein-coupled receptor (s) enhances the excitability of rat sensory neurons. J. Neurophysiol. 96, 1042–1052 [DOI] [PubMed] [Google Scholar]
- 50. Zhang Y. H., Nicol G. D. (2004) NGF-mediated sensitization of the excitability of rat sensory neurons is prevented by a blocking antibody to the p75 neurotrophin receptor. Neurosci. Lett. 366, 187–192 [DOI] [PubMed] [Google Scholar]
- 51. Zhang Y. H., Vasko M. R., Nicol G. D. (2006) Intracellular sphingosine 1-phosphate mediates the increased excitability produced by nerve growth factor in rat sensory neurons. J. Physiol. 575, 101–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tang N., Ong W. Y., Yeo J. F., Farooqui A. A. (2009) Anti-allodynic effect of intracerebroventricularly administered antioxidant and free radical scavenger in a mouse model of orofacial pain. J. Orofac. Pain 23, 167–173 [PubMed] [Google Scholar]
- 53. Collier H. O., Schneider C. (1972) Nociceptive response to prostaglandins and analgesic actions of aspirin and morphine. Nat. New Biol. 236, 141–143 [DOI] [PubMed] [Google Scholar]
- 54. Moncada S., Ferreira S. H., Vane J. R. (1975) Inhibition of prostaglandin biosynthesis as the mechanism of analgesia of aspirin-like drugs in the dog knee joint. Eur. J. Pharmacol. 31, 250–260 [DOI] [PubMed] [Google Scholar]
- 55. Ferreira S. H., Nakamura M., de Abreu Castro M. S. (1978) The hyperalgesic effects of prostacyclin and prostaglandin E2. Prostaglandins 16, 31–37 [DOI] [PubMed] [Google Scholar]
- 56. Williams T. J., Morley J. (1973) Prostaglandins as potentiators of increased vascular permeability in inflammation. Nature 246, 215–217 [DOI] [PubMed] [Google Scholar]
- 57. Willis A. L., Cornelsen M. (1973) Repeated injection of prostaglandin E2 in rat paws induces chronic swelling and a marked decrease in pain threshold. Prostaglandins 3, 353–357 [DOI] [PubMed] [Google Scholar]
- 58. Martin H. A., Basbaum A. I., Kwiat G. C., Goetzl E. J., Levine J. D. (1987) Leukotriene and prostaglandin sensitization of cutaneous high-threshold C- and A-delta mechanonociceptors in the hairy skin of rat hindlimbs. Neuroscience 22, 651–659 [DOI] [PubMed] [Google Scholar]
- 59. Schaible H. G., Schmidt R. F. (1988) Excitation and sensitization of fine articular afferents from cat's knee joint by prostaglandin E2. J. Physiol. 403, 91–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Davis K. D., Meyer R. A., Campbell J. N. (1993) Chemosensitivity and sensitization of nociceptive afferents that innervate the hairy skin of monkey. J. Neurophysiol. 69, 1071–1081 [DOI] [PubMed] [Google Scholar]
- 61. Rueff A., Dray A. (1993) Sensitization of peripheral afferent fibres in the in vitro neonatal rat spinal cord-tail by bradykinin and prostaglandins. Neuroscience 54, 527–535 [DOI] [PubMed] [Google Scholar]
- 62. Taiwo Y. O., Levine J. D. (1990) Direct cutaneous hyperalgesia induced by adenosine. Neuroscience 38, 757–762 [DOI] [PubMed] [Google Scholar]
- 63. Taiwo Y. O., Levine J. D. (1991) Further confirmation of the role of adenyl cyclase and of cAMP-dependent protein kinase in primary afferent hyperalgesia. Neuroscience 44, 131–135 [DOI] [PubMed] [Google Scholar]
- 64. Khasar S. G., Wang J. F., Taiwo Y. O., Heller P. H., Green P. G., Levine J. D. (1995) Mu-opioid agonist enhancement of prostaglandin-induced hyperalgesia in the rat: a G-protein beta gamma subunit-mediated effect? Neuroscience 67, 189–195 [DOI] [PubMed] [Google Scholar]
- 65. Pettus B. J., Kitatani K., Chalfant C. E., Taha T. A., Kawamori T., Bielawski J., Obeid L. M., Hannun Y. A. (2005) The coordination of prostaglandin E2 production by sphingosine-1-phosphate and ceramide-1-phosphate. Mol. Pharmacol. 68, 330–335 [DOI] [PubMed] [Google Scholar]
- 66. Karin M., Ben-Neriah Y. (2000) Phosphorylation meets ubiquitination: the control of NF-κB activity. Annu. Rev. Immunol. 18, 621–663 [DOI] [PubMed] [Google Scholar]
- 67. Neumann M., Naumann M. (2007) Beyond IκBs: alternative regulation of NF-κB activity. FASEB J. 21, 2642–2654 [DOI] [PubMed] [Google Scholar]
- 68. Kefaloyianni E., Gaitanaki C., Beis I. (2006) ERK1/2 and p38-MAPK signalling pathways, through MSK1, are involved in NF-κB transactivation during oxidative stress in skeletal myoblasts. Cell. Signal. 18, 2238–2251 [DOI] [PubMed] [Google Scholar]
- 69. Reber L., Vermeulen L., Haegeman G., Frossard N. (2009) Ser276 phosphorylation of NF-κB p65 by MSK1 controls SCF expression in inflammation. PLoS One 4, e4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wang Z., Ma W., Chabot J. G., Quirion R. (2010) Calcitonin gene-related peptide as a regulator of neuronal CaMKII-CREB, microglial p38-NFκB and astroglial ERK-Stat1/3 cascades mediating the development of tolerance to morphine-induced analgesia. Pain 151, 194–205 [DOI] [PubMed] [Google Scholar]
- 71. Saha R. N., Jana M., Pahan K. (2007) MAPK p38 regulates transcriptional activity of NF-κB in primary human astrocytes via acetylation of p65. J. Immunol. 179, 7101–7109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wang G., Silva J., Krishnamurthy K., Tran E., Condie B. G., Bieberich E. (2005) Direct binding to ceramide activates protein kinase Cζ before the formation of a pro-apoptotic complex with PAR-4 in differentiating stem cells. J. Biol. Chem. 280, 26415–26424 [DOI] [PubMed] [Google Scholar]
- 73. Kitano M., Hla T., Sekiguchi M., Kawahito Y., Yoshimura R., Miyazawa K., Iwasaki T., Sano H., Saba J. D., Tam Y. Y. (2006) Sphingosine 1-phosphate/sphingosine 1-phosphate receptor 1 signaling in rheumatoid synovium: regulation of synovial proliferation and inflammatory gene expression. Arthritis Rheum. 54, 742–753 [DOI] [PubMed] [Google Scholar]
- 74. Gutierrez G., Mendoza C., Montano L. F., Lopez-Marure R. (2007) Ceramide induces early and late apoptosis in human papilloma virus+ cervical cancer cells by inhibiting reactive oxygen species decay, diminishing the intracellular concentration of glutathione and increasing nuclear factor-κB translocation. Anticancer Drugs 18, 149–159 [DOI] [PubMed] [Google Scholar]
- 75. Sauane M., Su Z.-z., Dash R., Liu X., Norris J. S., Sarkar D., Lee S.-G., Allegood J. C., Dent P., Spiegel S., Fisher P. B. (2010) Ceramide plays a prominent role in MDA-7/IL-24-induced cancer-specific apoptosis. J. Cell. Physiol. 222, 546–555 [DOI] [PubMed] [Google Scholar]
- 76. Lei X., Zhang S., Bohrer A., Bao S., Song H., Ramanadham S. (2007) The group via calcium-independent phospholipase A2 participates in ER stress-induced INS-1 insulinoma cell apoptosis by promoting ceramide generation via hydrolysis of sphingomyelins by neutral sphingomyelinase. Biochemistry 46, 10170–10185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hung J.-H., Su I.-J., Lei H.-Y., Wang H.-C., Lin W.-C., Chang W.-T., Huang W., Chang W.-C., Chang Y.-S., Chen C.-C., Lai M.-D. (2004) Endoplasmic reticulum stress stimulates the expression of cyclooxygenase-2 through activation of NF-κB and pp38 mitogen-activated protein kinase. J. Biol. Chem. 279, 46384–46392 [DOI] [PubMed] [Google Scholar]
- 78. Joo J. H., Jetten A. M. (2008) NF-κB-dependent transcriptional activation in lung carcinoma cells by farnesol involves p65/RelA(Ser276) phosphorylation via the MEK-MSK1 signaling pathway. J. Biol. Chem. 283, 16391–16399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Tóth B., Balla A., Ma H., Knight Z. A., Shokat K. M., Balla T. (2006) Phosphatidylinositol 4-kinase IIIβ regulates the transport of ceramide between the endoplasmic reticulum and golgi. J. Biol. Chem. 281, 36369–36377 [DOI] [PubMed] [Google Scholar]
- 80. Ardail D., Popa I., Bodennec J., Famy C., Louisot P., Portoukalian J. (2002) Subcellular distribution and metabolic fate of exogenous ceramides taken up by HL-60 cells. Biochim. Biophys. Acta 1583, 305–310 [DOI] [PubMed] [Google Scholar]
- 81. Senkal C. E., Ponnusamy S., Bielawski J., Hannun Y. A., Ogretmen B. (2010) Antiapoptotic roles of ceramide-synthase-6-generated C16-ceramide via selective regulation of the ATF6/CHOP arm of ER-stress-response pathways. FASEB J. 24, 296–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Camandola S., Cutler R. G., Gary D. S., Milhavet O., Mattson M. P. (2005) Suppression of calcium release from inositol 1,4,5-trisphosphate-sensitive stores mediates the anti-apoptotic function of nuclear factor-κB. J. Biol. Chem. 280, 22287–22296 [DOI] [PubMed] [Google Scholar]
- 83. Bischof L. J., Kao C. Y., Los F. C., Gonzalez M. R., Shen Z., Briggs S. P., van der Goot F. G., Aroian R. V. (2008) Activation of the unfolded protein response is required for defenses against bacterial pore-forming toxin in vivo. PLoS Pathog. 4, e1000176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Martinon F., Chen X., Lee A.-H., Glimcher L. H. (2010) TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat. Immunol. 11, 411–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ichijo H., Nishida E., Irie K., ten Dijke P., Saitoh M., Moriguchi T., Takagi M., Matsumoto K., Miyazono K., Gotoh Y. (1997) Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 275, 90–94 [DOI] [PubMed] [Google Scholar]
- 86. Cheng J. K., Ji R. R. (2008) Intracellular signaling in primary sensory neurons and persistent pain. Neurochem. Res. 33, 1970–1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Schafers M., Lee D. H., Brors D., Yaksh T. L., Sorkin L. S. (2003) Increased sensitivity of injured and adjacent uninjured rat primary sensory neurons to exogenous tumor necrosis factor-alpha after spinal nerve ligation. J. Neurosci. 23, 3028–3038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Schafers M., Sommer C., Geis C., Hagenacker T., Vandenabeele P., Sorkin L. S. (2008) Selective stimulation of either tumor necrosis factor receptor differentially induces pain behavior in vivo and ectopic activity in sensory neurons in vitro. Neuroscience 157, 414–423 [DOI] [PubMed] [Google Scholar]