

Abstract
Fatostatin, a recently discovered small molecule that inhibits activation of sterol regulatory element-binding protein (SREBP), blocks biosynthesis and accumulation of fat in obese mice. The present study synthesized and evaluated a series of fatostatin derivatives. Our structure-activity relationships led to the identification of N-(4-(2-(2-propylpyridin-4-yl)thiazol-4-yl)phenyl)methanesulfonamide (24; FGH10019) as the most potent drug-like molecule among the analogs tested. Compound 24 has high aqueous solubility and membrane permeability, and may serve as a seed molecule for further development.
Introduction
Long-chain fatty acids are major sources of energy and important components of the lipids that compose cellular membranes. De novo lipid synthesis is activated in some types of cancers, and synthesized lipids are accumulated in the body in metabolic syndrome. Therefore, pharmacological intervention for de novo lipid synthesis may prove to be effective against a number of human diseases. The conversion of carbohydrates into lipids through de novo fatty acid involves at least twelve enzymatic reactions, and conversion through cholesterol synthesis involves at least twenty-three. Expression levels of the genes encoding these enzymes are controlled by transcription factors, which are designated sterol regulatory element-binding proteins (SREBPs).1, 2 SREBPs are synthesized as ER membrane-bound precursors, and must be proteolytically released by two proteases bound to the Golgi membrane – Site-1 (S1P) and Site-2 (S2P) proteases – to generate forms that activate transcription of target genes in the nucleus.1, 3 Activation of SREBPs is tightly regulated by negative feedback: sterols interact with SREBP cleavage-activating protein (SCAP), an ER membrane-bound escort protein of SREBPs, thereby retaining the SCAP/SREBP complex in the ER.4, 5 Binding of the sterol to SCAP promotes the interaction of SCAP with INSIG, a retention protein in the ER.6, 7 Thus, SREBPs are key lipogenic transcription factors that govern the homeostasis of fat metabolism, and SCAP plays a pivotal role in the regulation of SREBPs.
We previously described a synthetic diaryl thiazole molecule, fatostatin (molecule 1, Figure 1), and identified its target. Fatostatin (125B11) was originally discovered from a chemical library as a synthetic small molecule that inhibited insulin-induced adipogenesis and serum-independent growth of cancer cells in cell culture.8 The molecule was later shown to selectively reduce the expression of SREBP target genes by blocking the activation of SREBP transcription factors.9 The direct target of fatostatin appears to be SCAP; however, fatostatin does not affect the interaction of SCAP with SREBP-2 or insulin-induced gene (INSIG), and binds to SCAP at a site distinct from that of sterols.9 Thus, fatostatin is a unique small molecule that blocks the activation of SREBPs independently of INSIG and sterols. Fatostatin or its analogs may serve as research tools for investigating the SREBP/SCAP pathway.
Figure 1.
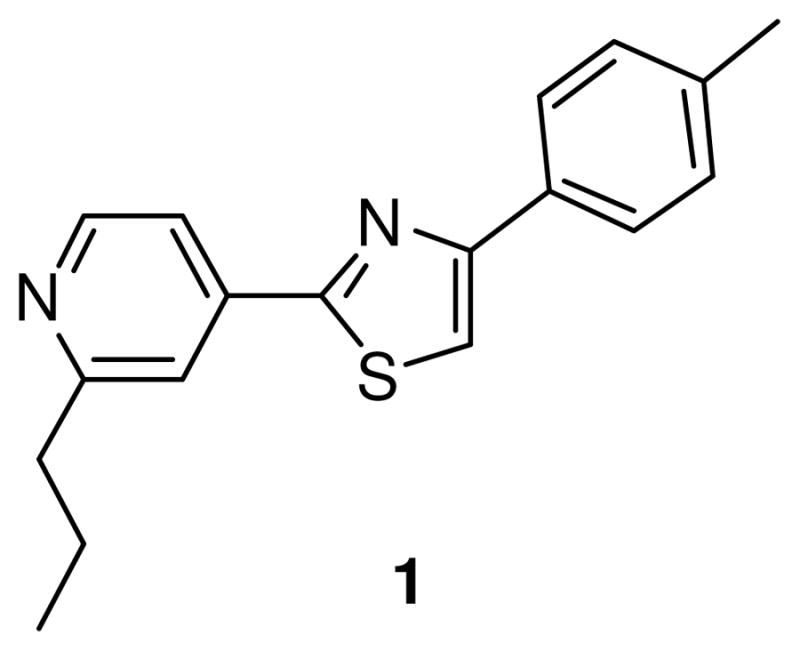
Structure of fatostatin (1).
The structure of fatostatin also provides a model that may help direct the design of molecules for pharmacological intervention against cancer progression and metabolic diseases, including fatty liver disease. In fact, fatostatin downregulated the expression of lipogenic enzymes and blocked increases in body weight, blood glucose, and hepatic fat accumulation in obese ob/ob mice, even under uncontrolled food intake.9 However, fatostatin showed only moderate potency in mice, and its utility was limited by low aqueous solubility. The present study synthesized a number of fatostatin derivatives and compared their potency and physicochemical properties, with the goal of identifying an analog with improved characteristics for use in further in vivo evaluation in a variety of disease models.
Chemistry
Fatostatin is composed of three aromatic rings: pyridine, thiazole and toluene. Four routes were used to synthesize fatostatin derivatives (Schemes 1–4). In the first route, a central thiazole ring was constructed by reacting various thioamide derivatives with α-bromoketone derivatives (Scheme 1). Thioamide 2 or 3 was coupled with an α-bromoketone precursor in warm ethanol to yield 4-phenyl-2-(pyridin-4-yl)thiazole derivatives (analogs 4–9). The products crystallized directly from the reaction mixtures were filtered to provide high yields of pure thiazoles. Analog 10 was synthesized by the reaction of 3 with 2-bromo-1-(thiophen-2- yl)ethanone. Diphenyl thiazole 12 was similarly synthesized from benzothioamide (11) and 2-bromo-1- p-tolylethanone.
Scheme 1.

Synthesis of analogs 4–10, 12.
Scheme 4.

Synthesis of analogs 20–26.a
aReagents and conditions: (a) acetone, benzaldehyde or cyclopropanecarboxaldehyde, AcOH, Na(AcO)3BH, CH2Cl2, room temperature, 19–21 h, 52–73% (b) acetyl chloride, methanesulfonyl chloride, thiophene sulfonylchloride or tosyl chloride, pyridine, CH2Cl2, 0°C to room temperature, 0.5–72 h, 14–87%
To prepare analog 15, 4- bromo-2-propylpridine (14) was first synthesized from 4-bromopyridine (13), as previously described.10 Suzuki coupling of bromide 14 with 4′-methyl-4- biphenylboronic acid produced analog 15 (Scheme 2).
Scheme 2.

Synthesis of analog 15.a
aReagents and conditions: (a) 4′-methyl-4- biphenylboronic acid, (Ph3P)4Pd, K2CO3, DMF, H2O, 80°C, 18 h, 52%
Phenol derivatives (Scheme 3) were prepared by alkaline hydrolysis of analog 6, producing the p-phenol derivative 16, or demethylation of methyl ether 8 and 9 by BBr3, producing the m-phenol derivative 17 and o-phenol derivative 18.
Scheme 3.

Synthesis of analogs 16–18.a
aReagents and conditions: (a) KOH aq. THF, MeOH, 60°C, 0.5 h, 80% (b) BBr3, CH2Cl2, 0°C to room temperature, 6–14 h, 16–40%
Amine, amide, and sulfonamide derivatives were also synthesized (Scheme 4). Isopropyl amine 20, benzyl amine 21, and cyclopropylmethyl amine 22 were prepared in high yields by a direct reductive amination reaction of acetone, benzaldehyde, or cyclopropanecarboxaldehyde with a previously prepared primary amine 19.11 Amine 19 was also reacted with acetyl chloride, methane sulfonylchloride, thiophene sulfonylchloride, or tosyl chloride in the presence of pyridine to yield acetoamide 23, methane sulfoneamide 24, thiophene sulfoneamide 25, and toluene sulfoneamide 26, respectively.
Results and Discussion
All of the synthesized fatostatin derivatives were assayed for their ability to inhibit the expression of a luciferase reporter gene, in which expression of luciferase was controlled by three repeats of sterol regulatory elements (SREs). This SREBP-responsive reporter construct was co-transfected into Chinese hamster ovary-K1 (CHOK1) cells with a control β-gal reporter gene in which the expression of β-gal was driven by a constitutively active actin promoter. Luciferase expression from the SREBP-responsive reporter gene, normalized to β-gal expression, was activated upon lipid depletion.9 Fatostatin decreased activation of the reporter gene with an IC50 value of 5.6 μM (Figure 2), which is consistent with results of previous studies.9
Figure 2.

Inhibitory activities of fatostatin (1) and analogs 10, 12, and 15 against the activation of SREBP in the luciferase reporter assay.
Replacement of any one of fatostatin’s three aromatic rings with simpler aromatic structures affected activity. For example, analog 10, a thiophene substitution, had an IC50 value of 14.7 μM in the reporter assay (Figure 2). Replacement of the 2-propylpridine group with phenyl group (analog 12) resulted in a 3.3-fold decrease in potency. Analog 15, which lacked the central thiazole of fatostatin, showed a large loss of activity, suggesting that the central thiazole moiety is important.
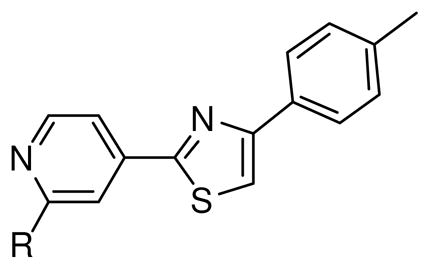
Changing the length of the alkyl chain at the 2-position of pyridine moiety also influenced activity of the fatostatin derivatives. Activity of the ethyl derivative (analog 4a) was almost the same as that of fatostatin (Table 1). However, removal of n-propyl group (analog 4b) resulted in reduced activity, suggesting that a lipophilic alkyl group at this position is essential.
Table 1.
Effects of pyridine group substitutions on the activation of SREBP.
 | ||
|---|---|---|
| Compound | R | IC50 (μM)a |
| 1 | n-Pr | 5.6 ± 2.9 |
| 4a | Et | 6.8 ± 3.4 |
| 4b | H | >30 |
All values are mean ± standard deviation, n ≥ 3.
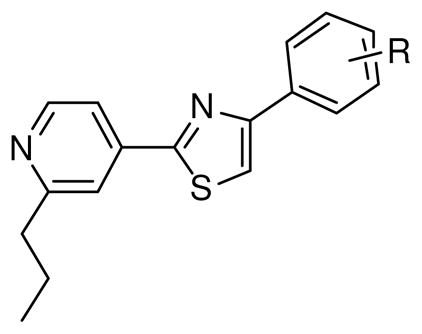
The methyl group of toluene is often oxidized in vivo by cytochrome P450. Therefore, the effects of substitutions of the toluene moiety of fatostatin were examined (analogs 5- 9 and 16–18). Removal of the methyl group in the toluene moiety (analog 5a) led to a fourfold decrease in potency (Table 2). Halogen substitutions (analogs 5b-d), which are likely to be more metabolically stable than fatostatin, showed moderate activity. Incorporation of a large benzoyl group (analog 6) resulted in potency similar to that of the halogenated derivatives. Hydrophilic substitutions, such as hydroxy and methoxy groups (analogs 7 and 16) also failed to increase activity.
Table 2.
Effects of phenyl group substitutions on the activation of SREBP.
 | ||
|---|---|---|
| Compound | R | IC50 (μM)a |
| 5a | H | 23.0 ± 5.6 |
| 5b | 4-F | 12.4 ± 3.1 |
| 5c | 4-Cl | 10.7 ± 0.9 |
| 5d | 4-Br | 9.8 ± 0.6 |
| 6 | 4-OBz | 7.7 ± 1.0 |
| 7 | 4-OMe | 13.0 ± 3.3 |
| 8 | 3-OMe | 10.2 ± 1.2 |
| 9 | 2-OMe | 3.2 ± 0.4 |
| 16 | 4-OH | 9.6 ± 1.2 |
| 17 | 3-OH | 9.5 ± 0.4 |
| 18 | 2-OH | 14.4 ± 0.2 |
All values are mean ± standard deviation, n ≥ 3.
Effects of substitution at 2- and 3- positions of the benzene ring were examined, as well. 3- Methoxy, 3-hydroxy, and 2-hydroxy derivatives (analogs 8, 17, and 18) were less potent than fatostatin. However, the 2-methoxy derivative (analog 9) exhibited a slight increase in activity (Table 2), suggesting that substitution of the toluene moiety of fatostatin might further optimize potency and aqueous solubility. Therefore, further efforts were focused on substitution at the 4- position of the toluene moiety.
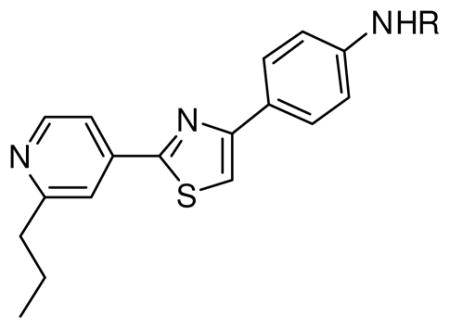
A variety of basic amine groups were introduced in the 4-position of the toluene moiety. The potency of an aniline derivative (analog 19) was similar to that of fatostatin, and secondary amine derivatives (analogs 20–22) showed slight improvement in activity (Table 3). Introduction of amide (analog 23) or carbamate (analog 27)9 did not significantly improve potency; however, addition of sulfonamide (analogs 24–26) increased activity. Analog 24, with methanesulfonamide, exhibited the most potent activity: an IC50 value of 0.7 μM, comparable to the potency of an endogenous inhibitor, 25- hydroxycholesterol, which had an IC50 value of 0.3 μM.12
Table 3.
Effects of N-substitution on the activation of SREBP.
 | ||
|---|---|---|
| Compound | R | IC50 (μM)a |
| 19 | H | 8.6 ± 2.0 |
| 20 | Isopropyl | 2.8 ± 0.4 |
| 21 | Benzyl | 3.0 ± 0.1 |
| 22 | Methylcyclopropyl | 4.9 ± 3.1 |
| 23 | Acetyl | 7.1 ± 1.5 |
| 24 | Methansulfonyl | 0.7 ± 0.2 |
| 25 | Thiophenesulfonyl | 0.9 ± 0.5 |
| 26 | Tosyl | 2.3 ± 0.0 |
| 27 | t-Butoxycarbonyl | 1.9 ± 0.2 |
| 25-hydroxy- cholesterol | 0.3 ± 0.0 | |
All values are mean ± standard deviation, n ≥ 3.
The inhibition of SREBP activation mediated by analog 24 was confirmed by Western blot analysis of SREBP-2 (Figure 3). Treatment of the CHO-K1 cells with analog 24 decreased the percentage of the mature form of SREBP-2 (68 kDa) at lower concentrations than treatment with fatostatin. Densitometric analysis of the gels indicated that the IC50 of analog 24 was approximately 1 μM, which is 5–10 times lower than the IC50 of fatostatin (~10 μM). The IC50 values were consistent with the values obtained via the reporter gene assays.
Figure 3.

Effects of fatostatin (1) and analog 24 on the proteolytic activation of SREBP-2 in cells. Top: western blot analysis of SREBP-2. CHO-K1 cells were treated with 1% (v/v) DMSO (vehicle), varied concentrations (1, 5, and 20 μM) of fatostatin (1) or compound 24, or sterols (10 μg mL−1 cholesterol or 1 μg mL−1 25- hydroxycholesterol). The precursor and mature forms of SREBP-2 were detected by an SREBP-2 antibody. The positions of the precursor and mature forms of SREBP- 2 are indicated. Bottom: quantification of the western blot analysis. Percentages of the mature form of SREBP-2 are shown. Values are means ± S.D. of three independent experiments.
Fatostatin selectively downregulates endogenous SREBP-responsive genes in cultured human prostate cancer cells.9 Real-time RT-PCR analysis of nine representative SREBPresponsive genes13 was carried out to determine the selectivity of analog 24. Transcription of all nine genes was downregulated by at least 25% (Figure 4). In contrast, treatment with analog 24 had no significant effect on transcription of three control genes unrelated to SREBP. Thus, the mode of action of analog 24 appears to be similar to that of fatostatin.
Figure 4.

Effects of analog 24 on the expression of SREBP-responsive genes in DU145 cells: mevalonate kinase, MVK; HMG-CoA reductase, HMGCR; ATP citrate lyase, ACL; insulin induced gene 1, INSIG1; mevalonate pyrophosphate decarboxylase, MVD; HMGCoA synthase 1, HMGCS; isopentenyl-diphosphate delta isomerase 1, IDI1; stearoyl-CoA desaturase, SCD1; LDL receptor, LDLR; ribosomal protein L13a, RPL13A; beta- 2 microglobulin, B2M; glyceraldehyde-3-phosphate dehydrogenase, GAPDH. Values are means ± SD of two independent experiments.
The in silico properties calculated for fatostatin and analogs 20 and 24 suggested that analog 24 has potential for further pharmaceutical development (see Supporting Information, Table S1). Typically, compounds that violate one or more of the “rule-of-five” – molecular weight >500 Da, logP >5, more than 5 H-bond donors, and more than 10 H-bond acceptors – are poorly absorbed.14 Fatostatin and analog 20 violated one “rule-of-five”; their clogP values were 5.72 and 5.82, respectively. Analog 24 had a clogP value of 3.97, and a polar surface area of 108.57 Å2. As expected, the in vitro aqueous solubility of analog 24 at pH 7 was more than 10 times higher than that of fatostatin or analog 20 (Table 4).
Table 4.
In vitro aqueous solubility at pH 3 and 7
| Compound | Solubility pH 3 (μM) | Solubility pH 7 (μM) |
|---|---|---|
| 1 | 80.0 | 2.0 |
| 20 | >225 | 3.0 |
| 24 | >225 | 33.0 |
We examined other physicochemical properties of fatostatin and analogs 20 and 24. The intrinsic clearance rates measured in mouse hepatocytes were 606, 272, and 137 ml min−1 kg−1, respectively, and the corresponding half-lives (t1/2) were 10.1, 22.4 and 44.4 min. Although fatostatin and analog 20 showed low metabolic stability in mouse hepatocytes, analog 24 was moderately stable.
Results of parallel artificial membrane permeability assays (PAMPA) indicated that passive permeability through an artificial membrane was low for fatostatin, moderate for analog 20, and high for analog 24 (Table 5). Thus, analog 24 is likely to be have greater permeability in vivo than fatostatin or analog 20. Warfarin (10 μM) was used as a control for the integrity of membrane. Retention of all compounds within the membrane was greater than 90%, probably due to the high lipophilicity of the compounds.
Table 5.
PAMPA permeability results for fatostatin (1) and analogs 20 and 24
| Compound | PAMPA Papp (10−6 cm/sec) | % Membrane retention | Reference ID | Reference PAMPA Papp (10−6 cm/sec) | Reference % membrane retention |
|---|---|---|---|---|---|
| 1 | 0.7555 | 94 | Warfarin | 4.11 | 0 |
| 20 | 1.059 | 93.95 | Warfarin | 5.34 | 0 |
| 24 | 14.98 | 97.35 | Warfarin | 5.58 | 0 |
We previously demonstrated that intraperitoneal injection of fatostatin blocked increases in body weight, blood glucose and hepatic fat accumulation in obese ob/ob mice, even under conditions of uncontrolled food intake.9 However, fatostatin was not suited for oral administration and further animal studies. To demonstrate oral availability of analog 24, we provided ob/ob mice with ad libitum access over an 8-wk period to normal chow that contained analog 24. The analog 24-treated chow was fed at a dose rate calculated to provide about 0.7 mg analog 24 per day, at about 23 mg/kg body weight, to 5-wk-old male ob/ob mice weighing an average of ~30 g. After 8 wk on the analog 24-treated chow, the mice gained 8–9 % less weight than control mice which were fed the same chow minus the compound (37.9 ± 0.9 vs. 41.4 ± 1.1 g/mouse, respectively) (Table 6). Food intake was slightly lower in the treated mice compared to controls (3.4 ± 0.54 vs. 3.6 ± 0.54 g/mouse per day.) Blood glucose was lower in the treated mice vs. controls (214 ± 16 vs 243.7 ± 27.6 mg/dl) (Table 6). The serum level of total cholesterol was lower in the treated mice compared to controls (220 ± 11 vs. 285 ± 8 mg/dl). There was a ~37% decrease in the level of LDL in the serum of treated mice compared to controls (50.2 ± 4.4 vs. 79.38 ± 2.85 mg/dl); however, no detectable change was observed in the HDL level in the serum of treated mice compared to controls (164.5 ± 5.3 vs. 164.83 ± 4.5 mg/dl) (Table 6). The triglyceride level in the liver tissues was significantly lower in the treated mice compared to controls, suggesting fatty liver conditions were ameliorated in the treated group (28 ± 1.0 vs. 42 ± 2.0 mg/g tissue). These results indicate that analog 24 is orally available in mice.
Table 6.
Effect of feeding ob/ob mice analog 24a
| Control | Treated | |
|---|---|---|
| Body weight (g/mouse) | 41.4 ± 1.1 | 37.9 ± 0.9* |
| Glucose (mg/dl) | 243.7 ± 27.6 | 214 ± 16# |
| Cholesterol (dl) | 285 ± 8 | 220 ± 11* |
| HDL (mg/dl) | 164.83 ± 4.5 | 164.5 ± 5.3 |
| LDL (mg/dl) | 79.38 ± 2.85 | 50.2 ± 4.4* |
| TG in liver (mg/g tissue) | 42 ± 2 | 28 ± 1* |
Male ob/ob mice were fed either normal chow (controls) or normal chow with compound analog 24 for 8 wk. Body weight and food intake was monitored and recorded daily. At the end of the experiments, serum constituents were determined and TG levels in liver were determined as we described previously.9
HDL: high-density lipoprotein; LDL: low-density lipoprotein; TG: triglycerides. Values are mean ± SE (n = 10):
P < 0.05;
P = 0.09.
Overall, analog 24, a methanesulfonamide derivative of fatostatin, exhibited the highest activity in a cell-based assay, and exhibited better in vitro and in vivo physicochemical properties than fatostatin or the other derivatives that were synthesized and evaluated. Analog 24 (FGH10019) is the most appropriate molecule for further testing in animal models, and this testing is currently under way.
Experimental Section
General
The general chemistry, experimental information, and syntheses of all other compounds are described in the Supporting Information. All tested compounds were >95% purity, as determined by examination of LC chromatogram (wavelength 235 nm) (Table S2) and/or combustion analysis (Table S3).
4-(2-Methoxyphenyl)-2-(2-propylpyridin-4-yl)thiazole (9)
A mixture of prothionamide (3) (700 mg, 3.88 mmol) and 2-bromo-1-(2-methoxyphenyl)ethanone (890 mg, 3.89 mmol) in ethanol (15 mL) was stirred for 0.5 h at 80°C, then cooled to 0°C. The yellow precipitate was filtered off and washed with cold ethanol. The residue was partitioned between EtOAc and saturated NaHCO3 solution. The aqueous phase was extracted with EtOAc. The combined extracts were dried over Na2SO4 and concentrated to produce compound 9 (742 mg, 62%) as a yellow foam. 1H NMR (300 MHz, CDCl3): δ 8.60 (d, J = 5.2 Hz, 1H), 8.38 (d, J = 7.7 Hz, 1H), 8.06 (s, 1H), 7.78 (s, 1H), 7.71 (d, J = 5.2 Hz, 1H), 7.33 (t, J = 7.7 Hz, 1H), 7.09 (t, J = 7.7 Hz, 1H), 7.01 (d, J = 7.7 Hz, 1H), 3.95 (s, 3H), 2.87 (t, J = 7.7 Hz, 2H), 1.83 (m, 2H), 1.00 (t, J = 7.4 Hz, 3H); m/z = 311 [M+H]+. Anal. (C18H18N2OS) C, H, N, S.
N-Isopropyl-4-(2-(2-propylpyridin-4-yl)thiazol-4-yl)aniline (20)
Acetone (2.5 mL, 34.5 mmol) and acetic acid (2.0 mL, 34.5 mmol) were added to a stirred solution of compound 19 (1.02 g, 3.45 mmol) in CH2Cl2 (20 mL). After stirring for 1 h, Na(AcO)3BH (1.5 g, 6.9 mmol) was added, and the reaction mixture was stirred for 20 h. The reaction mixture was poured into saturated NaHCO3 solution, and extracted with EtOAc. The combined extracts were dried over Na2SO4, and concentrated. Chromatography of the crude product was (SiO2, 4:1 hexane: EtOAc) produced compound 20 (845 mg, 73%) as a white foam. 1H NMR (300 MHz, CDCl3): δ 8.61 (d, J = 5.2 Hz, 1H), 7.81 (d, J = 8.5 Hz, 2H), 7.76 (d, J = 1.4 Hz, 1H), 7.68 (dd, J = 1.4, 5.2 Hz, 1H), 7.34 (s, 1H), 6.65 (d, J = 8.5 Hz, 2H), 3.70 (m, 1H), 2.86 (t, J = 7.6 Hz, 2H), 1.83 (m, 2H), 1.25 (d, J = 6.0 Hz, 6H), 1.02 (t, J = 7.4 Hz, 3H); m/z = 338 [M+H]+. Anal. (C20H23N3S) C, H, N, S.
N-(4-(2-(2-Propylpyridin-4-yl)thiazol-4-yl)phenyl)methanesulfonamide (24)
Methanesulfonyl chloride (0.23 mL, 2.97 mmol) was added to a stirred solution of compound 19 (800 mg, 2.71 mmol) and pyridine (0.66 mL, 8.1 mmol) in CH2Cl2 (20 mL) at 0°C. After stirring for 0.5 h, the reaction mixture was poured into 2 M citric acid solution and extracted with EtOAc. The combined extracts were washed with saturated NaHCO3 solution and brine, dried over Na2SO4, and concentrated to produce compound 24 (880 mg, 87%) as a yellow foam. 1H NMR (300 MHz, CD3OD): δ 8.55 (d, J = 5.2 Hz, 1H), 8.02 (d, J = 8.8 Hz, 2H), 7.95 (s, 1H), 7.90 (d, J = 1.9 Hz, 1H), 7.84 (dd, J = 1.9, 5.2 Hz, 1H), 7.34 (d, J = 8.8 Hz, 2H), 3.00 (s, 3H), 2.86 (t, J = 7.7 Hz, 2H), 1.80 (m, 2H), 1.01 (t, J = 7.3 Hz, 3H); m/z = 374 [M+H]+. Anal. (C18H19N3O2S2) C, H, N, S.
Supplementary Material
Acknowledgments
This work was supported in part by grants to M.U. from the Hoh-ansha Foundation and MEXT (Grant-in-Aid 21310140), and by grants to S.J.W. from the Hefni Technical Training Foundation and the National Institutes of Health (GM-63115). We also thank J. Sakai for an SREBP-2 antibody and a reporter gene construct, and T. Orihara, T. Morii, and T. Hasegawa for experimental support. S.K. is a postdoctoral fellow of JSPS. The Kyoto research group participates in the Global COE program “Integrated Materials Science” (#B-09).
Abbreviations
- SREBP
sterol regulatory element-binding protein
- S1P
site-1 protease
- S2P
site-2 protease
- SCAP
SREBP cleavage-activating protein
- CHO-K1
Chinese hamster ovary- K1
- PAMPA
parallel artificial membrane permeability assay
- MVK
mevalonate kinase
- HMGCR
HMG-CoA reductase
- ACL
ATP citrate lyase
- INSIG1
insulin-induced gene 1
- MVD
mevalonate pyrophosphate decarboxylase
- HMGCS1
HMG-CoA synthase 1
- IDI1
isopentenyl-diphosphate delta isomerase 1
- SCD1
stearoyl-CoA desaturase
- LDLR
low-density lipoprotein receptor
- RPL13A
ribosomal protein; L13a
- B2M
Beta-2 microglobulin
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- HDL
high-density lipoprotein
- TG
triglycerides
Footnotes
Conflict of interest statement: L.A.E., S.J.W., and M.U. have signed a scientific advisory agreement with a company founded to develop the technology described in this article.
Supporting Information Available: Supplementary data, synthesis of compounds 4-8, 10, 12, 15-18, 21-23, 25, and 26, supplementary biological assay methods. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89:331–340. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- 2.Osborne TF. Sterol regulatory element-binding proteins (SREBPs): key regulators of nutritional homeostasis and insulin action. J Biol Chem. 2000;275:32379–32382. doi: 10.1074/jbc.R000017200. [DOI] [PubMed] [Google Scholar]
- 3.Sakai J, Duncan EA, Rawson RB, Hua X, Brown MS, Goldstein JL. Sterolregulated release of SREBP-2 from cell membranes requires two sequential cleavages, one within a transmembrane segment. Cell. 1996;85:1037–1046. doi: 10.1016/s0092-8674(00)81304-5. [DOI] [PubMed] [Google Scholar]
- 4.Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell. 2006;124:35–46. doi: 10.1016/j.cell.2005.12.022. [DOI] [PubMed] [Google Scholar]
- 5.Hua X, Nohturfft A, Goldstein JL, Brown MS. Sterol resistance in CHO cells traced to point mutation in SREBP cleavage-activating protein. Cell. 1996;87:415–426. doi: 10.1016/s0092-8674(00)81362-8. [DOI] [PubMed] [Google Scholar]
- 6.Radhakrishnan A, Sun LP, Kwon HJ, Brown MS, Goldstein JL. Direct binding of cholesterol to the purified membrane region of SCAP: mechanism for a sterol-sensing domain. Mol Cell. 2004;15:259–268. doi: 10.1016/j.molcel.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 7.Yang T, Espenshade PJ, Wright ME, Yabe D, Gong Y, Aebersold R, Goldstein JL, Brown MS. Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell. 2002;110:489–500. doi: 10.1016/s0092-8674(02)00872-3. [DOI] [PubMed] [Google Scholar]
- 8.Choi Y, Kawazoe Y, Murakami K, Misawa H, Uesugi M. Identification of bioactive molecules by adipogenesis profiling of organic compounds. J Biol Chem. 2003;278:7320–7324. doi: 10.1074/jbc.M210283200. [DOI] [PubMed] [Google Scholar]
- 9.Kamisuki S, Mao Q, Abu-Elheiga L, Gu Z, Kugimiya A, Kwon Y, Shinohara T, Kawazoe Y, Sato S, Asakura K, Choo HY, Sakai J, Wakil SJ, Uesugi M. A small molecule that blocks fat synthesis by inhibiting the activation of SREBP. Chem Biol. 2009;16:882–892. doi: 10.1016/j.chembiol.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 10.Comins DL, Mantlo NB. Regiospecific alpha-alkylation of 4-chloro(bromo) pyridine. J Org Chem. 1985;50:4410–4411. [Google Scholar]
- 11.Bilgin AA. 2-Pyridylthiazoles II, synthesis and structure elucidations. Acta Pharm Turcica. 1988;30:133–138. [Google Scholar]
- 12.Adams CM, Reitz J, De Brabander JK, Feramisco JD, Li L, Brown MS, Goldstein JL. Cholesterol and 25-hydroxycholesterol inhibit activation of SREBPs by different mechanisms, both involving SCAP and Insigs. J Biol Chem. 2004;279:52772–52780. doi: 10.1074/jbc.M410302200. [DOI] [PubMed] [Google Scholar]
- 13.Horton JD, Shah NA, Warrington JA, Anderson NN, Park SW, Brown MS, Goldstein JL. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc Natl Acad Sci U S A. 2003;100:12027–12032. doi: 10.1073/pnas.1534923100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.