

Abstract
A gene cluster responsible for the biosynthesis of the antitumor agent cetoniacytone A was identified in Actinomyces sp. strain Lu 9419, an endosymbiotic bacteria isolated from the intestines of the rose chafer beetle (Cetonia aurata). The nucleotide sequence analysis of the 46 kb DNA region revealed the presence of 31 complete ORFs, including genes predicted to encode a 2-epi-5-epi-valiolone synthase (CetA), a glyoxalase/bleomycin resistance protein (CetB), an acyltransferase (CetD), an FAD-dependent dehydrogenase (CetF2), two oxidoreductases (CetF1 and CetG), two aminotransferases (CetH and CetM), and a pyranose oxidase (CetL). CetA has previously been demonstrated to catalyze the cyclization of sedoheptulose 7-phosphate to the cyclic intermediate, 2-epi-5-epi-valiolone. In this report, the glyoxalase/bleomycin resistance protein homolog CetB was identified as a 2-epi-5-epi-valiolone epimerase (EVE), a new member of the Vicinal Oxygen Chelate (VOC) superfamily. The 24 kDa recombinant histidine-tagged CetB was found to form a homodimer; each monomer contains two βαβββ scaffolds that form a metal binding site with two histidine and two glutamic acid residues. BLAST search using the newly isolated cet biosynthetic genes revealed an analogous suite of genes in the genome of Frankia alni ACN14a, suggesting that this plant symbiotic nitrogen-fixing bacterium is capable of producing a secondary metabolite related to the cetoniacytones.
Keywords: biosynthesis, aminocyclitol, cetoniacytone, epimerase, endosymbiotic, vicinal oxygen chelate
Introduction
Soil bacteria have long been recognized to be one of the major producers of secondary metabolites with diverse biological activities. They produce a wide variety of natural products including polyketides, polypeptides, aminoglycosides and aminocoumarin antibiotics. In addition to soil microorganisms, there is a growing body of evidence demonstrating that many secondary metabolites are produced by endosymbiotic bacteria living in plants (e.g., coronamycin),[1] insects (e.g., pederin),[2] and marine animals (e.g., bryostatin).[3] Among them are the cetoniacytones, a group of antibiotics produced by an endosymbiotic Actinomyces sp. strain Lu 9419 living in the intestines of the rose chaffer beetle (Cetonia aurata).[4] The cetoniacytones contain a unique C7N-aminocyclitol moiety as their central core structure (Scheme 1). In contrast to most of the secondary metabolites belonging to the C7N aminocyclitol family,[5] which normally have an alkylated nitrogen atom at the C-1 position (e.g., validamycin A), the amino group in the cetoniacytones is acetylated and located at the C-2 position. The same core structure is found in the anti-rheumatoid arthritis agents, the epoxyquinomicins, which were isolated from the culture broth of Amycolatopsis sp. strain MK 299-95F4.[6] To some extent, they also share similar structural features with the neuraminidase inhibitor, oseltamivir (Tamiflu®),[7] which is a semi-synthetic drug derived from shikimic acid that is widely used for the prevention and treatment of influenza.
Scheme 1.

Chemical structure of cetoniacytone A and related compounds.
Structurally, the core unit of cetoniacytone A is also similar to (+)-isoepoxidon, a polyketide-derived precursor of patulin.[8] However, preliminary feeding experiments using [U-13C]glycerol suggested that the core moiety of cetoniacytone is derived from the pentose phosphate pathway.[4] The pentose phosphate pathway has also been implicated with the biosynthesis of the antidiabetic agent acarbose[9] and the antifungal agent validamycin A, both of which have valienamine as a common core unit. More recent studies have demonstrated the involvement of sedoheptulose 7-phosphate and its cyclization product 2-epi-5-epi-valiolone in the biosynthesis of acarbose, validamycin, and pyralomicin.[10, 11] While the core structure of cetoniacytone is different from valienamine, labeling and coupling patterns from feeding experiments with [U-13C]glycerol are similar with those of the valienamine moiety of acarbose and validamycin A. Addition of sodium [1-13C]acetate to cultures of the producer led to the labeling of the carbonyl atom of the acetyl group of cetoniacytone A. This corresponds with the proposed pathway, in which the introduction of the acetyl side chain takes place during the late stages of biosynthesis.[4] Further feeding experiments with the cetoniacytone producer have also established the involvement of 2-epi-5-epi-valiolone, but not valienone, in the biosynthesis of cetoniacytone. In order to gain further information regarding the biosynthesis of cetoniacytone, genetic and biochemical approaches have been pursued. This paper describes the identification and functional analysis of the biosynthetic gene cluster of cetoniacytone A. Biochemical characterization of two dedicated enzymes for the formation of the aminocyclitol core unit provides evidence for the involvement of the gene cluster in cetoniacytone biosynthesis.
Results
Sequence analysis of the cetoniacytone A biosynthetic gene cluster
During our study on various sugar phosphate cyclases that are involved in the biosynthesis of bioactive secondary metabolites, we have previously identified a homolog of the 2-epi-5-epi-valiolone synthase gene (cetA) in the cetoniacytone producing strain Actinomyces sp. Lu 9419.[12] The gene was recombinantly expressed in Escherichia coli and the catalytic function of the protein was characterized. The identification of the cetA gene has enabled the further characterization of the 46 kb flanking region of the chromosome using chromosomal walking. The ORFs were identified using ORF finder analysis (NCBI) and analyzed with software in the BLAST server. The computer-aided analysis of the 46 kb sequence revealed the presence of 31 complete ORFs (Figure 1). According to the structure of cetoniacytone A and the BLAST results, 20.5 kb of the sequenced DNA encoding 17 ORFs is predicted to be involved in the biosynthesis of cetoniacytone A. Other orfs located on the outer region of the 20.5 kb DNA seem to be unrelated to the biosynthesis of cetoniacytone A.[13-16] The product of each orf and its proposed function are shown in Table 1.
Figure 1.
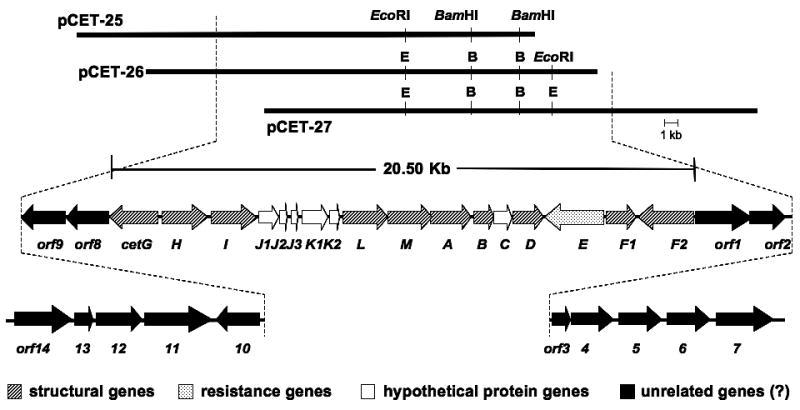
Genetic organization of the cetoniacytone gene cluster.
Table 1.
Deduced function of the cetoniacytone biosynthetic genes.
| gene | homology | species | Size (bp) | % identi | % sim |
|---|---|---|---|---|---|
| cetA | 2-epi-5-epi-valiolone synthase | Streptomyces hygroscopicus | 1146 | 54% | 71% |
| cetB | putative glyoxalase I | Frankia alni ACN14a | 552 | 76% | 83% |
| cetC | hypothetical protein | Frankia alni ACN14a | 495 | 54% | 71% |
| cetD | putative arylamine N-acetyltransferase | Streptomyces murayamaensis | 867 | 40% | 53% |
| cetE | drug resistance transporter | Burkholderia thailandensis E264 | 1860 | 42% | 56% |
| cetF1 | Putative oxidoreductase | Frankia alni ACN14a | 780 | 52% | 68% |
| cetF2 | FAD/FMN-dependent dehydrogenase | Brevibacterium linens BL2 | 1650 | 47% | 67% |
| cetG | glucose-methanol-choline oxidoreductase | Pseudomonas fluorescens | 1359 | 35% | 52% |
| cetH | adenosylmethionine-8-amino-7-oxononanoate aminotransferase | Magnetospirillum magneticum AMB-1 | 1299 | 33% | 50% |
| cetI | FAD-dependent oxidoreductase | Actinomadura kijaniata | 1278 | 34% | 51% |
| cetJ1 | hypothetical protein | Actinoplanes sp. A40644 | 570 | 59% | 72% |
| cetJ2 | hypothetical protein | Frankia alni ACN14a | 327 | 68% | 86% |
| cetJ3 | hypothetical protein | Frankia alni ACN14a | 402 | 59% | 67% |
| cetK1 | hypothetical protein | Frankia alni ACN14a | 720 | 61% | 74% |
| cetK2 | hypothetical protein | Frankia alni ACN14a | 297 | 57% | 68% |
| cetL | putative pyranose oxidase (glucose 2-oxidase) | Frankia alni ACN14a | 1248 | 66% | 76% |
| cetM | L-alanine:N-amidino-3-keto-scyllo-inosamine aminotransferase | Streptomyces griseus | 1224 | 54% | 66% |
| orf1 | ASPIC/UnbV homolog | Herpetosiphon aurantiacus ATCC 23779 | 1914 | 40% | 54% |
| orf2 | UnbU | Micromonospora chersina | 1062 | 46% | 61% |
| orf3 | putative ACP | Streptomyces coelicolor A3(2) | 198 | 42% | 65% |
| orf4 | putative 3-oxoacyl-ACP synthase | Streptomyces avermitilis MA-4680 | 1203 | 37% | 51% |
| orf5 | putative 3-oxoacyl ACP-reductase | Deinococcus geothermalis DSM 11300 | 1470 | 45% | 56% |
| orf6 | putative 3-oxoacyl ACP synthase | Zymomonas mobilis subsp. mobilis ZM4 | 1275 | 30% | 45% |
| orf7 | Hypothetical protein | N/A | 1932 | - | - |
| orf8 | TruD homolog – putative pseudouridylate synthase | Pseudomonas stutzeri A1501 | 1059 | 29% | 42% |
| orf9 | NikS – putative carboxylase | Streptomyces tendae | 1179 | 41% | 56% |
| orf10 | NocH (membrane transport protein) | Nocardia uniformis subsp. tsuyamanensis | 1221 | 31% | 46% |
| orf11 | putative ATPase | Saccharopolyspora erythraea NRRL 2338 | 3189 | 65% | 75% |
| orf12 | putative ATPase | Saccharopolyspora erythraea NRRL 2338 | 1632 | 48% | 61% |
| orf13 | putative chalcone/stilbene synthase | Saccharopolyspora erythraea NRRL 2338 | 612 | 73% | 79% |
| orf14 | Putative nitrite reductase | Saccharopolyspora erythraea NRRL 2338 | 2607 | 86% | 91% |
As previously reported, the deduced product of cetA showed significant similarity to 2-epi-5-epi-valiolone synthase, (54% identity, 71% similarity to ValA, and 56% identity, 72% similarity to AcbC).[12] CetB bears similarity with a putative glyoxalase I from Frankia alni ACN14a.[17] It also shows high similarity to ValD of the validamycin pathway.[18] Glyoxalase I (GLO) is a metalloenzyme that catalyzes the glutathione-dependent inactivation of toxic methylglyoxal by catalyzing the conversion of methylglyoxal to S-D-lactoylglutathione via a 1,2-hydrogen transfer.
CetD belongs to the family of arylamine N-acetyltransferases with the highest identity to a putative arylamine N-acetyltransferase from Streptomyces murayamaensis.[19, 20] The arylamine N-acetyltransferase (NAT) enzymes have been found in a broad range of both eukaryotic and prokaryotic organisms. The NAT enzymes catalyze the transfer of an acetyl group from acetyl-CoA onto the terminal nitrogen of a range of arylamine, hydrazine and arylhydrazine compounds, and are proposed to mediate the transfer of an acetyl group to the C-2 position of cetoniacytone (Scheme 2).
Scheme 2.

Proposed biosynthetic pathway to cetoniacytone A. Dashed-box indicates putative pathways that may involve the oxidoreductases CetF1, CetF2, CetG, and CetI, as well as the hypothetical proteins related to the cupin superfamily, CetC, CetJ1, CetK1 and CetK2.
CetE appears to belong to a drug resistance transporter family, bearing 42% identity and 56% similarity to a drug resistance transporter of Burkholderia thailandensis E264.[21] In Burkholderia thailandensis E264, the putative drug resistance transporter belongs to the major facilitator superfamily (MFS), which is one of the two largest families of membrane transporters known. Based on the high resolution structures of transporters, all members of the MFS share a similar structure, regardless of their low sequence identity.[22]
The deduced product of cetF1 shows significant similarity to a putative oxidoreductase of Frankia alni ACN14a,[17] whereas CetF2 shows significant similarity to the known protein FAD/FMN-containing dehydrogenase of Brevibacterium linens BL2.[23] Based on CDD (Conserved Domain Database) and COG (Clusters of Orthologous Groups) assignments and functional annotation, the gene product contains an FAD binding domain. This family of proteins consists of various enzymes that use FAD as a co-factor; most of the enzymes are similar to oxidoreductases such as vanillyl-alcohol oxidase (VAO). VAO catalyzes the oxidation of a wide variety of substrates, ranging from aromatic amines to 4-alkylphenols. Other members of this family include d-lactate dehydrogenase, which catalyses the conversion of d-lactate to pyruvate using FAD as a co-factor and radical oxidase, which oxidizes the reduced form of the mitomycins and is involved in mitomycin resistance.
CetG bears similarity to the glucose-methanol-choline (GMC) oxidoreductase from Pseudomonas fluorescens.[24] This family of proteins requires FAD as a cofactor. Similarly, CetI has high homology to the FAD-dependent oxidoreductase (KijD3) of Actinomadura kijaniata.[25] Together CetF2, CetG and CetI are proposed to be involved in several redox reactions during the biosynthesis. However, it is unclear at this point about the timing of these steps in cetoniacytone biosynthesis.
CetH is similar to adenosylmethionine-8-amino-7-oxononanoate aminotransferase from Magnetospirillum magneticum AMB-1.[26] It is proposed to mediate the transamination step at the C-2 position. However, another gene (cetM) in the cluster also encodes a protein that shares homology with aminotransferases, bearing high similarity to the L-alanine:N-amidino-3-keto-scyllo-inosamine aminotransferase from Streptomyces griseus.[27] It belongs to the DegT/DnrJ/EryC1/StrS aminotransferase family, which includes StsA, StsC and StsS from the streptomycin biosynthetic gene cluster. The aminotransferase activity was demonstrated for purified StsC protein as the l-glutamine:scyllo-inosose aminotransferase, which catalyses the first amino transfer in the biosynthesis of the streptidine unit of streptomycin.[27] Since both CetH and CetM encode an aminotransferase, these two genes may be redundant and only one is required during cetoniacytone biosynthesis.
CetL shares high sequence homology with a putative pyranose 2-oxidase from Frankia alni ACN14a. Pyranose 2-oxidase oxidizes d-glucose and other aldopyranoses regioselectively at C-2 to the corresponding 2-keto sugars.[28-31] In cetoniacytone biosynthesis, this enzyme may catalyze the oxidation of the C-2 hydroxyl group of 5-epi-valiolone to a keto group.
The deduced products of cetC, cetJ1, cetK1, and cetK2 are hypothetical proteins with unknown function.[32-35] These gene products are structurally related to the cupin superfamily of proteins, which is one of the most functionally diverse protein superfamilies. However, those involved in the biosynthesis of secondary metabolites are most related to hydroxylation, epoxidation, decarboxylation, dehydration, and halogenation reactions. Therefore, we predict that cetC, cetJ1, cetK1, and cetK2 are involved in cetoniacytone A biosynthesis (Scheme 2).
Attempts to inactivate the cet genes in strain Lu 9419 and to express the entire gene cluster in a heterologous host
To examine whether the identified 20.5 kb gene cluster is involved in the biosynthesis of cetoniacytone A, several strategies, e.g., gene disruption, heterologous expression of the entire gene cluster, and characterization of the individual genes in the cluster, have been attempted. In most cases, gene disruption is the method of choice because it tends to give more direct and persuasive results. Therefore, we set out to create mutants of the cetoniacytone producer in which one or more genes within the cet cluster is disrupted. However, the attempts were hampered by the lack of an appropriate selection marker for the knockout experiments, as strain Lu 9419 is literally resistant to all antibiotics commonly used in molecular genetics work, including apramycin, gentamicin, hygromycin, kanamycin, spectinomycin, streptomycin, thiostrepton, tetracycline, neomycin, puromycin, paromomycin, ampicillin, and chloramphenicol. Without selection markers, the mutant constructs could not be made and subsequently, the gene inactivation approach was abandoned.
We then turned our efforts to the expression of the entire cluster in a heterologous host. Because the genomic library of strain Lu 9419 was prepared using the E. coli-Streptomyces shuttle cosmid pOJ446, it is possible to transfer the cosmid clones directly into a Streptomyces host such as S. lividans. However, sequence analysis of the previously identified cosmid clones pCET25, pCET26, and pCET27, revealed that all three clones contain a truncated cetoniacytone cluster. In order to find cosmid clones that harbor the complete cluster, the genomic library from the cetoniacytone producer was re-screened using the cetA probe, from which an additional eight positive cosmids were identified. End sequencing of these eight positive clones showed only one clone (pCET2) that contained the entire cluster. Thus, pCET2 was transferred into S. lividans 1326 by the protoplast transformation method. While the transformation of S. lividans 1326 with pCET2 could be done seamlessly, the plasmid appeared to be quite unstable in the heterologous host, as no cetoniacytone production was detected and the plasmid isolated from the tranformants consistently showed a digestion pattern different from the original construct. Although a number of other natural product pathways have been successfully expressed using the pOJ446 shuttle vector, there are also numerous reports on the unstable nature of this vector.[36-38] The latter proved to be the case in our system.
Characterization of the 2-epi-5-epi-Valiolone Epimerase (CetB), a New Member of the Vicinal Oxygen Chelate (VOC) Superfamily
As both gene inactivation and heterologous expression of the entire cluster were unsuccessful, we set out to confirm the identity of the cluster by functionally characterizing recombinantly expressed proteins from the pathway. Previously, we demonstrated that CetA is a dedicated sugar phosphate cyclase that is involved in C7-cyclitol biosynthesis. In the current study, we explored the function of the putative glyoxalase gene (cetB), which is located downstream of cetA. Amino acid sequence analysis of CetB revealed a high similarity between CetB and ValD from the validamycin pathway, albeit CetB (183 aa) is much smaller than ValD (451 aa) and related proteins belonging to the VOC superfamily. The VOC superfamily includes a set of structurally related proteins that are able to catalyze a large range of divalent metal ion-dependent reactions. Members of this family include the glyoxalase I proteins (GLO),[39-44] the extradiol dioxygenases (DHBD),[45-47] the bleomycin resistance proteins (BRP),[48, 49] the methylmalonyl-CoA epimerases (MMCE),[50-52] and the fosfomycin resistance proteins (FOS).[53, 54] Further analysis of the amino acid sequence revealed that ValD contains two similar domains, each of which have high identity to CetB. Because the results of our previous feeding experiments with isotopically labeled intermediates suggested that the two pathways only share the first two biosynthetic steps (Scheme 2),[12] we hypothesized that CetB may be involved in the epimerization of 2-epi-5-epi-valiolone to 5-epi-valiolone.
To confirm the activity of CetB as a 2-epi-5-epi-valiolone epimerase, the cetB gene was subcloned and overexpressed in E. coli BL21(DE3)pLysS. Expression of cetB yielded large quantities of a 24 kDa soluble polyhistidine-tagged protein (Figure S1). The native protein exists as a homodimer, as estimated by nondenaturing-PAGE and high performance gel filtration chromatography, in which CetB eluted as a 48 KDa protein (Figure S2). As the potential substrate for CetB, 2-epi-5-epi-valiolone, was not readily available, characterization of CetB was carried out using a coupled enzyme assay using sedoheptulose 7-phosphate as substrate and the 2-epi-5-epi-valiolone synthase (CetA) as the first enzymatic step. Incubation of sedoheptulose 7-phosphate with CetA for 1 h followed by the addition of cell-free extracts containing CetB, indeed furnished 5-epi-valiolone, as monitored by TLC. GC-MS analysis of the silylated product of the reaction showed a retention time and mass fragmentation pattern consistent with that of the synthetic standard, tetra-TMS-5-epi-valiolone (Figure 2). In contrast, extracts from cells carrying the empty vector pRSET B incubated with sedoheptulose 7-phosphate plus CetA gave only the 2-epi-5-epi-valiolone product (data not shown).
Figure 2.
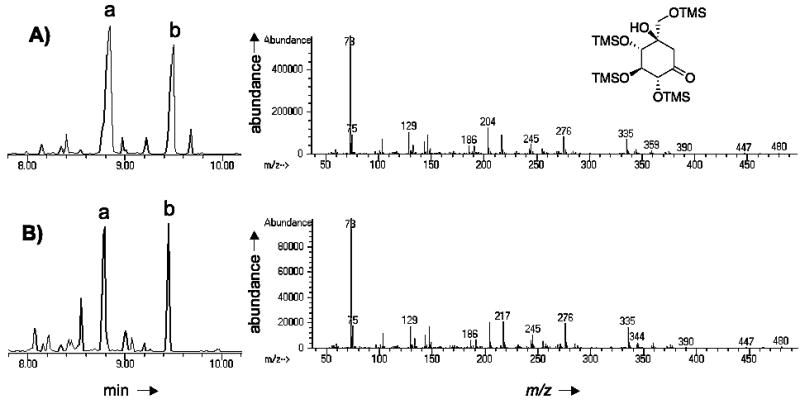
GC-MS analysis of CetB product. (A) GC-MS profile of silylated 5-epi-valiolone standard. (B) GC-MS profile of silylated products of CetB reaction. Left panel show the GC trace and right panel show the MS fragmentation pattern of the major peak eluting at 8.75–8.85 minutes. Peak a, tetra-TMS-5-epi-valiolone. Peak b, penta-TMS-5-epi-valiolone. MS fragmentation pattern of b not shown.
CetB is a dedicated 2-epi-5-epi-valiolone epimerase
To evaluate the substrate specificity of CetB, a number of compounds that have chemical structures similar to 2-epi-5-epi-valiolone, e.g., 1L-epi-2-inosose, d-(+)-gluconic acid delta-lactone, mannose, shikimic acid, dehydroshikimic acid, and aminodehydroshikimic acid were tested as substrates. Analysis of the reaction products by TLC revealed that CetB demonstrates restricted substrate specificity and only epimerizes 2-epi-5-epi-valiolone to 5-epi-valiolone.
Metal binding analysis of CetB
Many members of the VOC superfamily require divalent metal ions for activity. For instance, MMCE is activated by various divalent metal ions, with the most effective being Co2+ and Mn2+, whereas human GLO is a Zn2+-dependent metalloprotein. Surprisingly, E. coli GLO is inactive in the presence of Zn2+ but is active with Ni2+, Co2+, and Cd2+.[55] On the other hand, DHBD prefers Fe2+ for its activity and FosA is a Mn2+-dependent enzyme. Despite having different metal preferences, structural comparisons of VOC family members such as MMCE, DHBD, and GLO showed that they have almost identical metal binding sites with four conserved residues. Sequence comparisons of E. coli GLO with CetB, BE-Orf13,[35] and ValD showed that all of these proteins contain these conserved four residues (Figure 3). Because ValD consists of two similar GLO homologs, both the N-terminal half (ValD-N) and the C-terminal half (ValD-C) of the ValD were analyzed individually as a separate entry in the alignment. To determine the metal preference of CetB, we attempted to employ an assay system involving the CetA-CetB two enzyme reaction system. First, sedoheptulose 7-phosphate was incubated with CetA, in the presence of Co2+ for 2 h to provide 2-epi-5-epi-valiolone. This was followed by addition of EDTA or 1,10-phenanthroline to the reaction mixture to remove Co2+ and other background divalent ions. Subsequently, purified CetB (pretreated with 1,10-phenanthroline) was added into the reaction and the mixture was incubated for another 3 h with or without divalent metal ions. Surprisingly, TLC analysis of the reaction products suggested that even without addition of a metal ion, CetB retains strong epimerase activity, which suggests CetB may bind to its metal cofactor very tightly, such that EDTA and 1,10-phenanthroline are unable to remove the cofactor from the enzyme. In order to overcome this problem, the four putative metal binding residues of CetB were individually altered as single mutations (H14G, E76G, H99G, E151G) and double mutations (H14G/E76G, H14G/H99G, H14G/E151G, E76G/H99G, E76G/E151G, H99G/E151G) and the epimerase activity of the mutant enzymes was characterized (Figure 4A and B). The results showed that, in the absence of 1,10-phenanthroline, the activities of the single mutant variants were similar to that of the wild type (Figure 4A). However, when 1,10-phenanthroline was added into the reaction mixtures, the activity of the single mutant variants diminished (Figure 4C). Similar analysis of all of the double mutant variants revealed none had detectable activity even without 1,10-phenanthroline (Figure 4B). These results suggest that a single mutation at the metal-binding site of cetB only modestly distorts the metal-binding affinity of the protein, whereas a double mutation of cetB completely disrupted the active site and the metal-binding ability of the enzyme.
Figure 3.
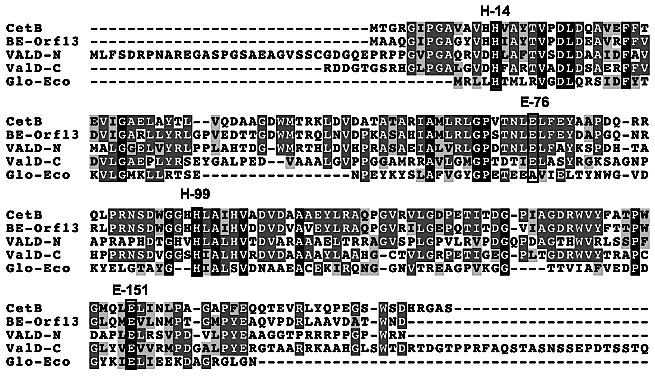
Sequence alignment of CetB, BE-Orf13, ValD-N, ValD-C, and E. coli GLO using ClustalW Basic Interface. The conserved metal binding sites are indicated in the frames. The glutamic acid residue corresponding to E-76 in E. coli GLO shifts by one residue.
Figure 4.

(A) Partial TLC of reaction products of CetB single mutants. 1, 5-epi-valiolone standard; 2, 2-epi-5-epi-valiolone standard; 3, CetA reaction only; 4, CetA+CetB; 5-8, CetA+CetB single mutants: 5, H14G; 6, H99G; 7, E76G; 8, E151G. (B) Partial TLC of reaction products of CetB double mutants. 1, 5-epi-valiolone standard; 2, 2-epi-5-epi-valiolone standard; 3, CetA reaction only; 4, CetA+CetB; 5, CetA+H14G/H99G CetB with 1,10-phenanthroline; 6-11, CetA+CetB double mutants without 1,10-phenanthroline: 6, H14G/H99G; 7, E76G/E151G; 8, H14G/E76G; 9, H14G/E151G; 10, H99G/E76G; 11, H99G/E151G. (C) Partial TLC of reaction products of CetB mutants with 1,10-phenanthroline. 1, 2-epi-5-epi-valiolone standard; 2, 5-epi-valiolone standard; 3, CetA reaction only; 4, CetA+CetB without 1,10-phenanthroline; 5, CetA+CetB with 1,10-phenanthroline; 6-15, CetA+CetB mutants with 1,10-phenanthroline: 6, H14G; 7, H99G; 8, E76G; 9, E151G; 10, H14G/H99G; 11, E76G/E151G; 12, H14G/E76G; 13, H14G/E151G; 14, H99G/E76G; 15, H99G/E151G. (D) Partial TLC of reaction products of H99G CetB with different metal ions. 1, 2-epi-5-epi-valiolone standard; 2-9, CetA+H99G CetB with 1,10-phenanthroline (1 mM) and then metal ions (1.5 mM each) or water (negative control) were added: 2, Fe2+; 3, Co2+; 4, Ca2+; 5, Mg2+; 6, Ni2+; 7, Zn2+; 8, Cu2+; 9, Mn2+; 10, water. Stars indicate the production of 5-epi-valiolone.
Because mutant H99G was found to have similar activity as the wild-type enzyme and yet the metal ion can be removed with 1,10-phenanthroline, mutant H99G was selected for further analysis of metal ion preference. To eliminate any possible interference from the CetA reaction, the substrate of CetB, 2-epi-5-epi-valiolone, was synthesized enzymatically in a coupled reaction using transketolase (EC 2.2.1.1) and ValA[56] with ribose 5-phosphate and hydroxypyruvate as substrates. 2-epi-5-epi-Valiolone was then purified and incubated with the H99G mutant protein (pretreated with 1,10-phenanthroline). Different metal ion solutions (either Co2+; Zn2+; Mg2+; Mn2+; Fe2+; Ni2+; or Cu2+) were added into the mixture. TLC analysis showed that only Co2+ and Ni2+ could effectively rescue the activity of H99G (Figure 4D). Furthermore, inductively-coupled plasma optical emission spectrometry (ICP-OES) was used to analyze samples of freshly purified CetB for the presence of tightly bound zinc, manganese, cobalt, nickel, iron, magnesium, and copper. The results indicated the presence of zinc (0.3 mol), nickel (0.2 mol), iron (0.2 mol), magnesium (0.1 mol), and manganese (0.05 mol) per mole of protein, which suggests that CetB, similar to MMCE and GLO, can strongly bind a number of different metal ions.[51, 57] However, some metals may be bound by the enzyme in an inactive form.[57] Interestingly, while cobalt could restore the catalytic activity of H99G, it appears to be present in wild-type CetB at concentrations below the minimum detectable level of the instrument (<0.05 ppm).
Comparison of the cetoniacytone cluster with an unknown gene cluster in the Frankia alni ACN14a genome
During the genetic analysis of the individual genes in the cetoniacytone A biosynthetic gene cluster, twelve genes were found to have high identity to genes in the Frankia alni ACN14a genome. Further analysis of the F. alni ACN14a genome revealed that the twelve homologous genes are also clustered together in the genome, indicating that Frankia alni ACN14a may have the capacity to produce secondary metabolites similar to cetoniacytone A (Figure 5). In addition to these twelve homologs, a halogenase was also found in this unknown cluster, which suggests that the compound may contain one or more halogen atoms. This finding provides new opportunities for the exploration of novel analogues of natural products in this plant symbiotic bacterium.
Figure 5.
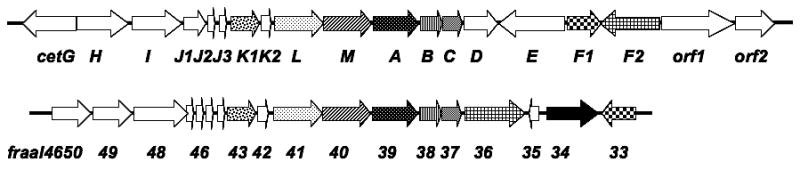
Comparison of the genetic organizations of the cet cluster with the unknown cluster in Frankia alni ACN14a genome.
Discussion
Traditionally, the C7N-aminocyclitol family of natural products was known to be a group of pseudoaminooligosaccharides with potent sugar hydrolase inhibitory activities. However, more recent discoveries indicate a broader scope to the structural diversity and biological activities of this class of natural products, yet little is known about their formation in nature. It was not until recently that a number of reports on the biosynthesis of select members of this class of natural products have been published, in which 2-epi-5-epi-valiolone was found to be a common precursor of the valienamine moiety of acarbose and validamycin A.[58] While cetoniacytone does not have a valienamine moiety in its structure, preliminary feeding experiments indicated that this unique compound is also derived from the same pathway.[4, 12] However, the non-incorporation of valienone and the unusual structure of cetoniacytone A imply a more complex biosynthetic pathway to cetoniacytone A, which may involve multiple redox and elimination reactions.
In acarbose and validamycin biosyntheses, the activity of a kinase is necessary for the activation of the cyclitol ring by phosphorylation. It is believed that most intermediates in the acarbose and validamycin biosynthetic pathways are utilized in their phosphate form.[56, 59, 60] The phosphorylation is catalyzed by the cyclitol-kinases, AcbM (in the acarbose pathway)[56] and ValC (in the validamycin pathway).[61] However, no similar kinase gene was found in the cetoniacytone cluster, suggesting that there may be no phosphorylation step necessary in cetoniacytone biosynthesis. In contrast to the validamycin pathway, results of feeding studies indicate that valienone is not involved in cetoniacytone biosynthesis, which suggests that the downstream modifications of 5-epi-valiolone in cetoniacytone biosynthesis are different from those in the validamycin pathway (Scheme 2). While a number of possibilities may apply for 5-epi-valiolone, the most likely scenario would be the oxidation of the C-2 hydroxyl group by the action of CetL (similar to pyranose 2-oxidase) followed by a transamination catalyzed by CetM to give 2-amino-5-epi-valiolone. Reduction of the C-1 ketone of 5-epi-valiolone to an alcohol is unlikely because it would eliminate the driving force for the subsequent dehydration reaction. A C-2/C-3 dehydration is also unlikely at this point, as this would cause the introduction of the amino group to be less feasible. Finally, oxidation of the C-4 hydroxyl group by CetL or other oxidoreductase (CetF2, CetG, or cetI) is possible, but subsequent oxidation at C-2 would then be energetically unfavorable. Previous studies have shown that strain Lu 9419 can produce both cetoniacytone A and cetoniacytone B, the deacetylated form of cetoniacytone A. When sodium acetate was added to the production medium, cetoniacytone A was isolated in greater yields. So, it was proposed that acetylation should be the last step in the pathway. However, establishing whether epoxidation occurs prior to C-2/C-3 double bond formation or vice versa cannot be confirmed based solely on the genetic information. Further studies are needed to reveal these unique aspects of the biosynthetic pathway.
While attempts to inactivate the cet genes in strain Lu 9419 and heterologous expression of the entire cluster in S. lividans were hampered by technical difficulties, characterizations of the 2-epi-5-epi-valiolone synthase (CetA) and the epimerase (CetB) provide evidence that suggests we have identified the cetoniacytone cluster. Both CetA and CetB were found to be dedicated proteins that only recognize their respective substrates. The same activities have also been observed with ValA and ValD, respectively. Detailed work on the involvement of ValD in the biosynthesis of validamycin A will be reported elsewhere. The identification of CetB and ValD as the epimerases that are responsible for the conversion of 2-epi-5-epi-valiolone to 5-epi-valiolone suggests that this is a common feature of the proposed pathways to a number of C7-cyclitol-containing natural products. Similar enzymes may be involved in the biosynthesis of the gabosines, BE-40644, and the epoxyquinomycins.
In silico comparison and secondary structure analyses revealed that CetB contains two βαβββ structural motifs common to members of the VOC superfamily. Although members of the VOC superfamily have diverse functions and are not highly related to each other at the level of the amino acid sequence, they share a characteristic common structural scaffold composed of βαβββ modules that are combined in several different ways. Structural comparisons, as well as phylogenetic analyses, strongly indicate that the modern VOC family of proteins represented by these enzymes evolved through rich evolutionary events that include multiple gene duplications, gene fusion, and domain swapping.[62] An important initial event is proposed to be the establishment of metal binding in an oligomeric ancestor protein prior to the first gene fusion.[62] With the characterization of CetB as a 2-epi-5-epi-valiolone epimerase (EVE), this enzyme, together with ValD and BE-Orf19, should be listed as the sixth class of the VOC superfamily, in addition to BRP, DHBD, GLO, FOS, and MMCE.
Finally, results of recent X-ray crystallographic analysis of GLO in complex with a transition state analogue and site-directed mutagenesis studies strongly support a base-mediated, proton-transfer mechanism in which the bound diastereomeric substrates undergo catalytic interconversion to product via a metal-coordinated, cis-enediolate intermediate.[63] Therefore, it is proposed that CetB adopts the same reaction mechanism as depicted in Scheme 3.
Scheme 3.

Proposed reaction mechanism of CetB.
Experimental Section
Bacterial strains and culture conditions
The bacterial strains and plasmids used in this study are listed in Table S1. Actinomyces sp. strain Lu 9419 was kindly provided by Prof. Axel Zeeck at the University of Göttingen. This strain was maintained on YMG agar and cultured in YMG liquid medium consisting of malt extract (1%), yeast extract (0.4%), and glucose (0.4%), pH = 7.3 at 30 °C. Oatmeal medium (1 L) consisting of oatmeal (2%) and trace element solution (2.5 mL) [each liter contains CaCl2•2H2O (3 g), Fe(III)-citrate (1 g), MnSO4 (0.2 g), ZnCl2 (0.1 g), CuSO4•5H2O (25 mg), Na2B4O7•10H2O (20 mg), CoCl2 (4 mg), Na2MoO4•2H2O (10 mg) ], and sodium acetate (1 g) as a supplement was used as the production medium for cetoniacytone A. E. coli strains XL-1-Blue and DH10B (Invitrogen) were used as hosts for library construction and routine subcloning, respectively. Over-expression of recombinant His-tagged proteins was carried out in E. coli BL21Gold(DE3)pLysS (Stratagene). E. coli VCS257 was used as a host for the preparation of pOJ446 cosmid library. Escherichia coli strains were cultured in Luria-Bertani Miller (LB) or LB medium containing betaine (2.5 mM) and sorbitol (1 M) (LBBS)[64] supplemented with appropriate amount of antibiotics whenever necessary. S. lividans 1326 was used as host for the heterologous expression of the cetoniacytone A biosynthetic gene cluster. S. lividans was routinely maintained on SFM agar at 30 °C and grown in YEME medium for protoplast preparation.[65] The production medium for S. lividans harboring the cetoniacytone A biosynthetic gene cluster is the same oatmeal medium as used for Actinomyces sp. strain Lu 9419.
Cetoniacytone A production and isolation
Actinomyces sp. strain Lu 9419 was maintained as a stock culture on YMG agar. Pre-cultures were prepared by inoculating YMG medium (50 mL) with a 1 cm2 piece of agar from 7 day-old cultures in a rotary shaker and incubated for 48 hours at 28 °C. The main culture containing oatmeal medium (150 mL) was inoculated with the pre-culture (15 mL) and incubated for 96 hours at 28 °C. At 48 hours, glucose (1 g L-1) was added into the culture to increase the yield of cetoniacytone A.
The harvested culture broths were separated from the mycelia by centrifugation (2500 rpm, 25 minutes) and the supernatant was passed through Amberlite XAD-2 (40/20). The column was washed twice with ddH2O and the more lipophilic constituents were eluted with methanol. The methanol extract was evaporated to dryness using a rotary evaporator. The crude residue was subjected to silica gel column chromatography (CH2Cl2/methanol 9:1) and subsequently to Sephadex LH-20 column chromatography (methanol). The fractions containing cetoniacytone A were identified by TLC and MS. The chemical structure was confirmed by 1H NMR in DMSO-d6 on a Bruker topspin 300 (300 MHz). Mass spectra were taken by ThermoFisher LC-Q advantage.
DNA isolation and manipulations
Routine genetic procedures such as plasmid DNA isolations, restriction endonuclease digestions, alkaline phosphatase treatments, DNA ligations, and other DNA manipulations were performed according to standard techniques.[66] DNA fragments were excised from agarose gels and residual agarose was removed with the QiaQuick Gel Extraction Kit (Qiagen). PCR was carried out using Pfx or Taq high fidelity or GC-rich DNA polymerase (Invitrogen) according to the manufacturer's protocol.
Isolation of the cetoniacytone A biosynthetic gene cluster
Construction of the genomic library of Actinomyces sp. Lu 9419 has been described in our previous paper.[12] Briefly, purified DNA was partially digested with dilute Sau3AI to yield 35-45 Kb fragments. The size-selected DNA was pooled and ligated to pOJ446 predigested with BamHI and HpaI. In vitro packaging was carried out using Gigapack III Gold packaging extract (Stratagene) to yield approximately 6,000 colonies.
The valA gene from S. hygroscopicus var. limoneus and the acbC gene from Actinoplanes sp. encoding 2-epi-5-epi-valiolone synthases were used as heterologous probes to identify the equivalent gene in the genomic DNA of Actinomyces sp. by means of DNA-DNA hybridization experiments. The valA gene was obtained from a valA expression vector[67] previously constructed in our lab by cutting it with restriction enzymes NdeI and EcoRI. The acbC gene was amplified using PCR reaction from plasmid pAS8/7[11] by primers AcbC-F and AcbC-R which are listed in Table S2. Southern blot analysis was performed on Hybond-N nylon membranes (Amersham) with digoxigenin-labeled probes using the DIG high prime DNA labeling and detection starter kit II (Roche). For this purpose, a 1.24 kb valA gene and a 1.15 kb acbC gene were labeled with digoxigenin-dUTP. Screening was performed by colony lifting methodology and nine positive clones were identified. Cosmid DNA from the nine positive clones was isolated and digested with BamHI. Subsequently, a second screening was performed by Southern analysis and three positive candidates were identified and sequenced revealing the putative cetoniacytone biosynthetic gene cluster.
DNA sequencing and analysis
DNA sequencing was done with Applied Biosystems Capillary 3730 Genetic Analyzer at the Center for Genome Research and Biocomputing (CGRB) Facilities of Oregon State University. ValA (DQ164098), acbC (Y18523) and aroB (P07547) sequences were retrieved from Genbank at the NCBI database. The complete genome sequence of Frankia alni ACN14a (NC008278) and the sequence of BE-40644 gene cluster (AB113568) were also obtained from the NCBI database. Deduced amino acid sequence alignments were performed with the ClustalW sequence analysis program. The genome walking approach was employed to sequence the cosmid containing the cetoniacytone biosynthetic gene cluster. Potential open reading frames (ORF) were identified using ORF finder and were searched for homology using the BLAST server.[68]
Cloning and recombinant expression of CetB
The cetB gene was amplified from cosmid DNA using the CetB-F2 and CetB-R2 primers (Table S2) using the Pfx polymerase (Invitrogen) according the manufacturers specifications. The resulting PCR fragment was digested with XhoI and EcoRI and subcloned in frame with the 6XHis-tag into the pRSET B expression vector (Invitrogen).
The recombinant plasmid was transformed into the expression host E. coli BL21(DE3)pLysS and cultured in LBBS medium (500 mL) supplemented with ampicillin (100 μg mL-1) and chloramphenicol (20 μg mL-1). When the cells reached an OD600 of 0.5 at 37 °C, the growth temperature was reduced to 25 °C and protein expression was induced by the addition of isopropyl β-d-1-thiogalactopyranoside (IPTG, 0.2 mM). Cells were harvested by centrifugation after 15 hours incubation and were washed three times with LB broth to remove excess sorbitol from the growth medium. Bacterial pellets were stored at −80 °C until further use.
Enzyme assay for the activity of CetB
To examine CetB enzyme activity, bacterial cell pellets were lysed in K2HPO4/KH2PO4 (pH 7.5, 25 mM) and NaCl (150 mM). The cell lysate was sonicated for two 30 sec bursts (Misonix, microson ultrasonic cell disruptor) and clarified by centrifugation to yield the cell free extract. This mixture was further purified using Ni-NTA agarose beads with elution buffer containing imidazole (300 mM). The purified protein was dialyzed in dialysis buffer [K2HPO4/KH2PO4 pH 7.5 (25 mM)] (2 L) and visualized using 10% SDS-PAGE followed by Coomassie staining. CetA was prepared as previously reported.[12]
The enzyme assay (100 μL) was performed at 30 °C for 3 h in phosphate buffer (pH 7.5, 25 mM) containing purified CetA (30 μL, 0.3 mg mL-1) and cell free extract (30 μL) or purified CetB (30 μL, 0.3 mg mL-1), sedoheptulose 7-phosphate (5 mM), CoCl2 (0.05 mM), NAD+ (1 mM), and KF (2 mM). The reaction products were extracted with methanol using the same procedure as CetA enzymatic assay.[12] The substrate and the reaction product were detected on silica gel TLC (SiO2, nBuOH/EtOH/H2O 9:7:4) followed by staining with cerium sulfate/phosphomolybdic acid solution. The reaction products were mixed with a few drops of SIGMA-SIL-A and dried under argon. The products were extracted with n-hexane and analyzed by GC-MS (Hewlett Packard 5890 series II gas chromatograph). CetB was also incubated with different substrates including 1L-epi-2-inosose, d-(+)-gluconic acid delta-lactone, mannose, shikimic acid, dehydroshikimic acid, and amino-dehydroshikimic acid. For these reactions, CetB cell free extract (30 μL) was incubated with substrate (5 mM), CoCl2 (0.05 mM), NAD+ (1 mM), and KF (2 mM) at 30 °C for 3 h in phosphate buffer (pH 7.5, 25 mM). For metal ion analysis, the H99G CetB was pretreated with 0.1 mM 1,10-phenanthroline and incubated with 2-epi-5-epi-valiolone (5 mM) in the presence of various metal ions (2 mM). After a 2 h incubation, the protein was precipitated by methanol (50 μL) and centrifuged at 12,000 rpm for 10 min. The supernatant was collected and dried in vacuo. The product was analyzed by TLC (SiO2, nBuOH/EtOH/H2O 9:7:4) and stained with cerium- and molybdate-containing reagent.
Synthesis of 2-epi-5-epi-valiolone and 5-epi-valiolone
2-epi-5-epi-Valiolone was synthesized enzymatically in a coupled reaction using transketolase (EC 2.2.1.1, Sigma) and ValA[56] with ribose 5-phosphate and hydroxypyruvate as the initial substrates. The enzyme reaction was performed at 30 °C in a total volume of 30 mL containing transketolase (0.5 units, Sigma), purified ValA (9 mg), hydroxypyruvate (10 mM, Sigma), ribose 5-phosphate (10 mM, Sigma), thiamine pyrophosphate (0.5 mM, Sigma), NAD+ (1 mM), MgCl2 (1 mM), CoCl2 (0.025 mM), NaF (2 mM), pH 7.6. The reaction was monitored by TLC. The mixture was then applied to Vivaspin 15R concentrator (VIVASCIENCE) with a membrane of 10,000 MWCO HY to remove the protein. The flow-through was lyophilized, dissolved in ddH2O (1 mL), and subjected to a Dowex 1×8 (Cl- form, Sigma) column chromatography. The column was eluted with water and the fractions containing 2-epi-5-epi-valiolone were pooled and lyophilized. 5-epi-Valiolone was synthesized according to procedures reported previously[69].
Gel filtration chromatography
CetB (1 mg mL-1) in potassium phosphate buffer (pH 7.4, 25 mM, 200 μL) was applied to a HiLoad 26/60 Superdex 75 prep grade gel filtration column (GE Healthcare) equilibrated with potassium phosphate buffer (pH 7.4, 25 mM), NaCl (150 mM), fitted to a System Gold HPLC work station (BECKMAN). The column flow rate was 0.5 mL min-1. Retention time of proteins, measured at 280 nm, was compared to those of protein standards. The protein size was estimated by making the standard curve of Kav to molecular weight. The Kav for the individual proteins was calculated as follows: Kav = (VR-VO)/(VC-VO) where VO = void volume of the column. The void volume was determined using Blue Dextran 2000. VR = elution volume of the proteins and VC = the geometric bed volume (320 mL).
CetB site-directed mutagenesis and metal cofactor analysis
Circular PCR was performed to create all the CetB single (H14G, E76G, H98G, E151G) and double mutants (H14G/E76G, H14G/H98G, H14G/E151G, E76G/H98G, E76G/E151G, H98G/E151G). The composition of the PCR was as follows: 10× Taq Hi-fidelity polymerase buffer (5 μL); dATP, dTTP, dGTP, and dCTP (125 nM each); primers (125 ng each); plasmid pRSET B carrying the cetB gene (10 ng); H2O (up to 49 μL); and Taq Hi-fidelity polymerase (Invitrogen) (1 μL, 2.5 U). The primers used for this study are listed in Table S2 (CetBMF1 and CetBMR1 for H14G; CetBMF2 and CetBMR2 for H98G; CetBMF3 and CetBMR3 for E76G; CetBMF4 and CetBMR4 for E151G). For the double mutants, the single mutants were used as templates and the same PCR strategy was used to create secondary mutations. Following PCR, the mixtures were incubated with DpnI (Promega) (1.5 μL, 15 U) for 3.0 h to selectively digest the methylated parent plasmids, and the resulting PCR products were analyzed by agarose gel (0.8%) electrophoresis. The products were transferred into DH10B competent E. coli cells with selection for resistance to ampicillin (100 μg mL-1). Successful mutagenesis was confirmed by DNA sequencing. Mutant plasmids were transformed into BL21(DE3)pLysS E. coli cells with selection for resistance to ampicillin (100 μg mL-1) and chloramphenicol (20 μg mL-1).
All enzymatic characterizations of CetB mutants were performed in a similar manner as that for CetB wild type. For the metal cofactor analysis, CetB mutant H98G was used as this protein binds less strongly to the metal cofactor. After 3 hours reaction of CetA with sedoheptulose 7-phosphate, 1,10-phenanthroline (1 mM) was added and the mixture incubated on ice for 20 min. Different metal ions were tested by adding solutions of their corresponding salts (CoCl2, ZnCl2, MgCl2, MnCl2, FeSO4, NiCl2, CuSO4) (1.5 mM) into the reaction mixture. The reaction was started by the addition of the CetB mutant H98G and allowed to incubate for 3 hours at 30 °C.
Metal ion analysis of CetB using inductively-coupled plasma optical emission spectrometry
Freshly purified CetB was dialyzed against metal-ion-free Tris/HCl buffer pH 7.5 (50 mM) and the sample containing 0.7 mg mL-1 of CetB was run on a Perkin Elmer Optima 2100DV inductively-coupled plasma optical emission spectrometer with a diode array detector. The dialysis buffer was used as negative control. Metal-ion-free buffers were prepared by extraction with a CHCl3 solution of dithizone (0.02%, w/v).
Protoplast transformation and heterologous expression of cetoniacytone A biosynthetic gene cluster
To prepare S. lividans protoplasts, spore suspension (0.1 mL) was added to YEME medium (25 mL) in a baffled flask and incubated for 36-40 h at 30 °C. The cells were harvested by centrifugation at 1000×g for 10 min and washed twice with sucrose (10.3%). The mycelia were then treated with lysozyme solution (1 mg mL-1 lysozyme in P buffer[65]) at 30 °C for 20 minutes. After lysozyme treatment, the cells were washed twice with P buffer to remove lysozyme and then were passed through sterilized cotton wool. The collected protoplasts were pelleted at 1000 × g for 7 min and resuspended in P buffer (1 mL). The protoplasts were stored at −80 °C until further use.
The entire cetoniacytone A gene cluster was cut out from the pCET2 using the SpeI and AflII restriction enzymes and ligated into pWUX12b at XbaI and AflII restriction site. pWUX12b is a pSET152 derivative with a bla gene inserted in ApaI site and pACYC Duet-1 multiple cloning site replaced from BamHI to EcoRV site. The resulting plasmid was designated pWUX-CET2. pWUX-CET2 was then transformed into three different S. lividans strains (1326, T7, TK24) by protoplasts transformation. All transformants were confirmed by PCR amplification of three fragments in the gene cluster (N-terminal, mid-section, and C-terminal PCR products) with three sets primers listed in Table S2 (LF, LR; CF, CR; and RF, RR). S. lividans harboring pWUX-Cet2 was grown in Erlenmeyer flasks containing 100 ml oatmeal medium and apramycin (50 μg mL-1). Fermentation was carried out in the same way as that of Actinomyces sp. (strain Lu 9419). Metabolites from culture broth (1 L) were extracted with Amberlite XAD-2. The crude extracts were examined by LC-MS.
Supplementary Material
Table 2. Comparison of the cet genes with genes of the unknown cluster in Frankia alni ACN14a genome.
| cet gene | fraal gene | Putative function | identity | similarity |
|---|---|---|---|---|
| cetJ1 | fraal4646 | hypothetical protein (cupin 2) | 63% | 75% |
| cetJ2 | fraal4645 | hypothetical protein | 68% | 86% |
| cetJ3 | fraal4644 | hypothetical protein | 59% | 67% |
| cetK1 | fraal4643 | hypothetical protein (cupin 2) | 61% | 74% |
| cetK2 | fraal4642 | hypothetical protein (cupin 2) | 57% | 68% |
| cetL | fraal4641 | glucose 2-oxidase | 66% | 76% |
| cetM | fraal4640 | aminotransferase | 69% | 80% |
| cetA | fraal4639 | 2-epi-5-epi-valiolone synthase | 71% | 84% |
| cetB | fraal4638 | 2-epi-5-epi-valiolone epimerase | 76% | 83% |
| cetC | fraal4637 | hypothetical protein (cupin 2) | 54% | 71% |
| cetF1 | fraal4633 | oxidoreductase | 52% | 68% |
| cetF2 | fraal4636 | secreted FAD-linked oxidase | 52% | 70% |
| - | fraal4634 | halogenase | - | - |
Acknowledgments
The authors thank William Austin for metal ion analysis, and Dr. Jongtae Yang and Valerie J. Peterson for technical help. This work was supported by the National Institutes of Health (Grant number AI061528) and by the Oregon State University College of Pharmacy General Research Fund.
Footnotes
Accession Number. The complete DNA and deduced protein sequences of cet genes reported in this paper have been deposited in GenBank under the accession number EF120454.
References
- 1.Gunatilaka AAL. J Nat Prod. 2006;69:509. doi: 10.1021/np058128n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Piel J. Proc Natl Acad Sci USA. 2002;99:14002. doi: 10.1073/pnas.222481399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hildebrand M, Waggoner LE, Liu H, Sudek S, Allen S, Anderson C, Sherman DH, Haygood M. Chem Biol. 2004;11:1543. doi: 10.1016/j.chembiol.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 4.Schlörke O, Krastel P, Muller I, Uson I, Dettner K, Zeeck A. J Antibiot. 2002;55:635. doi: 10.7164/antibiotics.55.635. [DOI] [PubMed] [Google Scholar]
- 5.Mahmud T. Nat Prod Rep. 2003;20:137. doi: 10.1039/b205561a. [DOI] [PubMed] [Google Scholar]
- 6.Matsumoto N, Tsuchida T, Sawa R, Iinuma H, Nakamura H, Naganawa H, Sawa T, Takeuchi T. J Antibiot. 1997;50:912. doi: 10.7164/antibiotics.50.912. [DOI] [PubMed] [Google Scholar]
- 7.Ward P, Small I, Smith J, Suter P, Dutkowski R. J Antimicrob Chemother. 2005;55 1:i5. doi: 10.1093/jac/dki018. [DOI] [PubMed] [Google Scholar]
- 8.Sekiguchi J, Gaucher GM. Can J Microbiol. 1979;25:881. doi: 10.1139/m79-131. [DOI] [PubMed] [Google Scholar]
- 9.Degwert U, van Hulst R, Pape H, Herrold RE, Beale JM, Keller PJ, Lee JP, Floss HG. J Antibiot. 1987;40:855. doi: 10.7164/antibiotics.40.855. [DOI] [PubMed] [Google Scholar]
- 10.Mahmud T, Tornus I, Egelkrout E, Wolf E, Uy C, Floss HG, Lee S. J Am Chem Soc. 1999;121:6973. [Google Scholar]
- 11.Stratmann A, Mahmud T, Lee S, Distler J, Floss HG, Piepersberg W. J Biol Chem. 1999;274:10889. doi: 10.1074/jbc.274.16.10889. [DOI] [PubMed] [Google Scholar]
- 12.Wu X, Flatt PM, Schlörke O, Zeeck A, Dairi T, Mahmud T. Chem Bio Chem. 2007;8:239. doi: 10.1002/cbic.200600446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zazopoulos E, Huang K, Staffa A, Liu W, Bachmann BO, Nonaka K, Ahlert J, Thorson JS, Shen B, Farnet CM. Nat Biotechnol. 2003;21:187. doi: 10.1038/nbt784. [DOI] [PubMed] [Google Scholar]
- 14.Lauer B, Russwurm R, Bormann C. Eur J Biochem. 2000;267:1698. doi: 10.1046/j.1432-1327.2000.01162.x. [DOI] [PubMed] [Google Scholar]
- 15.Kelly WL, Townsend CA. J Bacteriol. 2005;187:739. doi: 10.1128/JB.187.2.739-746.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lauer B, Russwurm R, Schwarz W, Kalmanczhelyi A, Bruntner C, Rosemeier A, Bormann C. Mol Gen Genet. 2001;264:662. doi: 10.1007/s004380000352. [DOI] [PubMed] [Google Scholar]
- 17.Methe BA, Nelson KE, Deming JW, Momen B, Melamud E, Zhang X, Moult J, Madupu R, Nelson WC, Dodson RJ, Brinkac LM, Daugherty SC, Durkin AS, DeBoy RT, Kolonay JF, Sullivan SA, Zhou L, Davidsen TM, Wu M, Huston AL, Lewis M, Weaver B, Weidman JF, Khouri H, Utterback TR, Feldblyum TV, Fraser CM. Proc Natl Acad Sci USA. 2005;102:10913. doi: 10.1073/pnas.0504766102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bai L, Li L, Xu H, Minagawa K, Yu Y, Zhang Y, Zhou X, Floss HG, Mahmud T, Deng Z. Chem Biol. 2006;13:387. doi: 10.1016/j.chembiol.2006.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaneko T, Nakamura Y, Sato S, Asamizu E, Kato T, Sasamoto S, Watanabe A, Idesawa K, Ishikawa A, Kawashima K, Kimura T, Kishida Y, Kiyokawa C, Kohara M, Matsumoto M, Matsuno A, Mochizuki Y, Nakayama S, Nakazaki N, Shimpo S, Sugimoto M, Takeuchi C, Yamada M, Tabata S. DNA Res. 2000;7:331. doi: 10.1093/dnares/7.6.331. [DOI] [PubMed] [Google Scholar]
- 20.Holton SJ, Dairou J, Sandy J, Rodrigues-Lima F, Dupret JM, Noble ME, Sim E. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2005;61:14. doi: 10.1107/S1744309104030659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paulsen IT, Press CM, Ravel J, Kobayashi DY, Myers GS, Mavrodi DV, DeBoy RT, Seshadri R, Ren Q, Madupu R, Dodson RJ, Durkin AS, Brinkac LM, Daugherty SC, Sullivan SA, Rosovitz MJ, Gwinn ML, Zhou L, Schneider DJ, Cartinhour SW, Nelson WC, Weidman J, Watkins K, Tran K, Khouri H, Pierson EA, Pierson LS, 3rd, Thomashow LS, Loper JE. Nat Biotechnol. 2005;23:873. doi: 10.1038/nbt1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vardy E, Arkin IT, Gottschalk KE, Kaback HR, Schuldiner S. Protein Sci. 2004;13:1832. doi: 10.1110/ps.04657704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raty K, Kantola J, Hautala A, Hakala J, Ylihonko K, Mantsala P. Gene. 2002;293:115. doi: 10.1016/s0378-1119(02)00699-6. [DOI] [PubMed] [Google Scholar]
- 24.Saugar I, Sanz E, Rubio MA, Espinosa JC, Jimenez A. Eur J Biochem. 2002;269:5527. doi: 10.1046/j.1432-1033.2002.03258.x. [DOI] [PubMed] [Google Scholar]
- 25.Zhang H, White-Phillip JA, Melancon CE, 3rd, Kwon HJ, Yu WL, Liu HW. J Am Chem Soc. 2007;129:14670. doi: 10.1021/ja0744854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsunaga T, Okamura Y, Fukuda Y, Wahyudi AT, Murase Y, Takeyama H. DNA Res. 2005;12:157. doi: 10.1093/dnares/dsi002. [DOI] [PubMed] [Google Scholar]
- 27.Ahlert J, Distler J, Mansouri K, Piepersberg W. Arch Microbiol. 1997;168:102. doi: 10.1007/s002030050475. [DOI] [PubMed] [Google Scholar]
- 28.Takakura Y, Kuwata S. Biosci Biotechnol Biochem. 2003;67:2598. doi: 10.1271/bbb.67.2598. [DOI] [PubMed] [Google Scholar]
- 29.Hallberg BM, Leitner C, Haltrich D, Divne C. Acta Crystallogr D Biol Crystallogr. 2004;60:197. doi: 10.1107/s0907444903024922. [DOI] [PubMed] [Google Scholar]
- 30.Hallberg BM, Leitner C, Haltrich D, Divne C. J Mol Biol. 2004;341:781. doi: 10.1016/j.jmb.2004.06.033. [DOI] [PubMed] [Google Scholar]
- 31.Kujawa M, Ebner H, Leitner C, Hallberg BM, Prongjit M, Sucharitakul J, Ludwig R, Rudsander U, Peterbauer C, Chaiyen P, Haltrich D, Divne C. J Biol Chem. 2006;281:35104. doi: 10.1074/jbc.M604718200. [DOI] [PubMed] [Google Scholar]
- 32.Gonzalez V, Santamaria RI, Bustos P, Hernandez-Gonzalez I, Medrano-Soto A, Moreno-Hagelsieb G, Janga SC, Ramirez MA, Jimenez-Jacinto V, Collado-Vides J, Davila G. Proc Natl Acad Sci USA. 2006;103:3834. doi: 10.1073/pnas.0508502103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sohng JK, Oh TJ, Lee JJ, Kim CG. Mol Cells. 1997;7:674. [PubMed] [Google Scholar]
- 34.Krugel H, Schumann G, Hanel F, Fiedler G. Mol Gen Genet. 1993;241:193. doi: 10.1007/BF00280217. [DOI] [PubMed] [Google Scholar]
- 35.Kawasaki T, Kuzuyama T, Furihata K, Itoh N, Seto H, Dairi T. J Antibiot. 2003;56:957. doi: 10.7164/antibiotics.56.957. [DOI] [PubMed] [Google Scholar]
- 36.Hong ST, Carney JR, Gould SJ. J Bacteriol. 1997;179:470. doi: 10.1128/jb.179.2.470-476.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thapa LP, Oh TJ, Lee HC, Liou K, Park JW, Yoon YJ, Sohng JK. Appl Microbiol Biotechnol. 2007;76:1357. doi: 10.1007/s00253-007-1096-4. [DOI] [PubMed] [Google Scholar]
- 38.Cone MC, Petrich AK, Gould SJ, Zabriskie TM. J Antibiot. 1998;51:570. doi: 10.7164/antibiotics.51.570. [DOI] [PubMed] [Google Scholar]
- 39.Ariza A, Vickers TJ, Greig N, Armour KA, Dixon MJ, Eggleston IM, Fairlamb AH, Bond CS. Mol Microbiol. 2006;59:1239. doi: 10.1111/j.1365-2958.2006.05022.x. [DOI] [PubMed] [Google Scholar]
- 40.Thornalley PJ. Biochem Soc Trans. 2003;31:1343. doi: 10.1042/bst0311343. [DOI] [PubMed] [Google Scholar]
- 41.Richter U, Krauss M. J Am Chem Soc. 2001;123:6973. doi: 10.1021/ja0105966. [DOI] [PubMed] [Google Scholar]
- 42.Himo F, Siegbahn PE. J Am Chem Soc. 2001;123:10280. doi: 10.1021/ja010715h. [DOI] [PubMed] [Google Scholar]
- 43.Cameron AD, Olin B, Ridderstrom M, Mannervik B, Jones TA. EMBO J. 1997;16:3386. doi: 10.1093/emboj/16.12.3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ridderstrom M, Cameron AD, Jones TA, Mannervik B. J Biol Chem. 1998;273:21623. doi: 10.1074/jbc.273.34.21623. [DOI] [PubMed] [Google Scholar]
- 45.Vetting MW, Wackett LP, Que L, Jr, Lipscomb JD, Ohlendorf DH. J Bacteriol. 2004;186:1945. doi: 10.1128/JB.186.7.1945-1958.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eltis LD, Bolin JT. J Bacteriol. 1996;178:5930. doi: 10.1128/jb.178.20.5930-5937.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kovaleva EG, Lipscomb JD. Science. 2007;316:453. doi: 10.1126/science.1134697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martin TW, Dauter Z, Devedjiev Y, Sheffield P, Jelen F, He M, Sherman DH, Otlewski J, Derewenda ZS, Derewenda U. Structure. 2002;10:933. doi: 10.1016/s0969-2126(02)00778-5. [DOI] [PubMed] [Google Scholar]
- 49.Dumas P, Bergdoll M, Cagnon C, Masson JM. EMBO J. 1994;13:2483. doi: 10.1002/j.1460-2075.1994.tb06535.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McCarthy AA, Baker HM, Shewry SC, Patchett ML, Baker EN. Structure. 2001;9:637. doi: 10.1016/s0969-2126(01)00622-0. [DOI] [PubMed] [Google Scholar]
- 51.Leadlay PF. Biochem J. 1981;197:413. doi: 10.1042/bj1970413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kuhnl J, Bobik T, Procter JB, Burmeister C, Hoppner J, Wilde I, Luersen K, Torda AE, Walter RD, Liebau E. FEBS J. 2005;272:1465. doi: 10.1111/j.1742-4658.2005.04579.x. [DOI] [PubMed] [Google Scholar]
- 53.Pakhomova S, Rife CL, Armstrong RN, Newcomer ME. Protein Sci. 2004;13:1260. doi: 10.1110/ps.03585004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fillgrove KL, Pakhomova S, Schaab MR, Newcomer ME, Armstrong RN. Biochemistry. 2007;46:8110. doi: 10.1021/bi700625p. [DOI] [PubMed] [Google Scholar]
- 55.He MM, Clugston SL, Honek JF, Matthews BW. Biochemistry. 2000;39:8719. doi: 10.1021/bi000856g. [DOI] [PubMed] [Google Scholar]
- 56.Zhang CS, Stratmann A, Block O, Bruckner R, Podeschwa M, Altenbach HJ, Wehmeier UF, Piepersberg W. J Biol Chem. 2002;277:22853. doi: 10.1074/jbc.M202375200. [DOI] [PubMed] [Google Scholar]
- 57.Clugston SL, Yajima R, Honek JF. Biochem J. 2004;377:309. doi: 10.1042/BJ20030271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mahmud T, Lee S, Floss HG. Chem Rec. 2001;1:300. doi: 10.1002/tcr.1015. [DOI] [PubMed] [Google Scholar]
- 59.Dong H, Mahmud T, Tornus I, Lee S, Floss HG. J Am Chem Soc. 2001;123:2733. doi: 10.1021/ja003643n. [DOI] [PubMed] [Google Scholar]
- 60.Minagawa K, Zhang Y, Ito T, Bai L, Deng Z, Mahmud T. Chem Bio Chem. 2007;8:632. doi: 10.1002/cbic.200600528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Minagawa K, Zhang Y, Ito T, Bai L, Deng Z, Mahmud T. Chem Bio Chem. 2007;8:632. doi: 10.1002/cbic.200600528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bergdoll M, Eltis LD, Cameron AD, Dumas P, Bolin JT. Protein Sci. 1998;7:1661. doi: 10.1002/pro.5560070801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cameron AD, Ridderstrom M, Olin B, Kavarana MJ, Creighton DJ, Mannervik B. Biochemistry. 1999;38:13480. doi: 10.1021/bi990696c. [DOI] [PubMed] [Google Scholar]
- 64.Kim CG, Yu TW, Fryhle CB, Handa S, Floss HG. J Biol Chem. 1998;273:6030. doi: 10.1074/jbc.273.11.6030. [DOI] [PubMed] [Google Scholar]
- 65.Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. Practical Streptomyces Genetics. 2nd. John Innes; 2000. [Google Scholar]
- 66.Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. 3rd. Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- 67.Yu Y, Bai L, Minagawa K, Jian X, Li L, Li J, Chen S, Cao E, Mahmud T, Floss HG, Zhou X, Deng Z. Appl Environ Microbiol. 2005;71:5066. doi: 10.1128/AEM.71.9.5066-5076.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. J Mol Biol. 1990;215:403. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 69.Mahmud T, Xu J, Choi YU. J Org Chem. 2001;66:5066. doi: 10.1021/jo0101003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.