

Positron emission tomography (PET) of labeled metabolites, drugs, proteins and nanomaterials[1-3] is rapidly emerging as a powerful imaging tool to detect and stage disease, to study human biology, to investigate pharmacokinetics and pharmacodynamics of new drugs, or to measure treatment efficacy in clinical trials.[4-8] 18F is one of the most commonly used isotopes for clinical imaging given its half-life, ease of production, wide availability, and compatibility with microfluidics syntheses.[9] Despite extensive use and well established procedures of labeling some small molecules, facile 18F platform-type universally adaptable labeling strategies are still largely missing. This is especially true for rapid labeling of small molecules that emerge from high throughput screens or for optimizing hybrid and modular imaging agents. Bioorthogonal chemistries represent one avenue to develop such generic labeling platforms.
To date, several bioorthogonal reactions have been described[10-15] but only few have been adapted for isotope labeling. The most popular ones is the 1,3-dipolar cycloaddition, “click” reaction, between azides and alkynes.[9, 16] These reactions were found to be particularly useful for in vitro 18F-fluorination of biomolecules and are now commonly used.[1, 17-20] Our search for alternative more rapid, selective, and chemically accessible coupling reactions without need for a catalyst led us to investigate the [4+2] inverse electron demand Diels-Alder cycloaddition using trans-cyclooctenes (TCO) and tetrazines (Tz).[21, 22, 22] Advantages of the TCO/Tz labeling strategy include a) fast reaction times in excess of 6,000 M -1s-1, b) high selectivity, c) no need for elevated temperatures or catalysts, d) biocompatible reaction conditions and e) activatable tetrazines.[22]
Here we extend our previous work on TCO and Tz chemistry[21-24] and show that the Diels-Alder cycloaddition can be used for rapid 18F labeling of drugs. This strategy allows one to piggy-back onto the development of the vast number of small molecule affinity ligands, peptides, and antibodies targeting different enzymes such as kinases[25-27], receptors and other proteins. Using PARP1 as a model target[28-31] and AZD2281 as a well developed nM affinity ligand, we show that the TCO/Tz strategy can be used to efficiently label the drug scaffold in very short times and at exceptionally high yields.
This contrasts to conventional synthesis of directly 18F-fluorinated AZD2281 derivatives where yields are much lower. We show the successful fully automated synthesis of a 18F-labeled TCO, its conjugation with a tetrazine-modified AZD2281, and compare affinities of the parent drugs and intermediates. The described platform-based methodology is modular and could easily be adapted to other molecularly targeted drug scaffolds of interest.
There are three generic 18F labeling strategies for AZD2281: a) de novo synthesis of the native compound substituting 18F for the aryl fluorine atom, b) conventional tosylation and subsequent fluorination of hydroxy-AZD2281 derivatives and c) prosthetic group labeling followed by conjugation to the parent compound. The first choice would result in a chemically identical drug but synthetic steps would be lengthy, require purification of intermediates, may not be feasible given the short half-life of 18F and would be impractical for repeat for on-demand synthesis in a clinical setting. We therefore focused on the prosthetic group approach using the TCO/Tz chemistry. Advantages over the copper catalyzed 1,3-dipolar cycloaddition include: a) no need for elevated temperatures, b) no need for removal of catalysts (important for human use) and c) faster kinetics, important when working with short-lived isotopes. While 18F labeling of either TCO or Tz is conceivable, preliminary experiments demonstrated the substituted 1,2,4,5-tetrazine moiety was not stable to commonly used mild nucleophilic fluorination conditions (tetrabutyl ammonium bicarbonate/potassium fluoride (pH 8.5, 40 °C). Therefore, focus was turned to the design and synthesis of an 18F-labeled TCO in which labeling would take place away from the cyclooctenyl ring due to susceptibility of that system to isomerization to bicyclo[3.3.0]octenes.[32] (Z)-2-(Cyclooct-4-enyloxy)acetic acid, 2, was prepared in 63% yield over two steps from commercially available 9-oxabicyclo[6.1.0]non-4-ene (Scheme 1). Carboxylic acid 2 was converted to (E)-2-(cyclooct-4-enyloxy)ethanol, 4, first by LiAlH4 reduction to give (Z)-2-(cyclooct-4-enyloxy)ethanol, 3, in 78% yield, followed by photochemical cis/trans isomerization and isolation of the (E)-isomers by the previously described cycle/trap method.[15] The major (E)-cyclooctyl stereoisomer was isolated by column chromatography and converted to the corresponding tosylate 5 in 84% yield. (E)-5-(2-Fluoroethoxy)cyclooct-1-ene, 619F, was prepared in 91% yield by the treatment of 5 with tetrabutylammonium fluoride (TBAF) in THF. All previously unknown compounds were fully characterized by 1H, 13C and 19F NMR.
Scheme 1.

Synthetic scheme for the synthesis of radiolabeled 18F-TCO, 618F; Reagents and conditions: a) NaH, ICH2CO2H, reflux, 4h, (THF); b) LiAlH4, 0 °C to rt, 24h, (Et2O); c) hν, rt, 8h, (Et2O/hexanes); d) TsCl, Et3N, rt, 2 h, (MeCN); e) TBAF, rt, 2 h, (THF).
As a precursor for the chemoselective reactive PARP1 inhibitor AZD2281-Tz 9, 4-[[4-fluoro-3-(4-(5-oxopentanamide) piperazine-1-carbonyl)phenyl]methyl]-2H-phthalazin-1-one, 7, was generated according to known literature procedures (Scheme 2).[24] This precursor was reacted with 8 [21] in the presence of polymer-supported dicyclohexylcarbodiimide (DCC)-beads to yield 9 as a pink solid. Cycloadduct 1019F was prepared by the addition of DMSO solutions of 9 and 619F at rt and subsequent HPLC purification.
Scheme 2.

Synthetic scheme for the synthesis of radiolabeled AZD2281-18F 1018F; Reagents and conditions: a) polystyrene-bound DCC, Et3N, rt, 7h, (DCM); b) rt, 3 min, (DCM).
Radiofluorination of 5 was performed following a modified procedure previously described for the 18F-labeling of 1-azido-2-(2-(2-18F-fluoroethoxy)ethoxy)ethane.[33] In brief, 18F-fluoride n.c.a. (18F-) in 18O-enriched H2O obtained from PETNET and tetrabutylammonium bicarbonate (nBu4NHCO3) were dried by azeotropic distillation of the acetonitrile/water mixture under reduced pressure and a stream of argon. Tosylate 5 in DMSO was added to the dried 18F-(n.c.a.)/(nBu4NHCO3) and heated to 90 °C for 10 minutes. Filtration of the reaction mixture through alumina-N removed unreacted 18F- prior to HPLC purification. Having a low UV absorbance, verification of the identity of desired 618F product was confirmed in a separate experiment by HPLC injection of 619F, fraction collection at the same elution as observed for radioactive 618F, and NMR analysis of the non-radioactive concentrate. HPLC purified 618F was isolated from the collected HPLC solvents by C18 solid phase extraction (SPE) and eluted with dichloromethane (DCM) to give 7.7 ± 3.4 mCi (n = 16) 618F in 44.7 ± 7.8% decay-corrected radiochemical yield (dcRCY) in an average time of 41 min from the start of drying of [18F]-F- (n.c.a.). Analytical HPLC demonstrated >93% radiochemical purity of 618F. Tetrazine 9 in DMSO was added to the 618F/DCM solution, stirred for 3 min and subjected to HPLC purification (Scheme 2). C18 SPE provided 1018F in 59.6 ± 5.0% isolated dcRCY (n = 3) with >96% radiochemical purity.
Compound 9 and AZD2281-Tz/trans-cyclooctene inverse electron demand Diels-Alder products 1019F, 1016O and 1018O were analyzed using HPLC-ESI/MS spectroscopy (Figure 2). Under the reaction conditions for this cycloaddition, it was found that the initial dihydropyridazine products underwent aromatization to the corresponding pyridazines. LC/MS data confirmed the aromatization of the formed heterocycle, giving m/z-values of 792.6, 790.5 and 792.6, respectively (calc. 792.4, 790.4 and 792.4). To verify their elemental composition, compounds 9, 1019F, 1016O and 1018O were also subjected to high resolution mass spectrometry. All measured values reflected the calculated masses. Furthermore, high resolution mass spectrometry allowed distinction between 1019F and 1018O, whose masses differ by 0.0085 g/mol, confirming the radioactive decay of 1018F to 1018O (Figure 2).
Figure 2.

a) LC-ESI/MS traces and b) High resolution mass spectra results for AZD2281-derivatives 9, 1019F, 1016O, 1018O; c) HPLC radiotraces of 18F-labeled compounds 618F and 1018F.
In addition to the above described cycloadducts, we also prepared fluorinated AZD2281 analogs (Scheme 3) using nucleophilic substitution. Two candidates were designed and synthesized based on known literature procedures.[34] 4-[[4-Fluoro-3-(piperazine-1-carbonyl)phenyl]methyl]-2H-phthalazin-1-one was acylated with 6-hydroxyhexanoic acid or hydroxyacetic acid to give 12 and 15. These hydroxy-AZD2281 derivatives were converted to the corresponding tosylates, 13 and 16. Attempts to fluorinate 13 resulted in decomposition of starting material while the reaction of tosylate 16 with sodium fluoride provided 1719F in 16% yield. Standard 18F-radiolabeling K222/K2CO3 conditions, even at low temperatures (40°C), resulted in decomposition of the starting sulfonate ester 16. Experiments were conducted to test the use of microwave heating at short time intervals with no positive results. Attempts to use the less alkaline tetrabutyl ammonium bicarbonate (nBu4NHCO3) as a phase transfer catalyst also resulted in decomposition of the starting materials and <1% radiochemical yields (RCY) of 1718F.
Scheme 3.

Synthetic scheme for the synthesis of conventionally fluorinated AZD2281-derivatives; Reagents and conditions: a) HBTU, Et3N, 6-hydroxyhexanoic acid or 2-hydroxyacetic acid, rt, 60 min, (DMF); b) TsCl, Et3N, rt, over night, (DCM).
The above results show that the chemoselective approach resulted in significantly higher yields of 18F labeled AZD2281 analogs and in much shorter time. The next question was therefore how the TCO/Tz ligand would affect target affinity. To assess this, a colorimetric assay was employed to measure PARP1 activity (Figure 3). The published value for AZD2281 is 5 nM,[34, 35] identical to what we observed in our assay. Conventionally fluorinated 1719F had an IC50 of 5.2 ± 1.1 nM, consistent with the small side group. Compound 9 showed an IC50 of 8.4 ± 1.3 nM, quite remarkable given the bulkier side chain. Cycloaddition fluorinated 1019F had an IC50 of 17.9 ± 1.1 nM (Figure 3), still in the low double digit nanomolar range and likely sufficient for imaging purposes. These findings are also in agreement with previous results showing that modification of AZD2281 at the piperazine-position only minimally perturbs the ability to bind PARP1.[24] In summary, these results show that the cycloaddition approach can achieve rapid and high yield fluorination yields under mild conditions. Introduction of the hexahydrocycloocta[d]pyridazine group seems to only minimally affect affinity of AZD2281 with PARP1. While this retention binding affinity may not be achieved by all targeted small molecules due to the relative size of the hyzahydrocycloocta[d]pyridazine linker, further research in modified linkers to distance binding moiety and pyrazine may be warranted. It is envisioned that this labelling strategy will also have particular utility for the radiofluorination of peptides, antibodies and nanomaterials where size of the hexahydrocycloocta[d]pyridazine linker will of lesser importance.
Figure 3.
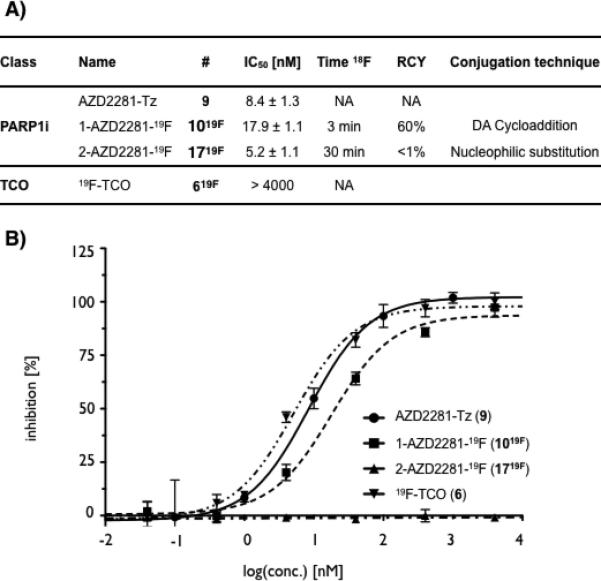
a) IC50s and radiochemical yields and b) IC50 curves for PARP1-inhibitors 9, 1019F, 1719F and trans-cyclooctene 619F.
Supplementary Material
Figure 1.

Structure of AZD2281
Acknowledgements
We thank Drs. Ralph Mazitschek and Neal Devaraj for helpful discussions. This research was supported in part by the NIH grants CA86355 and EB-010011. AT was supported by NIH grant NIGMS T32 GM008313. TR was supported by a grant from the Deutsche Akademie der Naturforscher Leopoldina.
References
- 1.Wang J, Sui G, Mocharla VP, Lin RJ, Phelps ME, Kolb HC, Tseng H-R. Angew. Chem. 2006;118:5402–5407. doi: 10.1002/anie.200601677. [DOI] [PubMed] [Google Scholar]; Angew. Chem., Int. Ed. 2006;45:5276–5281. doi: 10.1002/anie.200601677. [DOI] [PubMed] [Google Scholar]
- 2.Wu AM, Yazaki PJ, Tsai S, Nguyen K, Anderson AL, McCarthy DW, Welch MJ, Shively JE, Williams LE, Raubitschek AA, Wong JY, Toyokuni T, Phelps ME, Gambhir SS. Proc. Natl. Acad. Sci. U. S. A. 2000;97:8495–8500. doi: 10.1073/pnas.150228297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nahrendorf M, Zhang H, Hembrador S, Panizzi P, Sosnovik DE, Aikawa E, Libby P, Swirski FK, Weissleder R. Circulation. 2008;117:379–387. doi: 10.1161/CIRCULATIONAHA.107.741181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Avril N, Rosé CA, Schelling M, Dose J, Kuhn W, Bense S, Weber W, Ziegler S, Graeff H, Schwaiger M. J. Clin. Oncol. 2000;18:3495–3502. doi: 10.1200/JCO.2000.18.20.3495. [DOI] [PubMed] [Google Scholar]
- 5.Burns HD, et al. Proc. Natl. Acad. Sci. U. S. A. 2007;104:9800–9805. doi: 10.1073/pnas.0703472104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gambhir SS. Nat. Rev. Cancer. 2002;2:683–693. doi: 10.1038/nrc882. [DOI] [PubMed] [Google Scholar]
- 7.Willmann JK, van Bruggen N, Dinkelborg LM, Gambhir SS. Nat Rev Drug Discov. 2008;7:591–607. doi: 10.1038/nrd2290. [DOI] [PubMed] [Google Scholar]
- 8.Klimas MT. Mol Imaging Biol. 2002;4:311–337. doi: 10.1016/s1536-1632(02)00017-3. [DOI] [PubMed] [Google Scholar]
- 9.Lee CC, Sui G, Elizarov A, Shu CJ, Shin YS, Dooley AN, Huang J, Daridon A, Wyatt P, Stout D, Kolb HC, Witte ON, Satyamurthy N, Heath JR, Phelps ME, Quake SR, Tseng HR. Science. 2005;310:1793–1796. doi: 10.1126/science.1118919. [DOI] [PubMed] [Google Scholar]
- 10.Demko ZP, Sharpless KB. Angew. Chem. 2002;114:2214–2217. [Google Scholar]; Angew. Chem., Int. Ed. Engl. 2002;41:2110–2113. [Google Scholar]
- 11.Kolb HC, Finn MG, Sharpless KB. Angew. Chem. 2001;113:2056–2075. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int., Ed. Engl. 2001;40:2004–2021. [Google Scholar]
- 12.Agard NJ, Prescher JA, Bertozzi CR. J. Am. Chem. Soc. 2004;126:15046–15047. doi: 10.1021/ja044996f. [DOI] [PubMed] [Google Scholar]
- 13.Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, Bertozzi CR. Proc. Natl. Acad. Sci. U. S. A. 2007;104:16793–16797. doi: 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chang PV, Prescher JA, Sletten EM, Baskin JM, Miller IA, Agard NJ, Lo A, Bertozzi CR. Proc. Natl. Acad. Sci. U. S. A. 2010;107:1821–1826. doi: 10.1073/pnas.0911116107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Royzen M, Yap GP, Fox JM. J. Am. Chem. Soc. 2008;130:3760–3761. doi: 10.1021/ja8001919. [DOI] [PubMed] [Google Scholar]
- 16.Yi Sun E, Josephson LW, Ralph Molecular Imaging. 2006;5:122–128. [PubMed] [Google Scholar]
- 17.Devaraj NK, Keliher EJ, Thurber GM, Nahrendorf M, Weissleder R. Bioconjug. Chem. 2009;20:397–401. doi: 10.1021/bc8004649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nahrendorf M, Keliher E, Panizzi P, Zhang H, Hembrador S, Figueiredo JL, Aikawa E, Kelly K, Libby P, Weissleder R. JACC Cardiovasc Imaging. 2009;2:1213–1222. doi: 10.1016/j.jcmg.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Z-B, Wu Z, Chen K, Chin FT, Che X. Bioconjug. Chem. 2007;18:1987–1994. doi: 10.1021/bc700226v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shao F, Weissleder R, Hilderbrand SA. Bioconjug. Chem. 2008;19:2487–2491. doi: 10.1021/bc800417b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Devaraj NK, Weissleder R, Hilderbrand SA. Bioconjug. Chem. 2008;19:2297–2299. doi: 10.1021/bc8004446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Devaraj NK, Hilderbrand S, Upadhyay R, Mazitschek R, Weissleder R. Angew. Chem. 2010;122:2931–2934. doi: 10.1002/anie.200906120. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem., Int. Ed. Engl. 2010;49:2869–2872. [Google Scholar]
- 23.Devaraj NK, Upadhyay R, Haun JB, Hilderbrand SA, Weissleder R. Angew. Chem. 2009;121:7147–7150. doi: 10.1002/anie.200903233. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem., Int. Ed. Engl. 2009;48:7013–7016. doi: 10.1002/anie.200903233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reiner T, Earley S, Turetsky A, Weissleder R. ChemBioChem. 2010;11:2374–2377. doi: 10.1002/cbic.201000477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fabian MA, William H Biggs WH, III, Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG, Carter TA, Ciceri P, Edeen PT, Floyd M, Ford JM, Galvin M, Gerlach JL, Grotzfeld RM, Herrgard S, Insko DE, Insko MA, Lai AG, Lélias J-M, Mehta SA, Milanov ZV, Velasco AM, Wodicka LM, Patel HK, Zarrinkar PP, Lockhart DJ. Nature Biotechnology. 2005;23:329–336. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- 26.Goldstein DM, Gray NS, Zarrinkar PP. Nature Rev. Drug Discov. 2008;7:391–397. doi: 10.1038/nrd2541. [DOI] [PubMed] [Google Scholar]
- 27.Zhang J, Yang PL, Gray NS. Nature Rev. Cancer. 2009;9:28–39. doi: 10.1038/nrc2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 29.Farmer H, McCabe N, Lord CJ, Tutt ANJ, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NMB, Jackson SP, Smith GCM, Ashworth A. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 30.Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O'Connor MJ, Ashworth A, Carmichael J, Kaye SB, Schellens JH, de Bono JS. N. Eng. J. Med. 2009;361:123–134. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 31.Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. Nature Rev Cancer. 2010;10:293–301. doi: 10.1038/nrc2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Closson WD, Kwiatkowski GT. Tetrahedron Lett. 1966:6435–6440. [Google Scholar]
- 33.Nahrendorf M, Keliher E, Marinelli B, Waterman P, Feruglio PF, Fexon L, Pivovarov M, Swirski FK, Pittet MJ, Vinegoni C, Weissleder R. Proc. Natl. Acad. Sci. U. S. A. 2010;107:7910–7915. doi: 10.1073/pnas.0915163107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Menear KA, Adcock C, Boulter R, Cockcroft XL, Copsey L, Cranston A, Dillon KJ, Drzewiecki J, Garman S, Gomez S, Javaid H, Kerrigan F, Knights C, Lau A, Loh VMJ, Matthews IT, Moore S, O'Connor MJ, Smith GC, Martin NM. J. Med. Chem. 2008;51:6581–6591. doi: 10.1021/jm8001263. [DOI] [PubMed] [Google Scholar]
- 35.Ferraris DV. J. Med. Chem. 2010;53:4561–4584. doi: 10.1021/jm100012m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.