

Abstract
Posttraumatic Stress Disorder (PTSD) is an anxiety disorder which can develop as a result of exposure to a traumatic event and is associated with significant functional impairment. Family and twin studies have found that risk for PTSD is associated with an underlying genetic vulnerability and that more than 30% of the variance associated with PTSD is related to a heritable component. Using a fear conditioning model to conceptualize the neurobiology of PTSD, three primary neuronal systems have been investigated – the hypothalamic-pituitary-adrenal axis, the locus coeruleus-noradrenegic system, and neurocircuitry interconnecting the limbic system and frontal cortex. The majority of the initial investigations into main effects of candidate genes hypothesized to be associated with PTSD risk have been negative, but studies examining the interaction of genetic polymorphisms with specific environments in predicting PTSD have produced several positive results which have increased our understanding of the determinants of risk and resilience in the aftermath of trauma. Promising avenues of inquiry into the role of epigenetic modification have also been proposed to explain the enduring impact of environmental exposures which occur during key, often early, developmental periods on gene expression. Studies of PTSD endophenotypes, which are heritable biomarkers associated with a circumscribed trait within the more complex psychiatric disorder, may be more directly amenable to analysis of the underlying genetics and neural pathways and have provided promising targets for elucidating the neurobiology of PTSD. Knowledge of the genetic underpinnings and neuronal pathways involved in the etiology and maintenance of PTSD will allow for improved targeting of primary prevention amongst vulnerable individuals or populations, as well as timely, targeted treatment interventions.
Introduction
Posttraumatic Stress Disorder (PTSD) is classified as an Anxiety Disorder within DSM-IV. It is defined as the development of symptoms following exposure to an extreme traumatic event (criterion A). These symptoms are characterized as belonging to three separate but interrelated symptom clusters: re-experiencing, avoidance and numbing, and hyperarousal (DSM 1994). Individuals diagnosed with PTSD experience significant functional impairment, including increased risk for unemployment, disrupted relationships, and diminished physical health (Kessler 2000; Kubzansky, Koenen et al. 2007). While the lifetime prevalence of PTSD in adult Americans is estimated to be 6.8% (Kessler, Berglund et al. 2005), the conditional risk for PTSD following trauma exposure ranges from 5 – 31% (Kulka, Schlenger et al. 1990; Kessler, Sonnega et al. 1995; Breslau, Kessler et al. 1998; Adams and Boscarino 2006) with interpersonal and combat trauma associated with relatively greater risk. While an estimated 75% of the population has experienced a criterion A traumatic event (Breslau and Kessler 2001), only a minority of those individuals subsequently develop PTSD. This finding suggests that certain individuals have an underlying vulnerability to developing this disorder in the aftermath of trauma. Identifying those vulnerable individuals may allow for early and targeted intervention to prevent or reduce the symptoms and functional impairment associated with PTSD.
Family Studies of Heritability
If the risk for PTSD following traumatic exposure is associated with an underlying genetic vulnerability, it would be expected that biological relatives (family) of an individual with PTSD (proband) would have a higher risk of developing the disorder following trauma exposure than similarly traumatized non-relatives. Family studies of PTSD have demonstrated this finding. Specifically, PTSD diagnosis was more frequent in adult children of Holocaust survivors with PTSD as compared to children of Holocaust survivors without PTSD (Yehuda, Halligan et al. 2001). A similar finding has been reported in adult children of Cambodian refugees whose parents had PTSD (Sack, Clarke et al. 1995). A limitation of family studies is that PTSD cannot be assessed in those individuals who are not exposed to trauma, and therefore, it cannot be known whether they would have developed PTSD in response to trauma. This can have the effect of falsely lowering the estimated risk for PTSD. An even more significant limitation of family studies is that because family members share both genetic and environmental similarities, these studies cannot differentiate between a genetic versus environmental basis for the increased prevalence within a family pedigree.
Twin Studies of Heritability
Studies of twins (who share identical genetic inheritance) allow researchers to differentiate genetic from environmental influence on the development of a disorder. Twin studies have demonstrated that genetic factors influence the risk of exposure to traumatic events (Lyons, Goldberg et al. 1993; Stein, Jang et al. 2002). This finding, known as gene-environment correlation, may be related to the effect of genetics on temperament, anger and irritability. However, even after accounting for genetic impact on the likelihood of exposure to trauma, twin studies have demonstrated that genetics explains a significant proportion of the vulnerability to PTSD following trauma exposure. Examination of more than 3000 twin pairs from the Vietnam Twin Registry revealed that approximately 32-35% of the variance in PTSD symptoms could be attributed to genetic influences after controlling for combat exposure (True, Rice et al. 1993; Xian, Chantarujikapong et al. 2000). Similarly, Stein and others (2002) reported that amongst twin pairs in which each twin experienced a traumatic event, identical twins were significantly more likely to be matched on PTSD symptoms than fraternal twins. From these results, they calculated that 30% of the variance in response to trauma exposure could be attributed to heredity.
An ongoing concern for researchers investigating the underlying neurobiology of PTSD is the extent to which the biological markers identified in patients represent a consequence of the traumatic exposure and resultant PTSD versus a vulnerability factor that increases an individual's likelihood of developing PTSD subsequent to trauma exposure. Twin studies have been helpful in exploring this differentiation. In a study of hippocampal volume of Vietnam veterans with PTSD and their unaffected twins without combat exposure, both cohorts displayed smaller hippocampal volume as compared to twin pairs of Vietnam veterans without PTSD and their unaffected siblings (Gilbertson, Shenton et al. 2002). This finding suggests that reduced hippocampal volume may be a heritable, pre-existing vulnerability factor for developing PTSD following trauma exposure. A more recent study included twin pairs in which one twin was combat-exposed in Vietnam and diagnosed with PTSD, while the co-twin was not exposed to combat and not diagnosed with PTSD (“high risk” twin). In contrast, in a second twin pair, one twin was combat-exposed in Vietnam but did not develop PTSD and his sibling who was not exposed to combat and not diagnosed with PTSD (“low risk” twin). The authors found decreased gray matter volume in the pregenual anterior cingulate cortex only in the combat-exposed individuals with a PTSD diagnosis versus the other three groups of twins (Kasai, Yamasue et al. 2008). These data suggested that the lower gray matter volume in this region was a consequence of traumatic combat exposure and the subsequent development of PTSD. The current state of the field is that both of the above findings likely represent different aspects of the true phenomenology, such that: 1) following a significant trauma, PTSD is more likely to occur in those individuals who have a certain level of genetic and neural vulnerability, but that 2) there are almost certainly additional neurological / neuropathological sequelae that result after the onset of PTSD.
Twin studies have also revealed that genetic influences associated with risk for PTSD are common to other psychiatric disorders that are frequently comorbid with PTSD. As reviewed by Koenen et al. (2008), overlap with genes associated with major depressive disorder account for the majority of identified genetic variation in PTSD (Fu, Koenen et al. 2007; Koenen, Fu et al. 2008) Overlap common to panic disorder and generalized anxiety disorder account for approximately 60% of the genetic variance in PTSD (Chantarujikapong, Scherrer et al. 2001), and overlap with genetic influences common to alcohol, nicotine, and drug dependence account for greater than 40% (Xian, Chantarujikapong et al. 2000; Koenen, Hitsman et al. 2005). These findings suggests that the genetic determinants of risk for PTSD bi-directionally influence the risk for other commonly comorbid psychiatric disorders. It should be noted that these estimates are based on heritability studies and not on studies of specific genes. Only a small number of properly powered hypothesis-neutral gene discovery approaches (i.e., genome-wide association studies (GWAS)) have been performed on depression and substance use disorders, with no GWAS yet performed on these other anxiety disorders or PTSD. Thus, as more is understood about specific gene pathways involved in PTSD, anxiety, depression, and substance abuse, it is likely that we will gain a much greater understanding of the similarities and differences in the genetic architecture which may underlie these different stress and trauma-related disorders.
Candidate Genes and Neurobiological Pathways
While twin studies can indicate a heritable genetic risk for the development of PTSD, they cannot provide information on which specific genes confer that risk. Molecular genetic studies can be used to determine if specific genes influence risk or resilience for a specific disorder, such as PTSD. However, in the absence of large GWAS or similar hypothesis-neutral gene-discovery studies, knowledge of the underlying neurobiology of the disorder is needed in order to effectively identify appropriate candidate genes to study. When this knowledge is not directly available, translational models of the disorder or key components of the phenotype can be used to direct informed hypothesis generation.
Vulnerability to the development of PTSD after trauma exposure has been conceptualized to involve a fear conditioning process in which fear responses are exaggerated and/or resistant to extinction (Keane, Zimering et al. 1985; Amstadter, Nugent et al. 2009; Jovanovic and Ressler 2010). Within this classical conditioning model, a neutral (conditioned) stimulus (CS) that is temporally paired with an aversive (unconditioned) stimulus (US) acquires the ability to elicit a conditioned fear response that can be triggered when the individual subsequently encounters these or similar stimuli. With regard to the clinical features of PTSD, the traumatic event itself serves as the US and the sights, sounds, smells, and other contextual aspects of the ambient environmental stimuli that are present at the time of the trauma serve as CS to elicit conditioned fear responses.
While this process can serve an evolutionarily advantageous role by allowing an individual to identify and respond to threatening stimuli, excessive activation of conditioned fear responses to stimuli that are not accurately predictive of threat may have a role in the basis of PTSD. Consistent with this hypothesis, emotional and physiological reactivity to stimuli resembling the original traumatic event is a central characteristic of PTSD, can persist for an extended period of time following the traumatic exposure, and has been reliably replicated in the laboratory (Blanchard, Kolb et al. 1986; Pitman, Orr et al. 1987; Orr, Metzger et al. 2003).
The etiology and maintenance of this fear conditioning has been hypothesized to involve multiple interacting neurobiological systems, including the hypothalamic-pituitary-adrenal (HPA) axis, the locus coeruleus (LC)-noradrenergic systems, and connections between the limbic system and frontal cortex. The HPA axis is comprised of neural and endocrine structures that coordinate the hormonal response to stress. It also activates the LC-noradrenergic system (Claes 2004), which has been implicated in the over-consolidation of fear memories in the aftermath of traumatic exposure (O'Carroll, Drysdale et al. 1999; Southwick, Bremner et al. 1999; Southwick, Davis et al. 2002). The amygdala, part of the limbic system within the temporal lobe, plays a primary role in threat detection and elaborating conditioned and unconditioned fear responses, including behavioral responses and activation of the HPA axis (Davis 1992). The activity of the amygdala is regulated in a ‘top-down’ fashion by the medial prefrontal cortex which can function in an inhibitory manner to extinguish conditioned fear responses (Milad and Quirk 2002; Vidal-Gonzalez, Vidal-Gonzalez et al. 2006; Peters, Kalivas et al. 2009). Each of these neural systems interact in the development and maintenance of fear conditioning, and have been the focus on candidate genes studies into the neurobiology of PTSD as further described below (see Figure 1).
Figure 1. Fear neurocircuitry.
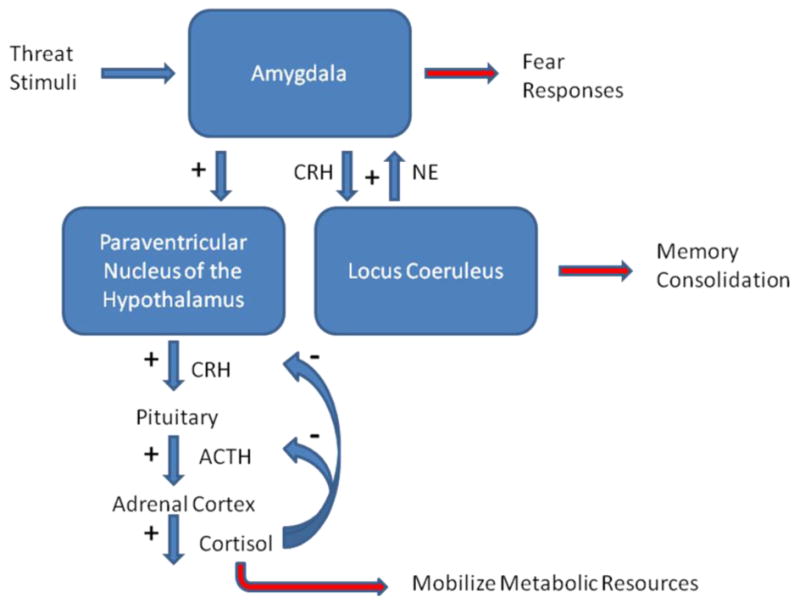
This schematic diagram illustrates the interactions between the Hypothalamic-Pituitary-Adrenal (HPA) axis, the locus coeruleus and the amygdala intrinsic to the fear neurocircuitry within the brain. In response to stimuli indicating potential threat, the amygdala is activated to elaborate the fear response. One target of the output of the amygdala is activation of the HPA axis at the level of the paraventricular nucleus of the hypothalamus, which leads to the release of Corticotrophin Releasing Hormone (CRH). This hormone activates the pituitary to release Adrenocorticotrophin Hormone (ACTH), which is released into the systemic circulation to stimulate the production and release of cortisol from the adrenal cortex. Cortisol, amongst other functions, serves to increase glucose availability to be utilized as a metabolic resource during times of stress. Feedback inhibition of the HPA axis is mediated in part by the binding of cortisol to glucocorticoid receptors (GRs) to limit excessive activation of the HPA axis. In addition to innervation of the HPA axis, the amygdala also activates the locus coeruleus, the primary noradrenergic nucleus in the brain, in a pathway which utilizes CRH neurotransmission. Norepinephrine released from the locus coeruleus has been hypothesized to play a role in the consolidation of fear memories and additionally projects to the amygdala to further stimulate its activation in a positive feedback fashion.
HPA Axis
The HPA axis coordinates the neuroendocrine response to stress. Neurons within the paraventricular nucleus (PVN) of the hypothalamus project to the median eminence where they release corticotrophin-releasing hormone (CRH) which subsequently binds to CRH1 receptors in the anterior pituitary gland promoting the secretion to adrenocorticotrophic hormone (ACTH). ACTH is released into the systemic circulation where it stimulates the production and release of cortisol from the adrenal cortex, which amongst other functions, serves to increase glucose availability to be utilized as an energy resource during times of stress (Charmandari, Tsigos et al. 2005). Feedback inhibition of the HPA axis is mediated in part by the binding of cortisol to glucocorticoid receptors (GRs) to limit stress-induced activation of the HPA axis.
While published reports are mixed, the majority of findings in patients with PTSD report elevated levels of CRH (Bremner, Licinio et al. 1997; Baker, West et al. 1999), lower basal cortisol levels (Yehuda, Southwick et al. 1990; Yehuda, Teicher et al. 1996; Thaller, Vrkljan et al. 1999; Bremner, Vermetten et al. 2007), and enhanced negative feedback suppression of the HPA axis by dexamethasone, a synthetic glucocorticoid (De Kloet, Vermetten et al. 2006; Yehuda 2009). Despite clear evidence of HPA axis perturbations in PTSD, initial investigations into main effects of candidate genes that impact on this system have not been very revealing. For example, a study of the glucocorticoid receptor gene demonstrated no significant association between two polymorphisms in this gene and a diagnosis of PTSD (Bachmann, Sedgley et al. 2005).
Given that PTSD is a psychiatric disorder that, by definition, requires exposure to a specific environmental event (the criterion A trauma), and that individuals vary in their phenotypic response to the event, with only a minority of individuals subsequently developing PTSD, the genetic basis of this disorder is likely to be better elucidated by studies of gene-environment (GxE) interactions rather than studies of the main effects of genes. Two studies have recently demonstrated an interaction between polymorphisms in FKBP5, a gene which impacts on HPA axis function by regulating GR activity, and childhood environment to predict severity of PTSD. FKBP5 is a co-chaperone protein that interacts with another molecular chaperone, hsp90, and is part of the mature GR heterocomplex (Hubler and Scammell 2004). It regulates GR sensitivity and nuclear translocation of GRs such that reduced activity of FKBP5 increases GR sensitivity (Figure 2). Polymorphisms in FKBP5 have previously been reported to be associated with peritraumatic dissociation in medically ill children, a symptom which is predictive of the development of PTSD (Koenen, Saxe et al. 2005). In a study of over 700 highly traumatized, inner city, African-American subjects recruited from primary care clinics (Binder, Bradley et al. 2008), four polymorphisms of the FKBP5 gene were demonstrated to interact with severity of childhood abuse to predict severity of adult PTSD symptoms (Table 1). Further, these polymorphisms were demonstrated to be functional, in that those individuals with both probable PTSD and the risk alleles demonstrated enhanced suppression of cortisol in response to dexamethasone (Binder, Bradley et al. 2008). A more recent study partially replicated these findings, reporting a significant interaction for one of the polymorphisms in the African-American population, such that the TT genotype was associated with the highest risk for PTSD amongst those individuals with a history of childhood adversity, while it was associated with the lowest risk for PTSD amongst those with no history of childhood adversity (Xie, Kranzler et al. 2010). While this GxE interaction was not significant in their European-American study population, alcohol dependence was found to interact with FKBP5 polymorphisms and childhood adversity to increase the risk for a diagnosis of PTSD. These data, along with a number of similar studies, suggest that a number of subject population characteristics as well as similar measurements of childhood and adult trauma will be critical factors in the robust identification of GxE effects.
Figure 2. Schematic of FKBP5 cellular function.
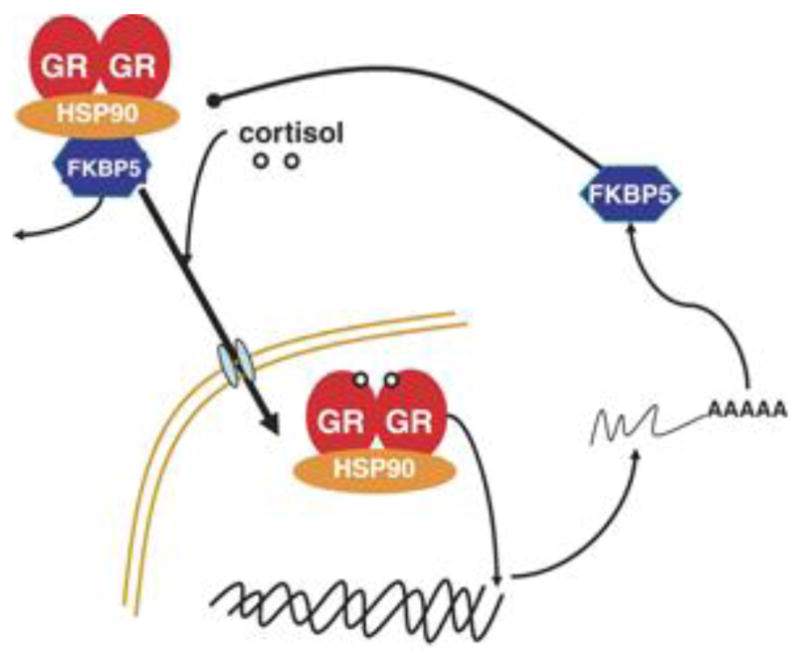
This diagram illustrates the function of FKBP5 as a co-chaperone which regulates glucocorticoid receptor (GR) binding and translocation within the nucleus. When sufficient cortisol is present, GRs dimerize and FKBP5 is displaced, allowing GR translocation and transcriptional activation. In this manner, reduced activity of FKBP5 is associated with increased sensitivity of GRs, as is demonstrated in PTSD. Heat shock protein 90 = hsp90, another molecular co-chaperone which interacts with FKBP5. Figure courtesy of Elisabeth Binder, MD PhD.
TABLE. Published Candidate Gene Association Studies of PTSD, Organized by Neurobiological Pathway.
| First Author | Year | Trauma Assessed | Gene Name (symbol) | Environmental Variable (if GxE) | Finding |
|---|---|---|---|---|---|
| HPA Axis | |||||
| Bachman | 2005 | Combat | Glucocorticoid Receptor (GCCR) | N/A | No significant association between GCCR polymorphisms and PTSD |
| Binder | 2008 | Various | FKBP5 | Childhood abuse | Four SNPs in the FKBP5 gene interact with severity of childhood abuse to predict severity of adult PTSD symptoms |
| Xie | 2010 | Childhood adversity | FKBP5 | Childhood adversity | Within the African-American population, one SNP in the FKBP5 gene is associated with the highest risk for PTSD amongst those with a history of childhood adversity, while it was associated with the lowest risk for PTSD amongst those with no history of childhood adversity. While this interaction was not significant in the European-American study population, alcohol dependence was found to interact with FKBP5 polymorphisms and childhood adversity to increase the risk for a diagnosis of PTSD. |
| Locus Coeruleus - Noradrenergic System | |||||
| Lappalainen | 2002 | Combat | Neuropeptide Y (NPY) | N/A | No significant association between NPY polymorphism and PTSD |
| Mustapic | 2007 | Combat | Dopamine Beta-hydroxylase (DBH) | N/A | No significant association between DBH polymorphisms and PTSD, but there was an association between one genotype and lower DBH activity in those with PTSD |
| Kolassa | 2010 | Rwandan genocide; Various | Catechol-O-methyltransferase (COMT) | Lifetime traumatic events | Carriers of the Val allele demonstrated the expected dose response relationship between higher number of lifetime traumatic events and a lifetime diagnosis of PTSD. However, homozygotes for the Met/Met genotype demonstrated a high risk for lifetime PTSD independent of the number of lifetime traumatic events experienced. |
| Limbic - Frontal System | |||||
| Comings | 1991 | Combat | Dopamine Receptor D2 (DRD2) | N/A | Excess D2A1 allele in PTSD cases |
| Comings | 1996 | Combat | Dopamine Receptor D2 (DRD2) | N/A | Excess D2A1 allele in PTSD cases (includes replication study) |
| Gelernter | 1999 | Combat | Dopamine Receptor D2 (DRD2) | N/A | No significant association between D2A1 allele and PTSD cases |
| Young | 2002 | Combat | Dopamine Receptor D2 (DRD2) | N/A | Excess D2A1 allele only in PTSD cases with harmful drinking |
| Voisey | 2008 | Combat | Dopamine Receptor D2 (DRD2) | N/A | Excess C allele in 975C>T in PTSD cases |
| Segman | 2002 | Various | Dopamine Transporter (DAT1) | N/A | Excess 9-repeat allele in PTSD cases |
| Lee | 2005 | Various | Serotonin Transporter (SLC6A4) | N/A | Excess s/s genotype in PTSD cases |
| Kilpatrick | 2007 | Hurricane | Serotonin Transporter (SLC6A4) | Hurricane exposure and social support | Significant association between s/s genotype and PTSD in adults with high hurrican exposure and low social support |
| Koenen | 2009 | Hurricane; Various | Serotonin Transporter (SLC6A4) | County-level environmental risks (crime and unemployment rates) | Significant interaction between genotype and county-level environmental risks such that s allele was associated with decreased risk of PTSD in low-risk environments but was associated with increased risk of PTSD in high-risk environments |
| Kolassa | 2010 | Rwandan genocide; Various | Serotonin Transporter (SLC6A4) | Lifetime traumatic events | S/s genotype associated with high risk for developing PTSD, even in response to a small number of traumatic events, whereas those with the l allele demonstrated an S-shaped dose-response curve with increasing rates of PTSD in response to an increasing number of traumatic life experiences |
| Lee | 2007 | Not specified | Serotonin 2A Receptor (5-HTR2A) | N/A | Excess GG genotype in female PTSD cases |
| Nelson | 2009 | Various | Gamma-aminobutyric Acid Receptor alpha 2 (GABARA2) | Childhood trauma | Three SNPs in GABARA2 interacted with childhood trauma exposure to predict PTSD in adulthood |
| Amstadter | 2009 | Hurricane; Various | Regulator of G-protein signaling 2 (RGS2) | Hurricane exposure, prior trauma, and social support | Significant association between CC genotype of RGS2 gene and posthurricane PTSD in adults exposed to high environmental stress (high exposure to hurricanes and low social support). CC genotype also associated with diagnosis of lifetime PTSD in adults with prior trauma exposure and low social support. |
| Lee | 2006 | Not specified | Brain-derived Neurotrophic Factor (BDNF) | N/A | No significant association between BDNF Val66Met polymorphism and PTSD |
| Zhang | 2006 | Not specified | Brain-derived Neurotrophic Factor (BDNF) | N/A | No significant association between BDNF polymorphisms and PTSD |
Expression of FKPB5 mRNA in the emergency room following a trauma was previously shown to be differentially expressed in subjects later developed PTSD (Segman, Shefi et al. 2005). More recently, FKBP5mRNA expression was found to be reduced in survivors of the World Trade Center attacks on 9/11/2001 who had a diagnosis of PTSD (Yehuda, Cai et al. 2009). Also STAT5B, a direct inhibitor of GR function, was found to have decreased expression as well in this study. Interestingly, given the high rate of comorbidity between PTSD and depression, another recent study demonstrated an interaction between these same four polymorphisms of the FKBP5 gene and high levels of childhood trauma to predict increased risk for suicide (Roy, Gorodetsky et al. 2010).
LC-Noradrenergic System
The LC is the primary noradrenergic nucleus within the mammalian brain. It receives neuronal input from and provides output to the hypothalamus, the amygdala, and the prefrontal cortex amongst other regions (Benarroch 2009). Activation of the LC can stimulate release of CRH from the hypothalamus and noradrenergic hyperactivity within the basolateral amygdala has been hypothesized to mediate the overconsolidation of fear memory in PTSD (Southwick, Bremner et al. 1999). Because cortisol can reduce noradrenergic activity, it has been proposed that lower levels of cortisol in individuals who develop PTSD fail to contain the increased noradrenergic response to traumatic stress. This increased noradrenergic response is thought to mediate the overconsolidation of fear memory and perhaps accounting for intrusive memories associated with PTSD, as well as in promoting conditioned fear responses to trauma memories and associated triggers (Yehuda, McFarlane et al. 1998; Yehuda 2002).
As with the HPA axis, studies of the main effects of candidate genes that impact on the LC-noradrenergic system have been largely negative. There was no significant association between polymorphisms in the gene for dopamine beta hydroxylase (DBH), an enzyme that catalyzes the conversion of dopamine to norepinephrine, and PTSD in combat veterans, although there was an association between one genotype and lower DBH activity in those individuals diagnosed with PTSD versus those without the diagnosis (Mustapic, Pivac et al. 2007). Neuropeptide Y (NPY) is colocalized with norepinephrine and has been reported to reduce the release of norepinephrine. It has been demonstrated to have anxiolytic properties in animal models, and levels of NPY have been found to be reduced in those with PTSD (Morgan III, Wang et al. 2000; Morgan 2002). However, in a 2002 study of a polymorphism within the NPY gene, no association was found with a diagnosis of PTSD (Lappalainen, Kranzler et al. 2002).
As noted above, studies of GxE interactions are likely to be more relevant and predictive of risk and resilience to trauma in the development of PTSD. Catechol-O-Methyltransferase (COMT) is an enzyme that catalyzes the extraneuronal breakdown of catecholeamines, including norepinephrine and dopamine. A functional polymorphism of this gene, in which valine (Val) has been substituted by methionine (Met) at codon 158 is associated with lower enzyme activity and slower breakdown of the catecholamines. In a study of survivors of the Rwandan Genocide, carriers of the Val allele demonstrated the expected dose response relationship between higher number of lifetime traumatic events and a lifetime diagnosis of PTSD. However, homozygotes for the Met/Met genotype demonstrated a high risk for lifetime PTSD independent of the number of lifetime traumatic events experienced (Kolassa, Kolassa et al. 2010). Interestingly, the Met/Met genotype, which is associated with higher levels of norephinephrine and dopamine, has also been associated with reduced extinction of conditioned fear responses. This extinction deficit may, in part, account for the mechanism by which this genotype is associated with a high risk for PTSD (Lonsdorf, Weike et al. 2009).
Limbic-Frontal System
The amygdala is composed of several nuclei located within the temporal lobe of the brain and is generally roughly divided into the basolateral nucleus (BLA) which receives the majority of the neuronal input and the central nucleus (CeA) which provides the majority of the output. The CeA projects to and activates both the HPA axis at the level of the hypothalamus and the LC-noradrenergic system. The BLA appears to be the locus for the comparison and development of associations between the unconditioned stimulus and conditioned stimulus in fear conditioning (Fanselow and LeDoux 1999). This nucleus projects to and activates the CeA, which then elaborates the fear response, including the behavioral responses to threat, elevated startle response, and activation of the HPA axis. Descending inhibitory inputs from the medial prefrontal cortex (mPFC) can regulate the transmission of excitatory neurotransmission from the BLA to the CeA and is thought to play a key role in the extinction of conditioned fear responses (Milad and Quirk 2002; Likhtik, Pelletier et al. 2005; Likhtik, Popa et al. 2008; Peters, Kalivas et al. 2009).
Given the extent and complexity of this limbic-frontal neurocircuitry, many different genes can impact on this system. Dopamine has been demonstrated to innervate the amygdala and animal models have reported alterations of this dopaminergic neurotransmission in response to stress (Puglisi-Allegra and Cabib 1997). Two studies reported a significant main effect of the D2A1 allele of the D2 dopamine receptor (DRD2) gene in association with a diagnosis of PTSD (Comings, Comings et al. 1991; Comings, Muhleman et al. 1996), while another study of this allele did not find a significant association (Gelernter, Southwick et al. 1999). However, the latter study did not assess for trauma exposure in the control group which may have had the effect of not identifying susceptible controls who would have developed PTSD if exposed to a traumatic event. Another study reported a significant association between the D2A1 allele and PTSD only amongst those individuals with PTSD who engaged in harmful drinking (Young, Lawford et al. 2002). A more recent study revealed a significant association between a different polymorphism in the DRD2 gene and PTSD in combat veterans (Voisey, Swagell et al. 2009). Finally, another study reported a significant association between a polymorphism in the dopamine transporter SLC6A3 gene and chronic PTSD versus trauma-exposed controls (Segman, Cooper-Kazaz et al. 2002).
Serotonin is another catecholamine neurotransmitter which plays an important role in regulating activity within the limbic-frontal system and has been reported to be involved in the inhibition of the development of fear memories (Rainnie 1999). A polymorphism of the serotonin 2A receptor gene has been found to be significantly associated with PTSD in Korean women (Lee, Kwak et al. 2007). More scientific attention has been focused on a polymorphism in the promoter region of the serotonin transporter (5-HTTLPR), in which the short (s) allele has been demonstrated to be less efficient than the long (l) allele and is also associated with decoupling of the circuit between the prefrontal cortex and the amygdala responsible for extinction of fear conditioning (Lesch, Bengel et al. 1996; Pezawas, Meyer-Lindenberg et al. 2005). An excess of the s/s genotype has been reported in a Korean population of PTSD patients relative to controls (Lee, Lee et al. 2005). Recent GxE studies of this polymorphism have been more revealing. Kilpatrick et al. (2007) reported that in a study of adults exposed to the 2004 Florida hurricanes, the s/s genotype was associated with an increased risk for PTSD only in those exposed to an environment of high stress (high exposure to hurricanes and low social support). An additional study in this population found a significant interaction between genotype of 5-HTTLPR and county-level environment, such that the s allele was associated with decreased risk of PTSD in a low-risk environment (low county-level crime and unemployment), but increased risk of PTSD in a high-risk environment (Koenen, Aiello et al. 2009). Replicating this finding in a different population (survivors of the Rwandan genocide), Kolassa et al. (2010) reported that those with the s/s genotype were at high risk for developing PTSD, even in response to a small number of traumatic events, whereas those with the l allele demonstrated an S-shaped dose-response curve with increasing rates of PTSD in response to an increasing number of traumatic life experiences. All three studies indicate risk associated with the s allele of 5-HTTLPR in association with exposure to environments of high stress/trauma.
Two additional GxE studies focused on genes which may have broad impact within the brain. Gamma-aminobutyric acid (GABA) is the major inhibitory neurotransmitter in the brain. A recent study reported a significant interaction between three single nucleotide polymorphisms in the GABA alpha-2 receptor gene and severity of childhood trauma in predicting adult PTSD in those homozygous for the risk alleles (Nelson, Agrawal et al. 2009). G-proteins regulate a wide variety of activities, receptors and transporters in the brain, and regulator of G-protein signaling 2 (RGS2) is a member of a protein family that facilitates the deactivation of G-proteins, thereby decreasing G protein-coupled receptor signaling (Neubig and Siderovski 2002). Amstadter et al. (2009), studying the hurricane-exposed population referenced above, found a significant association between a specific genotype of the RGS2 gene and posthurricane PTSD in adults exposed to high environmental stress (high exposure to hurricanes and low social support), as well as an association between this genotype and a diagnosis of lifetime PTSD in adults with prior trauma exposure and low social support.
Epigenetics
While an improvement over studying the main effects of genes alone, studies of GxE interaction do not explain the importance of developmental timing of stressor exposure to producing phenotypic changes associated with PTSD. However, investigation of the epigenetic modification of DNA can provide insight into this issue. Epigenetic modification describes an environmentally induced change in DNA which alters the function rather than the structure of a gene. These changes can be specific to critical developmental periods, can be stable, enduring, and site-specific, and may be intergenerationally transmitted (Meaney and Szyf 2005; Yehuda and Bierer 2009). The biological mechanism of epigenetic modification most often involves methylation of cytosine within a gene, which typically produces decreased transcription of that segment of DNA into RNA to reduce gene expression (Novik, Nimmrich et al. 2002).
A well described example of epigenetic modification in an animal model is provided by the work of Meaney and colleagues. In this rat model, maternal care characterized by higher amounts of licking and grooming produced lower levels of cortisol in the rat pups, but only if this variation in maternal care occurred during a specific, early developmental window. These rat pups also demonstrated enhanced suppression of cortisol in response to dexamethasone, as well as greater expression of the GR gene and a greater number of hippocampal GRs as a result of hypomethylation in promoter region of the hippocampal GR gene (Francis, Diorio et al. 1999; Weaver, Szyf et al. 2002). Further, by producing stable changes in stress response and behavior, these epigenetic changes and the associated phenotype were transmitted from one generation of female offspring to the next (Seckl and Meaney 2006; Weaver 2007). It is interesting to note that the neuroendocrine alterations described in this animal model parallel those of PTSD, with low basal cortisol and enhanced suppression of cortisol in response to a synthetic glucocorticoid. In fact, lower levels of GR mRNA have been demonstrated in the hippocampus of suicide victims who had a history of childhood abuse (McGowan, Sasaki et al. 2009). One caveat of these studies to date, is that it has not been possible to follow the state of methylation over time in a given individual or animal. Thus, although the interpretation is that there are early developmental methylation changes that are long-lasting and enduring, it cannot yet be ruled out that methylation may represent a state-dependent effect. In this alternative view, other causal factors in the adult animals and suicide victims (e.g. ongoing adult stress or depression) may lead to similar methylation effects as seen during development or in the previous generation, even if they were not maintained consistently over time in these individuals.
It can be hypothesized that the epigenetic-mediated changes in HPA axis reactivity could be associated with increased vulnerability to PTSD following subsequent traumatic exposure, perhaps by failing to constrain stress-induced elevations in noradrenergic neurotransmission leading to overconsolidation of fear memories, as well as resulting in increased physiologic arousal and distress. These findings could also help explain the mechanism by which early life trauma is a strongly validated risk factor for the subsequent development of PTSD in adulthood by recalibrating the set point and stress-responsivity of the HPA axis in an enduring manner that increases vulnerability to PTSD (Yehuda, Flory et al. 2010).
While there have been no published studies providing specific proof of the role of epigenetic modification in association with risk for PTSD, the results of several studies on intergenerational transmission of risk for PTSD could plausibly be explained by epigenetic mechanisms. Yehuda and colleagues (2007; 2008) have reported increased risk for PTSD, as well as low cortisol levels, in the offspring of female, but not male, Holocaust survivors with PTSD. As with the rat model, this increased risk may be produced by alternations in maternal care associated with the mother's PTSD, which may disrupt child-maternal attachment, a known risk factor for PTSD (Charuvastra and Cloitre 2008). These effects may also produce enduring changes in HPA axis responsivity to stressors that may influence risk for PTSD following subsequent traumatic exposure (Brand, Brennan et al. 2010).
Epigenetic mechanisms may also be relevant to the intrauterine environment. Pregnant mothers who developed PTSD as a result of exposure to the World Trade Center attacks of 9/11/2001, produced infants with lower salivary cortisol levels, but only if the traumatic exposure occurred during the third trimester of gestation (Yehuda, Engel et al. 2005). These changes may occur via transmission of hormonal responses to stress to the fetus in utero, leading to a reprogramming of glucocorticoid responsivity in the offspring (Levitt, Lindsay et al. 1996; Welberg, Seckl et al. 2000; Shoener, Baig et al. 2006; Uddin, Aiello et al. 2010).
Endophenotypes
PTSD is a complex diagnostic construct requiring exposure to a traumatic event and comprising a series of up to 17 symptoms organized into three symptom clusters. It is therefore difficult to tease out the multiple interacting genes likely to play a role in the heritability of this complex psychiatric disorder. Rather than examining the global diagnostic entity of PTSD, studying endophenotypes of the disorder may be more directly revealing. An endophenotype refers to a heritable, measurable biomarker associated with a deconstructed trait of a psychiatric illness, which is more directly amenable to genetic analysis (Gottesman and Gould 2003). Endophenotypes of PTSD may include dysregulation of the HPA axis, altered autonomic reactivity (as measured by galvanic skin response or heart rate variability), and an exaggerated startle response in the context of conditioned fear (Broekman, Olff et al. 2007).
The exaggerated startle response that is characteristic of PTSD can be studied in the context of the fear conditioning model by examining fear inhibition – the ability to suppress fear responses in the presence of safety cues. One of the primary laboratory models used for behavioral analysis of fear inhibition is the conditioned inhibition paradigm, in which the pairing of a conditioned stimulus (CS+) with an aversive stimulus (US) is intermingled with a separate stimulus (CS-) that is never paired with the US, and thus represents a safety signal. The acoustic startle response provides a useful behavioral outcome measure of fear in these paradigms as it is easily measured in the lab, is biologically relevant to PTSD, and is mediated by the output of the amygdala (LeDoux, Iwata et al. 1988; Davis 1992; Grillon 2008). This response is typically reported as fear-potentiated startle, which is the relative increase in the acoustic startle response elicited in the presence of the reinforced conditioned stimulus.
Myers and Davis (2004) developed an animal model using a conditional discrimination procedure that allows for the independent evaluation of excitation and inhibition of fear conditioning. A version of this paradigm studied in humans contains a danger signal (AX+), a safety signal (BX-), and a safety transfer test (combination of A and B, where B reduces fear to A). The A stimulus elicits fear potentiation of startle while the B stimulus elicits reduced startle compared to A (Jovanovic and Ressler 2010).
Conditioned inhibition of the acoustic startle response as a result of learning the safety signal has been demonstrated in healthy controls and in combat veterans with low levels of current PTSD symptoms (Jovanovic, Norrholm et al. 2009). In contrast, study subjects with high levels of PTSD symptoms were unable to reduce startle to AB trials. This has now been demonstrated in combat veterans from the United States, as well as veterans from Croatia (Jovanovic, Norrholm et al. 2007), and in a traumatized civilian population in urban Atlanta (Jovanovic, Norrholm et al. 2010). As these individuals with high levels of PTSD symptoms did not demonstrate fear potentiation to the danger signal (AX+) along, the results suggest a selective deficit in fear inhibition as a key trait of PTSD. It is unclear whether an impaired ability to inhibit fear responses is a risk factor for PTSD versus an acquired trait associated with the disorder.
As reviewed above, the neurocircuitry of fear inhibition involves the limbic-frontal neuronal systems. The amygdala is required for associative learning of conditioned fear and elaboration of the fear response. Specifically, output from the CeA to the pons in the midbrain mediates the fear-induced increase in the acoustic startle response. Descending inhibitory input from the mPFC to the amygdala is required for extinction or inhibition of conditioned fear responses. The key role of this circuit in regulating fear potentiated startle suggests that researchers should investigate genes known to impact on this system to assess the genetic basis of conditioned fear. As noted above a polymorphism in 5-HTTLPR is known to be associated with relative decoupling of mPFC inhibition from the amygdala (Pezawas, Meyer-Lindenberg et al. 2005) and interactions between this polymorphism and a history of trauma have been found to be predictive of PTSD. In addition, a recent study found great fear-potentiated startle in individuals with the s (risk) allele of this gene (Lonsdorf, Weike et al. 2009). It would be of interest to determine if those individuals with the s allele also demonstrate deficits in conditioned inhibition of fear.
On a molecular level, a significant amount of data implicate brain-derived neurotrophic factor (BDNF) in the plasticity underlying associative fear conditioning and extinction of conditioned fear through its TrkB receptor (Rattiner, Davis et al. 2004; Chhatwal, Stanek-Rattiner et al. 2006; Choi, Maguschak et al. 2010). Two studies of genetic polymorphisms in the BDNF gene found no significant association with PTSD (Lee, Kang et al. 2006; Zhang, Ozbay et al. 2006). However, as previously noted, studies of the main effects of genes on the outcome of PTSD have been of low yield. More interestingly, a very recent study demonstrated that humans and transgenic mice that carry the Met allele of the BDNF Met/Val polymorphism, both have a deficit in extinction of conditioned fear (Soliman, Glatt et al. 2010). Additionally, it would be very enlightening to examine the impact of polymorphisms in the BDNF gene on conditioned inhibition of the startle response. Finally, data also suggests that activation of GABA receptors within the amygdala is critically involved in the regulation of fear and its inhibition with extinction learning. As polymorphisms within the GABA alpha 2 receptor gene have been demonstrated to interact with childhood trauma to predict adult PTSD (Nelson, Agrawal et al. 2009), future studies should be directed to examine the impact of these genotypes on conditioned inhibition of the fear response.
Future Directions
While there are no agreed upon validated animals models of PTSD, there are established animal models of conditioned fear and fear inhibition amongst other biomarkers associated with PTSD as noted above. Human genetic investigations can be hampered by limitations such as sample size, statistical power and the inherent drawbacks of association studies. “Top down” translational studies, in which human genetic findings are validated by subsequent work in rodents, can address some of these methodological impediments. Alternatively, animal models can be used to identify genes of interest that might confer vulnerability to the disorder or mediate key clinical elements of the phenotype. In mice, for example, recent studies have shown that a quantitative trait locus on chromosome 1 is associated with anxiety-related phenotypes (Flint 2003). The principal quantitative trait gene for this linkage is RGS2, a specific genotype of which has been identified to interact with stress/trauma to predict PTSD in a human GxE study (Amstadter, Koenen et al. 2009).
Using a genome-wide association study (GWAS) avoids the need to have prior knowledge of a specific candidate gene; GWAS is emerging as an important complementary methodology for providing an unbiased approach to identifying genes of interest for further study. GWAS allows for the examination of the full genome of an individual and for comparison of genetic differences between those with versus without a specific disorder. Of course, similar to the above discussion on the importance of understanding environmental, population, and trauma variables relative to the study of genetic factors, it will be critical that the cases and controls are well matched for PTSD risk factors in such GWAS analyses. These studies offer an opportunity to identify novel risk variants for PTSD that in turn have the potential to inform our understanding of the etiology of the disorder. Further, they will help to identify which molecular and genetic pathways should be prioritized for mechanistic study in the animal models of PTSD endophenotypes. Early results have illustrated the feasibility and potential power of GWAS to identify biomarkers for anxiety-related behaviors and suggest its utility in future studies of PTSD. It should be noted that the utility of GWAS has been limited by issues such as the size of the sample population, relatively small effect sizes, and critically, lack of attention to matching for risk factors in case and control study populations. Nevertheless, there is great optimism that this methodology will ultimately lead to the discovery of novel loci for susceptibility to and symptomatology of anxiety disorders, such as PTSD (Norrholm and Ressler 2009).
Summary and Implications
PTSD is a prevalent anxiety disorder that can develop in the aftermath of exposure to a traumatic event. It is associated with significant adverse impact on occupational and social function, as well as physical and mental health and can become chronically disabling. The etiology and maintenance of PTSD is the result of multiple, complex interactions between genes and environmental influences. Understanding the nature of these influences, as well as their interaction, will be important to informing our understanding of risk and resilience to trauma in the development of PTSD. Such knowledge may allow improved targeting of primary prevention amongst vulnerable individuals or populations, as well as targeted treatment interventions, which may include early intervention for those most at risk in the aftermath of a trauma or pharmacologic interventions directed at specific genes or neurobiological pathways demonstrated to play a role in the development and/or symptomatic manifestations of PTSD. In addition, identifying specific modifiable environments associated with PTSD risk (such as social support or the use of safety signals) may broaden post-trauma intervention approaches to reduce risk for PTSD. While the literature on the genetics of PTSD is still relatively early, it is rapidly evolving with an increasing number of studies demonstrating important GxE interactions associated with risk for PTSD. It can be anticipated that in the coming years, the field will see advances in identifying novel genes associated with PTSD via methodologies such as GWAS and translational studies in animal models, as well as further elucidating the genetic underpinnings of PTSD endophenotypes, such as fear conditioning and extinction, and demonstrated epigenetic mechanisms of modifying gene expression in response to developmentally timed environmental exposures.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams R, Boscarino J. Predictors of PTSD and delayed PTSD after disaster: The impact of exposure and psychosocial resources. The Journal of nervous and mental disease. 2006;194(7):485. doi: 10.1097/01.nmd.0000228503.95503.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amstadter A, Koenen K, et al. Variant in RGS2 moderates posttraumatic stress symptoms following potentially traumatic event exposure. Journal of anxiety disorders. 2009;23(3):369–373. doi: 10.1016/j.janxdis.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amstadter A, Nugent N, et al. Genetics of PTSD: Fear conditioning as a model for future research. Psychiatric Annals. 2009;39(6):358. doi: 10.3928/00485713-20090526-01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann A, Sedgley T, et al. Glucocorticoid receptor polymorphisms and post-traumatic stress disorder. Psychoneuroendocrinology. 2005;30(3):297–306. doi: 10.1016/j.psyneuen.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Baker D, West S, et al. Serial CSF corticotropin-releasing hormone levels and adrenocortical activity in combat veterans with posttraumatic stress disorder. American Journal of Psychiatry. 1999;156(4):585. doi: 10.1176/ajp.156.4.585. [DOI] [PubMed] [Google Scholar]
- Benarroch E. The locus ceruleus norepinephrine system: Functional organization and potential clinical significance. Neurology. 2009;73(20):1699. doi: 10.1212/WNL.0b013e3181c2937c. [DOI] [PubMed] [Google Scholar]
- Binder E, Bradley R, et al. Association of FKBP5 polymorphisms and childhood abuse with risk of posttraumatic stress disorder symptoms in adults. Jama. 2008;299(11):1291. doi: 10.1001/jama.299.11.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard EB, Kolb LC, et al. Cardiac response to relevant stimuli as an adjunctive tool for diagnosing post-traumatic stress disorder in Vietnam veterans. Behavior Therapy. 1986;17:592–606. [Google Scholar]
- Brand S, Brennan P, et al. The impact of maternal childhood abuse on maternal and infant HPA axis function in the postpartum period. Psychoneuroendocrinology. 2010;35(5):686–693. doi: 10.1016/j.psyneuen.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremner D, Vermetten E, et al. Cortisol, dehydroepiandrosterone, and estradiol measured over 24 hours in women with childhood sexual abuse-related posttraumatic stress disorder. The Journal of nervous and mental disease. 2007;195(11):919. doi: 10.1097/NMD.0b013e3181594ca0. [DOI] [PubMed] [Google Scholar]
- Bremner J, Licinio J, et al. Elevated CSF corticotropin-releasing factor concentrations in posttraumatic stress disorder. American Journal of Psychiatry. 1997;154(5):624. doi: 10.1176/ajp.154.5.624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslau N, Kessler R. The stressor criterion in DSM-IV posttraumatic stress disorder: an empirical investigation. Biological Psychiatry. 2001;50(9):699–704. doi: 10.1016/s0006-3223(01)01167-2. [DOI] [PubMed] [Google Scholar]
- Breslau N, Kessler RC, et al. Trauma and posttraumatic stress disorder in the community: the 1996 Detroit Area Survey of Trauma. Archives of General Psychiatry. 1998;55(7):626–632. doi: 10.1001/archpsyc.55.7.626. [DOI] [PubMed] [Google Scholar]
- Broekman B, Olff M, et al. The genetic background to PTSD. Neuroscience & Biobehavioral Reviews. 2007;31(3):348–362. doi: 10.1016/j.neubiorev.2006.10.001. [DOI] [PubMed] [Google Scholar]
- Chantarujikapong S, Scherrer J, et al. A twin study of generalized anxiety disorder symptoms, panic disorder symptoms and post-traumatic stress disorder in men. Psychiatry Research. 2001;103(2-3):133–145. doi: 10.1016/s0165-1781(01)00285-2. [DOI] [PubMed] [Google Scholar]
- Charmandari E, Tsigos C, et al. Endocrinology of the stress response1. Physiology. 2005;67 doi: 10.1146/annurev.physiol.67.040403.120816. [DOI] [PubMed] [Google Scholar]
- Charuvastra A, Cloitre M. Social bonds and posttraumatic stress disorder. Annual review of psychology. 2008;59:301. doi: 10.1146/annurev.psych.58.110405.085650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhatwal JP, Stanek-Rattiner L, et al. Amygdala BDNF signaling is required for consolidation but not encoding of extinction. Nat Neurosci. 2006;9:870–872. doi: 10.1038/nn1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi D, Maguschak K, et al. Prelimbic cortical BDNF is required for memory of learned fear but not extinction or innate fear. Proceedings of the National Academy of Sciences. 2010;107(6):2675. doi: 10.1073/pnas.0909359107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claes J. Corticotropin-releasing hormone (CRH) in psychiatry: from stress to psychopathology. Annals of medicine. 2004;36(1):50–61. doi: 10.1080/07853890310017044. [DOI] [PubMed] [Google Scholar]
- Comings D, Comings B, et al. The dopamine D2 receptor locus as a modifying gene in neuropsychiatric disorders. Jama. 1991;266(13):1793. [PubMed] [Google Scholar]
- Comings DE, Muhleman D, et al. Dopamine D2 receptor (DRD2) gene and susceptibility to posttraumatic stress disorder: a study and replication. Biological Psychiatry. 1996;40(5):368–372. doi: 10.1016/0006-3223(95)00519-6. [DOI] [PubMed] [Google Scholar]
- Davis M. The role of the amygdala in fear and anxiety. Annual Review of Neuroscience. 1992;15(1):353–375. doi: 10.1146/annurev.ne.15.030192.002033. [DOI] [PubMed] [Google Scholar]
- De Kloet C, Vermetten E, et al. Assessment of HPA-axis function in posttraumatic stress disorder: pharmacological and non-pharmacological challenge tests, a review. Journal of Psychiatric Research. 2006;40(6):550–567. doi: 10.1016/j.jpsychires.2005.08.002. [DOI] [PubMed] [Google Scholar]
- DSM, I. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 1994 [PubMed] [Google Scholar]
- Fanselow M, LeDoux J. Why we think plasticity underlying Pavlovian fear conditioning occurs in the basolateral amygdala. Neuron. 1999;23:229–232. doi: 10.1016/s0896-6273(00)80775-8. [DOI] [PubMed] [Google Scholar]
- Flint J. Analysis of quantitative trait loci that influence animal behavior. Journal of Neurobiology. 2003;54(1):46–77. doi: 10.1002/neu.10161. [DOI] [PubMed] [Google Scholar]
- Francis D, Diorio J, et al. Nongenomic transmission across generations of maternal behavior and stress responses in the rat. Science. 1999;286(5442):1155. doi: 10.1126/science.286.5442.1155. [DOI] [PubMed] [Google Scholar]
- Fu Q, Koenen K, et al. Differential etiology of posttraumatic stress disorder with conduct disorder and major depression in male veterans. Biological Psychiatry. 2007;62(10):1088–1094. doi: 10.1016/j.biopsych.2007.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelernter J, Southwick S, et al. No association between D2 dopamine receptor (DRD2) “A” system alleles, or DRD2 haplotypes, and posttraumatic stress disorder. Biological Psychiatry. 1999;45(5):620–625. doi: 10.1016/s0006-3223(98)00087-0. [DOI] [PubMed] [Google Scholar]
- Gilbertson M, Shenton M, et al. Smaller hippocampal volume predicts pathologic vulnerability to psychological trauma. Nature Neuroscience. 2002;5(11):1242–1247. doi: 10.1038/nn958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman I, Gould T. The endophenotype concept in psychiatry: etymology and strategic intentions. American Journal of Psychiatry. 2003;160(4):636. doi: 10.1176/appi.ajp.160.4.636. [DOI] [PubMed] [Google Scholar]
- Grillon C. Models and mechanisms of anxiety: evidence from startle studies. Psychopharmacology. 2008;199(3):421–437. doi: 10.1007/s00213-007-1019-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubler T, Scammell J. Intronic hormone response elements mediate regulation of FKBP5 by progestins and glucocorticoids. Cell Stress Chaperones. 2004;9(3):243. doi: 10.1379/CSC-32R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic T, Norrholm S, et al. Impaired fear inhibition is a biomarker of PTSD but not depression. Depression and Anxiety. 2010;27(3):244–251. doi: 10.1002/da.20663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic T, Norrholm S, et al. Posttraumatic stress disorder may be associated with impaired fear inhibition: relation to symptom severity. Psychiatry Research. 2009;167(1-2):151–160. doi: 10.1016/j.psychres.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic T, Norrholm SD, et al. Conditioned and external fear inhibition in combat-related PTSD in Croatian war veterans. 37th Annual Meeting of the Society for Neuroscience; San Diego, CA. 2007. [Google Scholar]
- Jovanovic T, Ressler K. How the neurocircuitry and genetics of fear inhibition may inform our understanding of PTSD. American Journal of Psychiatry. 2010;167(6):648. doi: 10.1176/appi.ajp.2009.09071074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai K, Yamasue H, et al. Evidence for acquired pregenual anterior cingulate gray matter loss from a twin study of combat-related posttraumatic stress disorder. Biological Psychiatry. 2008;63(6):550–556. doi: 10.1016/j.biopsych.2007.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane TM, Zimering RT, et al. A behavioral formulation of posttraumatic stress disorder in Vietnam veterans. The Behavior Therapist. 1985;8:9–12. [Google Scholar]
- Kessler R. Posttraumatic stress disorder: the burden to the individual and to society. Journal of Clinical Psychiatry. 2000;61:4–14. [PubMed] [Google Scholar]
- Kessler RC, Berglund P, et al. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Archives of General Psychiatry. 2005;62(6):593–602. doi: 10.1001/archpsyc.62.6.593. [DOI] [PubMed] [Google Scholar]
- Kessler RC, Sonnega A, et al. Posttraumatic stress disorder in the national comorbidity survey. Archives of General Psychiatry. 1995;52:1048–1060. doi: 10.1001/archpsyc.1995.03950240066012. [DOI] [PubMed] [Google Scholar]
- Kilpatrick DG, Koenen KC, et al. The Serotonin Transporter Genotype and Social Support and Moderation of Posttraumatic Stress Disorder and Depression in Hurricane-Exposed Adults. Am J Psychiatry. 2007;164(11):1693–1699. doi: 10.1176/appi.ajp.2007.06122007. [DOI] [PubMed] [Google Scholar]
- Koenen K, Aiello A, et al. Modification of the association between serotonin transporter genotype and risk of posttraumatic stress disorder in adults by county-level social environment. American journal of epidemiology. 2009;169(6):704. doi: 10.1093/aje/kwn397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenen K, Fu Q, et al. Common genetic liability to major depression and posttraumatic stress disorder in men. Journal of affective disorders. 2008;105(1-3):109. doi: 10.1016/j.jad.2007.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenen K, Hitsman B, et al. A twin registry study of the relationship between posttraumatic stress disorder and nicotine dependence in men. Archives of General Psychiatry. 2005;62(11):1258. doi: 10.1001/archpsyc.62.11.1258. [DOI] [PubMed] [Google Scholar]
- Koenen K, Nugent N, et al. Gene-environment interaction in posttraumatic stress disorder. European archives of psychiatry and clinical neuroscience. 2008;258(2):82–96. doi: 10.1007/s00406-007-0787-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenen KC, Saxe G, et al. Polymorphisms in FKBP5 are associated with peritraumatic dissociation in medically injured children. Mol Psychiatry. 2005;10(12):1058–1059. doi: 10.1038/sj.mp.4001727. [DOI] [PubMed] [Google Scholar]
- Kolassa I, Ertl V, et al. Association study of trauma load and SLC6A4 promoter polymorphism in posttraumatic stress disorder: evidence from survivors of the Rwandan genocide. J Clin Psychiatry. 2010;71(5):543. doi: 10.4088/JCP.08m04787blu. [DOI] [PubMed] [Google Scholar]
- Kolassa I, Kolassa S, et al. The Risk of Posttraumatic Stress Disorder After Trauma Depends on Traumatic Load and the Catechol-O-Methyltransferase Val158Met Polymorphism. Biological Psychiatry. 2010;67(4):304–308. doi: 10.1016/j.biopsych.2009.10.009. [DOI] [PubMed] [Google Scholar]
- Kubzansky L, Koenen K, et al. Prospective study of posttraumatic stress disorder symptoms and coronary heart disease in the Normative Aging Study. Archives of General Psychiatry. 2007;64(1):109. doi: 10.1001/archpsyc.64.1.109. [DOI] [PubMed] [Google Scholar]
- Kulka RA, Schlenger WE, et al. Trauma and the Vietnam War generation: Report of findings from the national Vietnam Veterans Readjustment Study. New York: New York, Brunner/Mazel; 1990. [Google Scholar]
- Lappalainen J, Kranzler H, et al. A functional neuropeptide Y Leu7Pro polymorphism associated with alcohol dependence in a large population sample from the United States. Archives of General Psychiatry. 2002;59(9):825. doi: 10.1001/archpsyc.59.9.825. [DOI] [PubMed] [Google Scholar]
- LeDoux JE, Iwata J, et al. Different projections of the central amygdaloid nucleus mediate autonomic and behavioral correlates of conditioned fear. Journal of Neuroscience. 1988;8:2517–2529. doi: 10.1523/JNEUROSCI.08-07-02517.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Kang R, et al. No association between the brain derived neurotrophic factor gene Val66Met polymorphism and post traumatic stress disorder. Stress and Health. 2006;22(2):115–119. [Google Scholar]
- Lee H, Kwak S, et al. Association between serotonin 2A receptor gene polymorphism and posttraumatic stress disorder. PSYCHIATRY INVESTIGATION. 2007;4(2):104. [Google Scholar]
- Lee H, Lee M, et al. Influence of the serotonin transporter promoter gene polymorphism on susceptibility to posttraumatic stress disorder. Depression and Anxiety. 2005;21(3):135–139. doi: 10.1002/da.20064. [DOI] [PubMed] [Google Scholar]
- Lesch KP, Bengel D, et al. Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science. 1996;274(5292):1527–1531. doi: 10.1126/science.274.5292.1527. [DOI] [PubMed] [Google Scholar]
- Levitt N, Lindsay R, et al. Dexamethasone in the last week of pregnancy attenuates hippocampal glucocorticoid receptor gene expression and elevates blood pressure in the adult offspring in the rat. Neuroendocrinology. 1996;64(6):412–418. doi: 10.1159/000127146. [DOI] [PubMed] [Google Scholar]
- Likhtik E, Pelletier JG, et al. Prefrontal control of the amygdala. The Journal of Neuroscience. 2005;25(32):7429. doi: 10.1523/JNEUROSCI.2314-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Likhtik E, Popa D, et al. Amygdala intercalated neurons are required for expression of fear extinction. Nature. 2008;454(7204):642–645. doi: 10.1038/nature07167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonsdorf TB, Weike AI, et al. Genetic gating of human fear learning and extinction: Possible implications for gene-environment interaction in anxiety disorder. Psychological Science. 2009;20(2):198–206. doi: 10.1111/j.1467-9280.2009.02280.x. [DOI] [PubMed] [Google Scholar]
- Lyons M, Goldberg J, et al. Do genes influence exposure to trauma? A twin study of combat. American journal of medical genetics. 1993;48(1):22–27. doi: 10.1002/ajmg.1320480107. [DOI] [PubMed] [Google Scholar]
- McGowan P, Sasaki A, et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nature Neuroscience. 2009;12(3):342–348. doi: 10.1038/nn.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meaney M, Szyf M. Maternal care as a model for experience-dependent chromatin plasticity? TRENDS in Neurosciences. 2005;28(9):456–463. doi: 10.1016/j.tins.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Milad MR, Quirk GJ. Neurons in medial prefrontal cortex signal memory for fear extinction. Nature. 2002;420(6911):70–74. doi: 10.1038/nature01138. [DOI] [PubMed] [Google Scholar]
- Morgan C. Neuropeptide-Y, cortisol, and subjective distress in humans exposed to acute stress: replication and extension of previous report. Biological Psychiatry. 2002;52(2):136–142. doi: 10.1016/s0006-3223(02)01319-7. [DOI] [PubMed] [Google Scholar]
- Morgan C, III, Wang S, et al. Plasma neuropeptide-Y concentrations in humans exposed to military survival training. Biological Psychiatry. 2000;47(10):902–909. doi: 10.1016/s0006-3223(99)00239-5. [DOI] [PubMed] [Google Scholar]
- Mustapic M, Pivac N, et al. Dopamine beta-hydroxylase (DBH) activity and-1021C/T polymorphism of DBH gene in combat-related post-traumatic stress disorder. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics. 2007;144(8):1087–1089. doi: 10.1002/ajmg.b.30526. [DOI] [PubMed] [Google Scholar]
- Myers KM, Davis M. AX+, BX- discrimination learning in the fear-potentiated startle paradigm: possible relevance to inhibitory fear learning in extinction. Learn Mem. 2004;11(4):464–475. doi: 10.1101/lm.74704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson E, Agrawal A, et al. Association of childhood trauma exposure and GABRA2 polymorphisms with risk of posttraumatic stress disorder in adults. Molecular psychiatry. 2009;14(3):234–235. doi: 10.1038/mp.2008.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubig R, Siderovski D. Regulators of G-protein signalling as new central nervous system drug targets. Nature Reviews Drug Discovery. 2002;1(3):187–197. doi: 10.1038/nrd747. [DOI] [PubMed] [Google Scholar]
- Norrholm S, Ressler K. Genetics of anxiety and trauma-related disorders. Neuroscience. 2009;164(1):272–287. doi: 10.1016/j.neuroscience.2009.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novik K, Nimmrich I, et al. Epigenomics: genome-wide study of methylation phenomena. Current issues in molecular biology. 2002;4:111–128. [PubMed] [Google Scholar]
- O'Carroll R, Drysdale E, et al. Stimulation of the noradrenergic system enhances and blockade reduces memory for emotional material in man. Psychological Medicine. 1999;29(5):1083–1088. doi: 10.1017/s0033291799008703. [DOI] [PubMed] [Google Scholar]
- Orr SP, Metzger LJ, et al. Physiologic responses to sudden, loud tones in monozygotic twins discordant for combat exposure: association with posttraumatic stress disorder. Arch Gen Psychiatry. 2003;60(3):283–288. doi: 10.1001/archpsyc.60.3.283. [DOI] [PubMed] [Google Scholar]
- Peters J, Kalivas PW, et al. Extinction circuits for fear and addiction overlap in prefrontal cortex. Learn Mem. 2009;16(5):279–288. doi: 10.1101/lm.1041309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezawas L, Meyer-Lindenberg A, et al. 5-HTTLPR polymorphism impacts human cingulate-amygdala interactions: a genetic susceptibility mechanism for depression. Nature. 2005;8(6):828–834. doi: 10.1038/nn1463. [DOI] [PubMed] [Google Scholar]
- Pitman RK, Orr SP, et al. Psychophysiologic assessment of posttraumatic stress disorder imagery in Vietnam combat veterans. Arch Gen Psychiatry. 1987;44(11):970–975. doi: 10.1001/archpsyc.1987.01800230050009. [DOI] [PubMed] [Google Scholar]
- Puglisi-Allegra S, Cabib S. Psychopharmacology of dopamine: the contribution of comparative studies in inbred strains of mice. Progress in neurobiology. 1997;51(6):637–661. doi: 10.1016/s0301-0082(97)00008-7. [DOI] [PubMed] [Google Scholar]
- Rainnie D. Serotonergic modulation of neurotransmission in the rat basolateral amygdala. Journal of neurophysiology. 1999;82(1):69. doi: 10.1152/jn.1999.82.1.69. [DOI] [PubMed] [Google Scholar]
- Rattiner L, Davis M, et al. Brain-derived neurotrophic factor and tyrosine kinase receptor B involvement in amygdala-dependent fear conditioning. Journal of Neuroscience. 2004;24(20):4796. doi: 10.1523/JNEUROSCI.5654-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Gorodetsky E, et al. Interaction of FKBP5, a stress-related gene, with childhood trauma increases the risk for attempting suicide. Neuropsychopharmacology. 2010;35(8):1674–1683. doi: 10.1038/npp.2009.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sack W, Clarke G, et al. Posttraumatic stress disorder across two generations of Cambodian refugees. Journal of the American Academy of Child & Adolescent Psychiatry. 1995;34(9):1160–1166. doi: 10.1097/00004583-199509000-00013. [DOI] [PubMed] [Google Scholar]
- Seckl J, Meaney M. Glucocorticoid “programming” and PTSD risk. Annals of the New York Academy of Sciences. 2006;1071(1):351–378. doi: 10.1196/annals.1364.027. [DOI] [PubMed] [Google Scholar]
- Segman R, Cooper-Kazaz R, et al. Association between the dopamine transporter gene and posttraumatic stress disorder. Molecular psychiatry. 2002;7(8):903. doi: 10.1038/sj.mp.4001085. [DOI] [PubMed] [Google Scholar]
- Segman RH, Shefi N, et al. Peripheral blood mononuclear cell gene expression profiles identify emergent post-traumatic stress disorder among trauma survivors. Mol Psychiatry. 2005;10(5):500–513. 425. doi: 10.1038/sj.mp.4001636. [DOI] [PubMed] [Google Scholar]
- Shoener J, Baig R, et al. Prenatal exposure to dexamethasone alters hippocampal drive on hypothalamic-pituitary-adrenal axis activity in adult male rats. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology. 2006;290(5):R1366. doi: 10.1152/ajpregu.00757.2004. [DOI] [PubMed] [Google Scholar]
- Soliman F, Glatt C, et al. A genetic variant BDNF polymorphism alters extinction learning in both mouse and human. Science. 2010;327(5967):863. doi: 10.1126/science.1181886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southwick S, Bremner J, et al. Role of norepinephrine in the pathophysiology and treatment of posttraumatic stress disorder. Biological Psychiatry. 1999;46(9):1192–1204. doi: 10.1016/s0006-3223(99)00219-x. [DOI] [PubMed] [Google Scholar]
- Southwick S, Davis M, et al. Relationship of enhanced norepinephrine activity during memory consolidation to enhanced long-term memory in humans. American Journal of Psychiatry. 2002;159(8):1420. doi: 10.1176/appi.ajp.159.8.1420. [DOI] [PubMed] [Google Scholar]
- Stein M, Jang K, et al. Genetic and environmental influences on trauma exposure and posttraumatic stress disorder symptoms: a twin study. American Journal of Psychiatry. 2002;159(10):1675. doi: 10.1176/appi.ajp.159.10.1675. [DOI] [PubMed] [Google Scholar]
- Thaller V, Vrkljan M, et al. The potential role of hypocortisolism in the pathophysiology of PTSD and psoriasis. Collegium antropologicum. 1999;23(2):611–620. [PubMed] [Google Scholar]
- True W, Rice J, et al. A twin study of genetic and environmental contributions to liability for posttraumatic stress symptoms. Archives of General Psychiatry. 1993;50(4):257. doi: 10.1001/archpsyc.1993.01820160019002. [DOI] [PubMed] [Google Scholar]
- Uddin M, Aiello A, et al. Epigenetic and immune function profiles associated with posttraumatic stress disorder. Proceedings of the National Academy of Sciences. 2010;107(20):9470. doi: 10.1073/pnas.0910794107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal-Gonzalez I, Vidal-Gonzalez B, et al. Microstimulation reveals opposing influences of prelimbic and infralimbic cortex on the expression of conditioned fear. Learning & Memory. 2006;13(6):728–733. doi: 10.1101/lm.306106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voisey J, Swagell C, et al. The DRD2 gene 957C> T polymorphism is associated with posttraumatic stress disorder in war veterans. Depress Anxiety. 2009;26(1):28–33. doi: 10.1002/da.20517. [DOI] [PubMed] [Google Scholar]
- Weaver I. Epigenetic programming by maternal behavior and pharmacological intervention. Nature versus nurture: let's call the whole thing off. Epigenetics: official journal of the DNA Methylation Society. 2007;2(1):22. doi: 10.4161/epi.2.1.3881. [DOI] [PubMed] [Google Scholar]
- Weaver I, Szyf M, et al. From maternal care to gene expression: DNA methylation and the maternal programming of stress responses. Endocrine Research. 2002;28(4):699–699. doi: 10.1081/erc-120016989. [DOI] [PubMed] [Google Scholar]
- Welberg L, Seckl J, et al. Inhibition of 11 -hydroxysteroid dehydrogenase, the foeto-placental barrier to maternal glucocorticoids, permanently programs amygdala GR mRNA expression and anxiety-like behaviour in the offspring. European Journal of Neuroscience. 2000;12(3):1047–1054. doi: 10.1046/j.1460-9568.2000.00958.x. [DOI] [PubMed] [Google Scholar]
- Xian H, Chantarujikapong SI, et al. Genetic and environmental influences on posttraumatic stress disorder, alcohol and drug dependence in twin pairs. Drug and Alcohol Dependence. 2000;61(1):95–102. doi: 10.1016/s0376-8716(00)00127-7. [DOI] [PubMed] [Google Scholar]
- Xie P, Kranzler H, et al. Interaction of FKBP5 with Childhood Adversity on Risk for Post-Traumatic Stress Disorder. Neuropsychopharmacology. 2010;35(8):1684–1692. doi: 10.1038/npp.2010.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yehuda R. Post-traumatic stress disorder. New England Journal of Medicine. 2002;346(2):108. doi: 10.1056/NEJMra012941. [DOI] [PubMed] [Google Scholar]
- Yehuda R. Status of Glucocorticoid Alterations in Post traumatic Stress Disorder. Annals of the New York Academy of Sciences. 2009;1179(1):56–69. doi: 10.1111/j.1749-6632.2009.04979.x. [DOI] [PubMed] [Google Scholar]
- Yehuda R, Bell A, et al. Maternal, not paternal, PTSD is related to increased risk for PTSD in offspring of Holocaust survivors. Journal of Psychiatric Research. 2008;42(13):1104–1111. doi: 10.1016/j.jpsychires.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yehuda R, Bierer L. The relevance of epigenetics to PTSD: Implications for the DSM-V. Journal of Traumatic Stress. 2009;22(5):427–434. doi: 10.1002/jts.20448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yehuda R, Cai G, et al. Gene Expression Patterns Associated with Posttraumatic Stress Disorder Following Exposure to the World Trade Center Attacks. Biol Psychiatry. 2009 doi: 10.1016/j.biopsych.2009.02.034. [DOI] [PubMed] [Google Scholar]
- Yehuda R, Engel S, et al. Transgenerational effects of posttraumatic stress disorder in babies of mothers exposed to the World Trade Center attacks during pregnancy. Journal of Clinical Endocrinology & Metabolism. 2005;90(7):4115. doi: 10.1210/jc.2005-0550. [DOI] [PubMed] [Google Scholar]
- Yehuda R, Flory J, et al. Putative biological mechanisms for the association between early life adversity and the subsequent development of PTSD. Psychopharmacology. 2010:1–13. doi: 10.1007/s00213-010-1969-6. [DOI] [PubMed] [Google Scholar]
- Yehuda R, Halligan S, et al. Relationship of parental trauma exposure and PTSD to PTSD, depressive and anxiety disorders in offspring. Journal of Psychiatric Research. 2001;35(5):261–270. doi: 10.1016/s0022-3956(01)00032-2. [DOI] [PubMed] [Google Scholar]
- Yehuda R, McFarlane A, et al. Predicting the development of posttraumatic stress disorder from the acute response to a traumatic event. Biological Psychiatry. 1998;44(12):1305–1313. doi: 10.1016/s0006-3223(98)00276-5. [DOI] [PubMed] [Google Scholar]
- Yehuda R, Southwick S, et al. Low urinary cortisol excretion in patients with posttraumatic stress disorder. The Journal of nervous and mental disease. 1990;178(6):366. doi: 10.1097/00005053-199006000-00004. [DOI] [PubMed] [Google Scholar]
- Yehuda R, Teicher M, et al. Parental posttraumatic stress disorder as a vulnerability factor for low cortisol trait in offspring of holocaust survivors. Archives of General Psychiatry. 2007;64(9):1040. doi: 10.1001/archpsyc.64.9.1040. [DOI] [PubMed] [Google Scholar]
- Yehuda R, Teicher M, et al. Cortisol regulation in posttraumatic stress disorder and major depression: a chronobiological analysis. Biological Psychiatry. 1996;40(2):79–88. doi: 10.1016/0006-3223(95)00451-3. [DOI] [PubMed] [Google Scholar]
- Young R, Lawford B, et al. Harmful drinking in military veterans with post-traumatic stress disorder: association with the D2 dopamine receptor A1 allele. Alcohol and Alcoholism. 2002;37(5):451. doi: 10.1093/alcalc/37.5.451. [DOI] [PubMed] [Google Scholar]
- Zhang H, Ozbay F, et al. Brain derived neurotrophic factor (BDNF) gene variants and Alzheimer's disease, affective disorders, posttraumatic stress disorder, schizophrenia, and substance dependence. Am J Med Genet B Neuropsychiatr Genet. 2006;141B(4):387–393. doi: 10.1002/ajmg.b.30332. [DOI] [PMC free article] [PubMed] [Google Scholar]