

Abstract
An SAR study on the Dmt-substituted enkephalin-like tetrapeptide with a N-phenyl-N-piperidin-4-yl propionamide moiety at C-terminal was performed, and has resulted in highly potent ligands at μ and δ opioid receptors. In general, ligands with the substitution of D-Nle2 and halogenation of the aromatic ring of Phe4 showed highly increased opioid activities. Ligand 6 with good biological activities in vitro demonstrated potent in vivo antihyperalgesic and antiallodynic effects in the tail-flick assay.
Introduction
Many studies have shown that co-administration of a small amount of a δ opioid agonist can reduce serious side effects of morphine, a μ agonist, such as tolerance, while it increases the potency and efficacy.1-3 Furthermore several studies have observed synergistic antinociceptive effects between μ agonists and δ agonists.4 These observations imply that the modulation of both μ and δ receptors can allow treatment of pain with lower doses of μ agonists, therefore lessening μ receptor-mediated side effects. Our group previously sought to design nonselective bifunctional ligands possessing μ and δ agonist activities and as a result developed the very potent μ and δ opioid ligand 12 (Ki = 0.36 nM and 0.38 nM at hDOR and rMOR, respectively; IC50 = 1.8 nM and 8.5 nM in MVD and GPI assays, respectively) with the C-terminus of an enkephalin-like tetrapeptide (H-Dmt-DAla-Gly-Phe-OH) linked to a N-phenyl-N-piperidin-4-yl propionamide moiety which is a part of the fentanyl structure.5 We found that the propionamide moiety in the ligand resulted in a great increase in lipophilicity (aLogP = 2.96) which can improve the bioavailability of the ligand.
Despite their high potency and important role in combating pain, natural opioid peptides are limited as clinically viable drugs because of poor bioavailability, mainly due to their inability to penetrate the blood-brain barrier (BBB) and rapid degradation in vivo by peptidases. Increasing the lipophilicity can improve analgesia with a concomitant enhancement of bioavailability since the lipophilicity is a key factor in determining the rate at which a drug crosses the BBB. Methylation and halogenation enhance the lipophilicity and cell permeability by reducing overall hydrogen bonding. For example, addition of a chlorine on the Phe4 residue of [D-Pen2,D-Pen5]enkephalin (DPDPE) led to a significant increase in cell permeability in both in vivo and in vitro studies. 6-7
Pursuing enhanced bioavailability at the μ and δ receptors by increasing lipophilicity, a systematic Structure-Activity Relationship (SAR) study on the ligand 12 was performed (Figure 1). The propionamide moiety and the Dmt1 residue were conserved to increase cell permeability and potency, respectively. As a result, a series of highly lipophilic (aLogP: 3.01 - 4.74 in Table 1) nonselective μ and δ opioid agonists were obtained.
Figure 1. Design of opioid ligands.

Table 1. Structure and analytical data of opioid ligands.
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| no | 1 | 2 | 3 | 4 | aLogP11 | molecular formula | HRMSa (M-TFA + H)+ | HPLCb (tR, min) | ||
| Observed | calcd | A | B | |||||||
| 1 | Dmt | DNle | Gly | Phe | 3.66 | C42H56N6O6 | 741.4325 | 741.4340 | 19.3 | 10.8 |
| 2 | Dmt | DNle | Phe | 4.20 | C40H53N5O5 | 684.4109 | 684.4125 | 19.8 | 11.9 | |
| 3 | Dmt | DAla | Phe | 3.52 | C37H47N5O5 | 642.3665 | 642.3655 | 18.3 | 8.9 | |
| 4 | Dmt | DNle | Gly | Phe(Cl) | 4.18 | C42H55ClN6O6 | 775.3995 | 775.3950 | 20.7 | 12.5 |
| 5 | Dmt | DNle | Gly | Phe(F) | 3.74 | C42H55FN6O6 | 759.4247 | 759.4245 | 20.0 | 12.0 |
| 6 | Dmt | DAla | Gly | Phe(Cl) | 3.47 | C39H49ClN6O6 | 733.3494 | 733.3480 | 19.2 | 10.3 |
| 7 | Dmt | DNle | Phe(Cl) | 4.74 | C40H52ClN5O5 | 718.3713 | 718.3735 | 22.2 | 14.0 | |
| 8 | Dmt | DAla | Phe(Cl) | 3.97 | C37H46ClN5O5 | 676.3272 | 676.3266 | 19.8 | 11.0 | |
| 9 | Dmt | DAla | Gly | Phe(F) | 3.01 | C39H49FN6O6 | 717.3797 | 717.3776 | 17.7 | 9.5 |
| 10 | Dmt | DTic | Gly | Phe(F) | 4.02 | C46H53FN6O6 | 805.4055 | 805.4089 | 20.3 | 12.0 |
| 11 | Dmt | DTic | Gly | Phe(Cl) | 4.46 | C46H53ClN6O6 | 821.3799 | 821.3793 | 21.2 | 13.1 |
| 12 | Dmt | DAla | Gly | Phe | 2.96 | C39H50N6O6 | 699.3852 | 699.3870 | 20.1 | 12.7 |
FAB-MS (JEOL HX110 sector instrument) or MALDI-TOF.
Performed on a Hewlett Packard 1100 [C-18, Microsorb-MV™, 4.6 mm × 250 mm, 5 μm]. (A)10-90% of acetonitrile containing 0.1% TFA within 40 min and up to 100% within an additional 5 min, 1 mL/min, (B) 25-65% of acetonitrile containing 0.1% TFA within 20 min and up to 100% within an additional 5 min, 1 mL/min.
Results and Discussion
As published before, the ligands designed were prepared by stepwise solution-phase peptide syntheses using Nα-Boc-chemistry and the crude products were isolated by preparative RP-HPLC to afford > 98% pure compounds in 40-50% overall yields.5 During the chain elongations, pure peptide intermediates were obtained by simple precipitation from appropriate organic solvents, usually diethyl ether. Their in vitro bioassays were carried out as previously described.8
Opioid binding affinities at the human δ opioid receptor (hDOR) and the rat μ opioid receptor (rMOR) were determined by competition analyses against [3H]DPDPE (δ) and [3H]DAMGO (μ) using membrane preparations from transfected HN9.10 cells that constitutively express the respective receptors. Opioid agonist efficacy was examined by monitoring [35S]GTP-γ-S binding. Functional assays to evaluate their opioid agonist activities were performed using the stimulated isolated mouse vas deferens (MVD, δ) and guinea pig ileum (GPI, μ) bioassays. In general, the functional assay results correlated with those from the [35S]GTP-γ-S binding assay.
Our SAR study showed that replacement of DAla2 with DNle2, and halogenation at the 4 position of the aromatic ring of Phe4, increased opioid activitiy at both receptors in vitro, which was in agreement with the other observations, 9-10 along with increased lipophilicity (aLogP increase: 0.7 for DNle, 0.5 for Cl).11 First of all, replacement of DAla in 12 with DNle led to ligand 1, which showed increased opioid agonist activities [IC50 = 0.69 nM (3-fold increase) in the MVD assay; IC50 = 1.6 nM (5-fold increase) in the GPI assay] and efficacies [EC50 = 0.10 nM (8-fold increase) with Emax = 47% at hDOR; EC50 = 0.11 nM (8-fold increase) with Emax = 77% at rMOR in the [32S]GTP-γ–S assay], whereas its binding affinities at both receptors were not affected distinctly. The same trend was observed in the other ligands in which the DAla residue was replaced by DNle. The DNle substitution caused greater effects on the functional activities and efficacies than on the binding affinities, suggesting that the change of functional efficiency was due to increased lipophilicity.
To reduce pharmacophore size but increase lipophilicity, the Gly3 residue in ligands 1, 4, 6 and 12 was truncated, yielding ligands 2, 7, 8 and 3, respectively. The resulting ligands had decreased bioactivities at both receptors but raised μ selectivity because δ receptor activity was further reduced. Even though 3 had increased binding affinity (Ki = 0.15 nM) and agonist activity (IC50 = 3.6 nM) at the μ opioid receptor 2-fold by the truncation, in general, the truncation of Gly was not tolerated for both opioid receptors. It has been known that the relative proximity of the Phe4 aromatic ring and Tyr1 aromatic ring is critical for δ versus μ receptor selectivity.11 In our case, shortening their proximity by the truncation of Gly3 turned out to reduce δ receptor activity much more than μ receptor activity and subsequently to increase μ selectivity. All the Gly truncated ligands showed μ opioid receptor selectivity in the binding and functional assays.
All of the ligands with Cl (or F) at the 4′ position of the aromatic ring of Phe4 showed highly increased opioid activity in both binding and functional assays. Ligand 5, in which DNle2 and Phe(F)4 were substituted, showed the most potent subnanomolar opioid agonist activity (IC50 = 0.37 nM and 0.26 nM in the MVD and GPI assays, respectively) with excellent efficacy (EC50 = 0.07 nM, Emax = 48% at hDOR; EC50 = 0.29 nM, Emax = 98% at rMOR) at both receptors. The binding affinity at the μ receptor was subnanomolar (Ki = 0.02 nM) and the Emax at rMOR in the [35S]GTP-γ-S binding assay was 98% which represents high efficacy at the receptor. These results suggest that Cl (or F) in the opioid ligands could increase cell permeability and enhance potency which is more pronounced for the μ receptor, thus producing very well balanced (IC50δ/IC50μ < 2 in ligands 4, 5, and 9 mixed agonist activities for both receptors, respectively. On the contrary, the p-Cl substitution in the Gly truncated ligands 7 and 8 decreased opioid activity in all of the assays, especially the functional assays, up to 47-fold (IC50 = 250 nM and 220 nM for 7, IC50 = 180 nM and 93 nM for 8 at MVD and GPI, respectively), but retained μ opioid receptor selectivity in ligands 2 and 3.
Dmt-Tic is a highly potent selective δ opioid antagonist, and this modification has been recognized as leading to significant alterations in opioid activities and selectivities.12 In utilizing this potent pharmacophore to alter agonist functions at both opioid receptors, the Tic residue was modified to D-Tic in ligands 10 and 11. These ligands showed biological activities analogous to the other ligands, which confirmed that the constrained and very hydrophobic D-Tic residue could be replaced for the flexible D-Nle and D-Ala residues in the enkephalin structure. Both ligands showed high δ opioid receptor selectivities (δ/μ=0.03 for 9, and 0.04 for 10) in the functional assays, while ligand 10 gave the most potent agonist activity at the δ opioid receptor (IC50 = 0.029 nM in the MVD). No δ opioid antagonist activity was observed for either of the D-Tic-containing ligands.
Based on the in vitro bioassay results showing balanced high μ and δ opioid activities (Ki = 0.14 nM, EC50 = 0.16 nM at hDOR; IC50 = 0.70 nM in MVD, Ki = 0.14 nM, EC50 = 0.31 nM at rDOR; IC50 = 2.6 nM in GPI), ligand 6 was chosen for in vivo assays. Neuropathic pain was induced by L5/L6 spinal nerve ligation (SNL) in male Sprague Dawley rats.13 Thermal hypersensitivity and tactile allodynia were determined by paw withdrawal latencies to infrared radiant heat and paw withdrawal thresholds from von Frey filaments, respectively, which were applied to the plantar aspect of the hind paw. The time course of paw withdrawal latency or threshold after intrathecal microinjections of 6 at 3μg/5μL, 10μg/5μL, and 30μg/5μL were examined (Figure 2 and Figure 3). A dose-response curve was generated at the time of peak effect (30 min). The results showed that 6 attenuates tactile allodynia and thermal hyperalgesia when administered via i.th. injection. The L5/L6 SNL model was used to evaluate efficacy against tactile allodynia or thermal hyperalgesia as compared to baseline. Significant efficacy for 6 was observed at doses of 3, 10 and 30μg. A dose response curve was generated at peak effect which occurred 30 min after i.th. administration. The A50 values at this time point were 11.0 (6.5-21.1 μg; 95% CI for allodynia) and 1.04 (1.0-1.2 μg/5μL; 95% CI for hyperalgesia). No effect was seen in vehicle treated animals.
Figure 2.
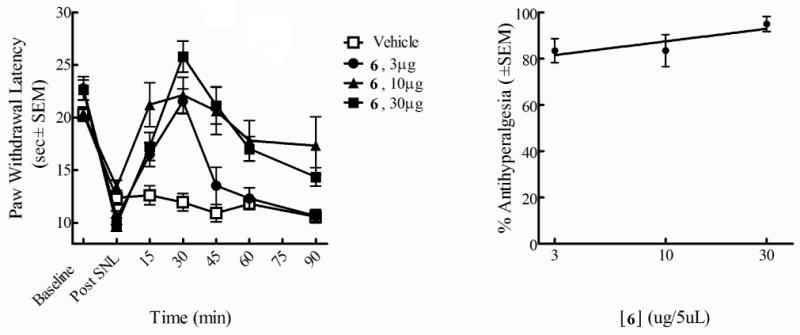
Antihyperalgesic effects of 6 (i.th.) using radiant heat in L5/L6 SNL-operated male SD rats. Thermal hypersensitivity by paw withdrawal (left) Antinociceptive dose-response curve at 30 min (right)
Figure 3.
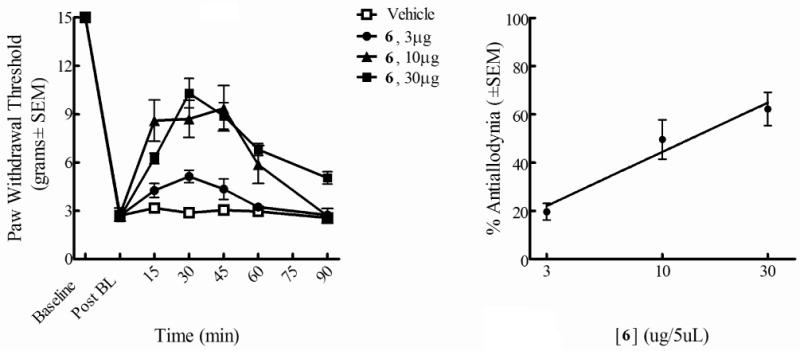
Antiallodynic effects of 6 (i.th.) using von Frey filaments in L5/L6 SNL-operated male SD rats. Tactile allodynia by paw withdrawal thresholds (left) Antinociceptive dose-response curve at30 min (right)
Conclusions
In the present study, a systematic SAR study on the enkephalin analogue 12 with conservation of the propionamide moiety and Dmt1 residue resulted in the development of nonselective potent opioid agonists for μ and δ receptors with high lipophilicity. These analogues can produce the desired physiological effects without many of the undesirable side effects of selective μ opioid agonist, for example, 5 (Ki = 0.40 nM and 0.02 nM at hDOR and rMOR; IC50 = 0.37 nM and 0.26 nM in MVD and GPI, respectively). In summary, the substitution of D-Nle2 and the halogenation of the aromatic ring of Phe4 increased opioid activities at both receptors along with the lipophilicity. Ligand 6 which possessed highly potent opioid activities at both receptors (Ki = 0.14 nM at hDOR and rMOR; IC50 = 0.70 nM and 2.6 nM in MVD and GPI assays, respectively) showed potent antihyperalgesic and antiallodynic effects in the in vivo assays, demonstrating that increase of lipophilicity can be a good approach to develop a potent ligand with good bioavailability.
Experimental Section
All amino acid derivatives were purchased from Novabiochem, ChemImpex International, and RSP. N-phenyl-N-piperidin-4-yl-propionamide was prepared as previously described.14 Coupling reactions were monitored by TLC using the following solvent systems: (1) CHCl3:MeOH:AcOH = 90:10:3 with ninhydrin spray used for detection. Analytical HPLC was performed on a Hewlett Packard 1090 [C-18, Vydac, 4.6 × 250 mm, 5 μm] and preparative RP-HPLC on Hewlett Packard 1100 [C-18, Microsorb-MV™, 10 × 250 mm, 10 μm]. 1H NMR spectra were recorded on a Bruker Advance-300 spectrometer. Mass spectra were taken in the positive ion mode under ESI methods.
Ligands 1-12
These ligands were prepared by stepwise synthesis using Nα-Boc chemistry starting from N-phenyl-N-piperidin-4-yl-propionamide. N-phenyl-N-piperidin-4-yl-propionamide (1 eq) and Nα–Boc-Phe-OH (1.1 eq) were dissolved in DMF and cooled in an ice bath for 10 min. Bop (1.1 eq), HOBt (1.1 eq), and NMM (2 eq) were added to the reaction mixture and stirred for 2 h at rt. After checking for the disappearance of the starting amine by TLC, the reaction mixture was concentrated under reduced pressure. The concentrated mixture was diluted with EtOAC and washed with 5% NaHCO3, 5% citric acid, brine, and water consecutively. The organic layer was dried under anhydrous Na2SO4 and filtered. The solution was concentrated under reduced pressure and triturated with diethyl ether to give a solid. The Nα-Boc group was deprotected by 100% TFA at 0 °C for 20 mins. After completion of the chain elongation by subsequent coupling and deprotection, the mixtures were evaporated under reduced pressure. The residues were solidified by diethyl ether and purified by preparative RP-HPLC (20 – 50% acetonitrile within 20 min for 1, 6 and 7, 20 - 60% acetonitrile within 15 min for 5 and 10) to give pure ligands as white powders in overall yields of 20 - 42%. The purity of the ligands was determined as ≥ 95% by analytical HPLC (10-90% acetonitrile in 40 min, 25-65% acetonitrile in 20 min). For analytical data, see Supporting Information.
Radioligand Labeled Binding Assay, [35S]GTP-γ-S binding Assay, GPI and MVD in Vitro Bioassay
The methods were carried out as previously described.8
In Vivo Assay
Animals
The experiments contained herein were carried out using male Sprague Dawley rats (250-350g; Harlan; Indianapolis, IN). All animals were maintained on a 12/12 h light/dark cycle (lights on at 07:00 am) and provided food and water ad libitum except as noted during the experimental procedures. All the experiments were performed under a protocol approved by Institutional Animal Care and Use Committee (IACUC) of the University of Arizona, and in accordance with policies and guidelines for the care and use of laboratory animals as adopted by International Association for the Study of Pain (IASP) and the National Institutes of Health (NIH). L5/L6spinal nerve ligation surgery. SNL injury was induced as described by Chung and colleagues.13 Intrathecal catheter surgery. All rats were prepared for intrathecal (i.th.) drug administration by placing anesthetized (ketamine/xylazine 100 mg/kg, i.p.) animals in a stereotaxic head holder. The cisternum magnum was exposed, an incision was made and animals were implanted with a catheter (PE: 10, 8 mm) that terminated in the lumbar region of the spinal cord. The animals were allowed to recover 5-7 days post-surgery before any pharmacological manipulations were made. Drug Administration. 6 was dissolved in 100% methanol. For i.th. drug administration, 5 μL of drug was injected followed by a 9 μL saline flush. Testing took place 15, 30, 45, 60, 75 and 90 min after drug injection and dose–response curves were generated from data gathered at the time of peak effect.
Behavioral Assessment
Thermal Hypersensitivity
Thermal hypersensitivity was assessed using the rat plantar test (Ugo Basile, Italy) following a modified method of Hargreaves et al.15 Paw withdrawal latencies were recorded in seconds. An automatic cut off point of 33 sec was set to prevent tissue damage. The apparatus was calibrated to give a paw withdrawal latency of approximately 20 sec on the uninjured paw. The radiant heat source was activated with a timer and focused onto the plantar surface of the hindpaw. Paw-withdrawal latency was determined by a motion detector that halted both heat source and timer when the paw was withdrawn.
Mechanical Hypersensitivity
The assessment of mechanical hypersensitivity consisted of measuring the withdrawal threshold of the paw ipsilateral to the site of nerve injury in response to probing with a series of calibrated von Frey filaments. Each filament was applied perpendicularly to the plantar surface of the ligated paw of rats kept in suspended wire-mesh cages. Measurements were taken both before and after administration of drug or vehicle. The withdrawal threshold was determined by sequentially increasing and decreasing the stimulus strength (‘up–down’ method) analyzed using a Dixon non-parametric test16 and expressed as the mean withdrawal threshold.
Supplementary Material
Table 2. Bioactivities of the Opioid Ligands.
| no | hDORa [3H]DPDPE b | rMORa [3H]DAMGO c | [35S]GTP-γ-S binding | IC50 (nM)e | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| hDORd | rMORd | |||||||||||
| LogIC50f,g | (Ki, nM)h | LogIC50f,g | (Ki, nM)h | LogEC50f | EC50 (nM)i | Emax (%)j | LogEC50f | EC50(nM)i | Emax (%)j | MVD(δ) | GPI(μ) | |
| 1 | -9.42±0.08 | 0.18 | -9.06±0.06 | 0.39 | -10.0±0.21 | 0.10 | 47 | -9.95±0.20 | 0.11 | 77 | 0.69±0.09 | 1.6±0.1 |
| 2 | -8.62±0.08 | 1.1 | -9.16±0.10 | 0.36 | -8.55±0.07 | 2.8 | 61 | -8.73±0.16 | 1.9 | 29 | 8.5±1.7 | 4.7±1.2 |
| 3 | -8.44±0.10 | 1.7 | -9.47±0.20 | 0.15 | - | nr | - | -9.03±0.19 | 0.94 | 33 | 15±1 | 3.6±0.8 |
| 4 | -9.73±0.04 | 0.08 | -9.71±0.03 | 0.10 | -10.19±0.20 | 0.07 | 37 | -9.85±0.43 | 0.14 | 58 | 1.9±0.5 | 1.3±0.3 |
| 5 | -9.07±0.05 | 0.40 | -10.50±0.10 | 0.02 | -10.15±0.16 | 0.07 | 48 | -9.53±0.33 | 0.29 | 98 | 0.37±0.12 | 0.26±0.14 |
| 6 | -9.52±0.08 | 0.14 | -9.41±0.06 | 0.14 | -9.79±0.25 | 0.16 | 58 | -9.46±0.13 | 0.31 | 55 | 0.70±0.38 | 2.6±0.8 |
| 7 | -7.40±0.09 | 19 | -7.97±0.11 | 5.0 | -7.52±0.12 | 30 | 36 | -7.72±0.25 | 19 | 15 | 250±90 | 220±40 |
| 8 | -8.49±0.11 | 1.6 | -9.13±0.04 | 0.33 | -7.59±0.46 | 26 | 27 | -8.33±0.33 | 4.7 | 29 | 180±50 | 93±30 |
| 9 | -10.19±0.10 | 0.03 | -10.81±0.07 | 0.01 | -9.92±0.25 | 0.12 | 23 | -9.16±0.21 | 0.69 | 50 | 1.8±0.8 | 1.0±0.1 |
| 10 | -8.97±0.14 | 0.48 | -9.15±0.13 | 0.35 | -10.07±0.37 | 0.09 | 24 | -9.14±0.23 | 0.72 | 51 | 0.029±0.007 | 0.96±0.13 |
| 11 | -9.59±0.06 | 0.11 | -9.55±0.04 | 0.15 | -9.69±0.17 | 0.20 | 31 | -10.23±0.27 | 0.06 | 37 | 0.21±0.06 | 4.8±0.7 |
| 12 | -9.10±0.10 | 0.36 | -9.08±0.22 | 0.38 | -9.12±0.17 | 0.77 | 24 | -9.05±0.18 | 0.88 | 50 | 1.8±0.2 | 8.5±3.3 |
| YDAGF-NH2 | -6.14±0.16 | 300 | -8.24±0.13 | 2.8 | -6.72±0.17 | 190 | 44 | -7.98±0.22 | 13 | 99 | 120±10 | 47±9 |
| DAMGO | - | - | - | - | - | - | - | -7.44± 0.19 | 37 | 150 | - | - |
| DPDPE | - | - | - | - | 8.80 ± 0.25 | 1.6 | 69 | - | - | - | - | - |
Competition analyses were carried out using membrane preparations from transfected HN9.10 cells that constitutively expressed the respective receptor types.
Kd = 0.50 ± 0.1 nM.
Kd = 0.85 ± 0.2 nM.
Expressed in CHO cells.
Concentration at 50% inhibition of muscle contraction in electrically stimulated isolated tissues.
Logarithmic values determined from the non-linear regression analysis of data collected from at least two independent experiments.
Competition against radiolabeled ligand.
Antilogarithmic value of the respective IC50.
Antilogarithmic value of the respective EC50.
Net total bound/basal binding × 100 ± SEM. nr: no response
Acknowledgments
The work was supported by grants from the USDHS, National Institute on Health (DA-13449 and DA-06284). We thank Margie Colie for assistance with the manuscript.
Abbreviations
- BBB
blood-brain barrier
- Boc
tert-butyloxycarbonyl
- BOP
(benzotriazole-1-yloxy)-tris(dimethylamino)-phosphonium hexafluorophosphate
- CHO
Chinese hamster ovary
- DMF
N,N-dimethylformamide
- hDOR
human δ opioid receptor
- DPDPE
c[D-Pen2,D-Pen5]enkephalin
- DAMGO
[D-Ala2,NMePhe4,Gly5-ol]enkephalin
- Dmt
2,6-dimethyltyrosine
- GPI
guinea pig ileum
- HOBt
1-hydroxybenzotriazole
- IACUC
Institutional Animal Care and Use Committee
- IASP
International Association for the Study of Pain
- i.th
intrathecal
- rMOR
rat μ opioid receptor
- MVD
mouse vas deferens
- NMM
N-methylmorpholine
- RP-HPLC
reverse phase high performance liquid chromatography
- SAR
structure-activity relationships
- SNL
spinal nerve ligation
- TFA
trifluoroacetic acid
- Tic
1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid
Footnotes
Supporting Information Available: 1H NMR data of the ligands 1-12 is available free of charge via the Internet at http://pubs/acs/org.
References
- 1.Zhao GM, Wu D, Soong Y, Shimoyama M, Berezowska I, Schiller PW, Szeto HH. Profound spinal tolerance after repeated exposure to a highly selective μ-opioid peptide agonist: role of δ-opioid receptors. J Pharmacol Exp Ther. 2002;302:188–196. doi: 10.1124/jpet.302.1.188. [DOI] [PubMed] [Google Scholar]
- 2.Jiang Q, Mosberg HI, Porreca F. Modulation of the potency and efficacy of mu-mediated antinociception by delta agonists in the mouse. J Pharmacol Exp Ther. 1990;254:683–689. [PubMed] [Google Scholar]
- 3.Vaught JL, Takemori AE. Differential effects of leucine and methionine enkephalin on morphine-induced analgesia, acute tolerance and dependence. J Pharmacol Exp Ther. 1979;208:86–90. [PubMed] [Google Scholar]
- 4.Porreca F, Takemori AE, Sultana M, Portoghese PS, Bowen WD, Mosberg HI. Modulation of mu-mediated antinociception in the mouse involves opioid delta-2-receptors. J Pharmacol Exp Ther. 1992;263:147–152. [PubMed] [Google Scholar]
- 5.Lee YS, Petrov R, Park C, Ma SW, Davis P, Lai J, Porreca F, Hruby VJ. Development of novel enkephalin analogues that have enhanced opioid activities at both μ and δ opioid receptors. J Med Chem. 2007;50:5528–5532. doi: 10.1021/jm061465o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weber SJ, Greene DL, Sharma SD, Yamamura HI, Krammer TH, Burks TF, Hruby VJ, Hersh LB, Davis TP. Distribution and analgesia of [3H][D-Pen2,D-Pen5]enkephalin and two halogenated analogs after intravenous administartion. J Pharmacol Exp Ther. 1991;259:1109–1117. [PubMed] [Google Scholar]
- 7.Weber SJ, Abbruscato TJ, Brownson EA, Lipkowski AW, Polt R, Misicka A, Haaseth RC, Bartosz H, Hruby VJ, Davis TP. Assessment of an in vitro blood-brain barrier model using several [Met5]enkephalin opioid analogs. J Pharmacol Exp Ther. 1993;266:1649–1655. [PubMed] [Google Scholar]
- 8.Lee YS, Agnes RS, Davis P, Ma SW, Lai J, Porreca F, Hruby VJ. Design and Synthesis of Novel Hydrazide Linked Bifunctional Peptides as δ/μ Opioid Receptor Agonists and CCK-1/CCK-2 Receptor Antagonists. J Med Chem. 2006;49(5):1773–1780. doi: 10.1021/jm05085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gesellchen PD, Shuman RT. Pharmacologically active peptides. US4265808 US. 1981:13.
- 10.Schiller PW. In: The Peptides: Analysis, Synthesis, Biology (Opioid Peptides: Biology, Chemistry and Genetics) Udentriend S, Meienhotfer J, editors. Vol. 6. Academic Press; New York: 1984. pp. 220–268. [Google Scholar]
- 11.http://www.vcclab.org/lab/alogps/
- 12.Bryant SD, Jinsmaa Y, Salvadori S, Okada Y, Lazarus LH. Dmt and opioid peptides: a potent alliance. Biopolymers. 2003;71:86–102. doi: 10.1002/bip.10399. [DOI] [PubMed] [Google Scholar]
- 13.Chung JM, Kim SH. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–363. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- 14.Lee YS, Nyberg J, Moye S, Agnes RS, Davis P, Ma SW, Lai J, Porreca F, Vardanyan R, Hruby VJ. Understanding the structural requirements of 4-anilidopiperidine analogues for biological activities at m and d opioid receptors. Bioorg Med Chem Lett. 2007;17:2161–2165. doi: 10.1016/j.bmcl.2007.01.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hargreaves KM, Dubner R, Brown F, Flores C, Joris J. A new and sensitive methods for measuring thermal nociception in cutaneous hyperalgesis. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- 16.Chaplan SR, Bach Fw, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Meth. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.