

Abstract
Protein kinase C βII (PKCβII) levels increase in the myocardium of patients with end-stage heart failure (HF). Also targeted overexpression of PKCβII in the myocardium of mice leads to dilated cardiomyopathy associated with inflammation, fibrosis and myocardial dysfunction. These reports suggest a deleterious role of PKCβII in HF development. Using a post-myocardial infarction (MI) model of HF in rats, we determined the benefit of chronic inhibition of PKCβII on the progression of HF over a period of 6 weeks after the onset of symptoms and the cellular basis for these effects. Four weeks after MI, rats with HF signs that were treated for 6 weeks with the PKCβII selective inhibitor (βIIV5-3 conjugated to TAT47–57 carrier peptide) (3 mg/kg/day) showed improved fractional shortening (from 21% to 35%) compared to control (TAT47–57 carrier peptide alone). Formalin-fixed mid-ventricle tissue sections stained with picrosirius red, haematoxylin and eosin and toluidine blue dyes exhibited a 150% decrease in collagen deposition, a two-fold decrease in inflammation and a 30% reduction in mast cell degranulation, respectively, in rat hearts treated with the selective PKCβII inhibitor. Further, a 90% decrease in active TGFβ1 and a significant reduction in SMAD2/3 phosphorylation indicated that the selective inhibition of PKCβII attenuates cardiac remodelling mediated by the TGF-SMAD signalling pathway. Therefore, sustained selective inhibition of PKCβII in a post-MI HF rat model improves cardiac function and is associated with inhibition of pathological myocardial remodelling.
Keywords: protein kinase; PKCβII inhibitor peptide; cardiac remodelling; heart failure; myocardial infarction; mast cells, myocardial fibrosis; inflammation
Introduction
Protein kinase C (PKC) isozymes emerge as important potential therapeutic targets in chronic cardiovascular diseases [1] and in particular, heart failure (HF) [2]. However, individual PKC isozymes play different roles in the pathogenesis of cardiac diseases [3–14]. For instance, PKCδ activation augments cardiac reperfusion damage after acute myocardial infarction (MI) and inhibition of PKCδ with δV1–1, a specific peptide inhibitor of PKCδ, reduces infarct size and cardiac dysfunction in a porcine model [4]; an effect that is supported by data from a recent clinical trial (phase IIa) in humans [15]. However, treating hypertensive rats exhibiting HF with δV1–1 did not improve cardiac function whereas V1–2, a specific PKC inhibitor peptide, prevented HF progression in these hypertensive animals [6]. Therefore, different PKC isozymes have unique role in various cardiac diseases and the use of isozyme selective pharmacological agents is more useful than using general PKC inhibitors or activators in assessing the role of PKC in these pathologies.
Cardiac remodelling is an essential component in the process of HF progression [16]. It involves both adaptive and maladaptive changes in cardiac myocyte size, fibrosis, vascular remodelling and inflammation, which change over time. Impairment of contractility and relaxation in the post-MI myocardium causes overload and stress to individual cells and leads to myocardial cell damage, adverse remodelling and further loss of function. It has long been suggested that decreasing the excessive remodelling events of the failing myocardium should decrease myocardial dysfunction [17].
In an earlier study, we reported that PKCβI and βII levels are elevated in the failing hearts of rats with sustained hypertension, and that angiotensin receptor blocker treatment improved cardiac function presumably by attenuating PKCβI and βII [5]. Further, in a recent study, we found that both acute and chronic inhibition of PKCβII improved cardiac contractility and calcium handling in rats with hypertension-induced cardiac dysfunction as well Langendorff preparation (under Revision). Here we focused on the effects of PKCβII inhibition on cardiac fibrosis and inflammation in this post-MI HF model.
Materials and methods
Treatment protocol for post-MI HF
The left anterior descending (LAD) coronary artery of male Wistar rats was occluded to induce MI, as described elsewhere [18] (see also Fig. 1A). Four weeks later, rats showing HF signs [fractional shortening (FS) <25] were treated with βIIV5-3 (3 mg/kg/day) or with an equimolar concentration of TAT47–57 carrier peptide for 6 weeks. Both the TAT peptide and the PKCβII inhibitory peptide were delivered systemically using subcutaneous Alzet osmotic pumps between the upper dorsal part of the rats. The sham-operated rats were treated with TAT to serve as a negative control. Percent fractional shortening was estimated by echocardiography using M-mode measurements before and after treatment. The animal protocols were approved by the Stanford University Institutional Animal Care and Use Committee and by the Ethical Committee of the School of Physical Education and Sport of the University of São Paulo, Brazil. The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health.
Fig 1.
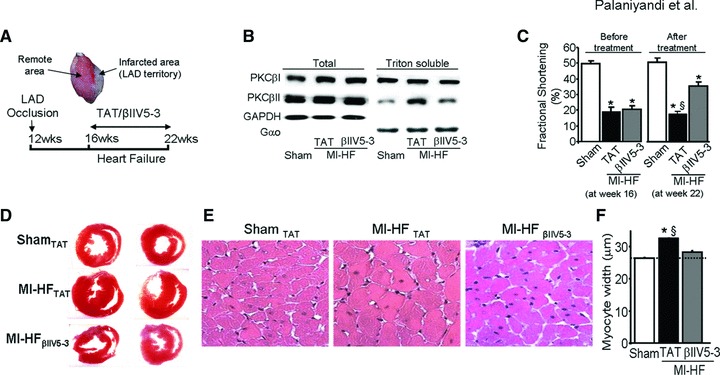
(A) Heart failure (HF) induction and treatment protocol. Twelve-week-old rats were subjected to myocardial infarction (MI) by left anterior descending coronary artery ligation. Four weeks after MI induction, the rats were treated with the PKCβII-specific inhibitor, βIIV5-3, or with control (TAT-carrier peptide). Peptide treatment was continuous (for 6 weeks) using subcutaneous Alzet pump delivery at 3/mg/kg/day. (B) Representative blots showing the levels of cardiac PKCβI and βII in the total and particulate fraction (triton soluble) from sham-operated normal rats or post-MI HF rats treated with TAT or βIIV5-3. Note an increase in PKCβII but not PKCβI in the particulate fraction of failed hearts and a selective loss of PKCβII increase in this fraction from rats treated with βIIV5-3. Association of PKC with the particulate fraction is a measure of PKC activation [36]. The level of cellular PKC distribution between the total and particulate fractions was normalized using GAPDH and Gαo, respectively. (C) Fractional shortening was measured and plotted as graphs before (at week 16) and after treatment (at week 22), n = 6 per group. (D) Cardiac morphology using haematoxylin and eosin stained cardiac slices. (E) Representative photomicrographs of each group (as labelled) depicting cardiomyocyte hypertrophy (increase in cell size). (F) Quantitative analyses of cardiomyocyte width, n = 6 per group. *P < 0.05 sham; §P < 0.05 versus post-MI HF treated with βIIV5-3. Data were analysed by the anova with a post hoc testing by Bonferroni.
Cardiovascular measurements
Heart rate (HR) and systolic blood pressure (BP) were determined non-invasively using a computerized tail-cuff system (BP-2000, Visitech System, Apex, NC, USA). Non-invasive cardiac function was assessed by two-dimensional guided M-mode echocardiography, in halothane-anesthetized control and HF rats, before and after experimental period. Transthoracic echocardiography was performed using an Acuson Sequoia model 512 echocardiography machine (Acuson Corporation, Mountain View, CA, USA) equipped with a 14 MHz linear transducer.
Histopathology
After measuring cardiac function in vivo, hearts were isolated, weighed and the left ventricle was excised. The ratio between left ventricle weight and body weight (LVW/BW) was calculated. Mid-ventricles were then fixed with 10% formalin in PBS, embedded in paraffin and several transverse sections were prepared.
Evaluation of cardiomyocyte hypertrophy
Paraffin-embedded cardiac sections of the myocardium were dewaxed using series of xylene and ethanol and further rehydrated. Then, these sections were stained with haematoxylin and eosin. Haematoxylin and eosin stained slides were used to measure myocyte width, an index of hypertrophied cardiomyocytes, using light microscopy with 400× magnification. At least 100 cardiomyocytes were counted for each group and three sections per group were analysed. Only those cells that had the nucleus at their centre were used for these measurements. These analyses were carried out in a blinded fashion using a computer-assisted morphometric system (Leica Quantimet 520, Cambridge Instruments, UK) [19].
Evaluation of myocardial fibrosis
Paraffin-embedded mid-ventricle sections of the myocardium were dewaxed by series of xylene and ethanol, and then rehydrated. Further, these sections were stained with picrosirius red; collagen deposition, an index of myocardial fibrosis, was observed and quantified by using a computer-assisted morphometric system (Leica Quantimet 500, Cambridge, UK) [20]. At least 20 fields per slide were counted for each group.
Evaluation of infiltration of inflammatory cells
Formalin-fixed, paraffin-embedded cardiac tissue sections were used to study myocardial inflammation. After dewaxing and rehydration, these sections were stained with haematoxylin and eosin. Haematoxylin and eosin staining was used to identify and to quantify the infiltration of polymorphonuclear cells; identification of these cells was guided by their localization and morphology of their nuclei. The photomicrographs of haematoxylin and eosin stained sections (400×) were captured using a camera attached to the microscope. Then, at least 15 fields per slide were observed for each group to evaluate the infiltration of inflammatory cells.
Evaluation of mast cell density and degranulation
Toluidine blue was used to stain mast cells, as described previously [21]. Briefly, paraffinized sections of myocardium were dewaxed using xylene and ethanol, then rehydrated and incubated with 0.05% (w/v) toluidine blue for 30 min. followed by counterstaining with 0.01% (w/v) eosin for 1 min. Decreased violet-coloured metachromatic staining in the mast cells and increased staining in their immediate surrounding was used to identify degranulated mast cells. Mast cell density was quantified by counting the number of toluidine blue-positive mast cells per field. At least 20 fields per slide were counted for each group.
Immunoblotting
Protein lysate was prepared by homogenizing left ventricular tissue in 1 ml of lysis buffer containing protease and phosphatase inhibitors. Samples were centrifuged at 3000 rpm for 10 min. at 4°C and supernatants were stored at −80°C until use. Total protein concentration of samples was measured by the Bradford method. Thirty micrograms of protein sample was applied to each well.
For Western blot experiments, proteins were separated by SDS-PAGE and identified with anti-TGFβ1 (transforming growth factor), PKCβI, PKCβII and Gαo rabbit polyclonal antibodies and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mouse monoclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA). The bound antibody was visualized with horseradish peroxidase (HRP)-coupled secondary antibody. GAPDH was used as a housekeeping marker for comparison.
Immunohistochemistry
Formalin-fixed, paraffin-embedded cardiac tissue sections were used for immunohistochemical staining. After dewaxing and rehydration, these slides were incubated with primary antibody (anti-phospho SMAD2/3 rabbit polyclonal antibody; dilution 1:100, Santa Cruz) at 4°C, the slides were washed in Tris-buffered saline and respective HRP-conjugated secondary antibody at a dilution of 1:500 (Santa Cruz) was added and incubated at room temperature for 45 min. The immunostaining was visualized by diaminobenzidine tetrahydrochloride which yielded a brown colour, and counterstained with haematoxylin. Then, the sections were viewed with a high power light microscope with camera, and phospho SMAD2/3-positive nuclei were counted in blinded fashion. At least 10 high power fields were counted from each slide.
Statistical analysis
Data are presented as mean ± standard error of mean (S.E.M.). One-way anova followed by Bonferroni’s post hoc test was used to compare the effect of PKCβII on PKC translocation, fractional shortening, heart weight/body-weight, cardiomyocyte diameter, collagen deposition, active/latent TGFβI and mast cell density and degranulation. Student’s t-test (one-tailed distribution/two-sample equal variance) was used to compare the effect of PKCβII versus TAT control on infarcted area. Statistical significance was considered achieved when the value of P was <0.05.
Results
PKCβII inhibition by βIIV5-3 attenuates cardiac hypertrophy in post-MI-induced end-stage HF
Sustained treatment of rats with the PKCβII-specific inhibitor, βIIV5-3 [22] selectively inhibited PKCβII activity as determined by a reduction in translocation of this isozyme to the particulate fraction; this treatment had no effect on PKCβI translocation in MI HF rats (Fig. 1B). Also, sustained treatment with βIIV5-3 did not affect the level or translocation of other PKC isozymes (data not shown). Echocardiographic analysis of a group of rats subjected to MI demonstrated a significant improvement in fractional shortening with βIIV5-3 treatment as compared with TAT treatment (Fig. 1C, P < 0.05). Fractional shortening was 21 ± 2% before βIIV5-3 treatment began and after 6 weeks of treatment with βIIV5-3, fractional shortening increased to 35 ± 2%. In contrast, in post-MI HF rats treated with TAT, fractional shortening did not improve; it was 19 ± 3% before treatment and 18 ± 2% after 6 weeks treatment with the control peptide, TAT (Fig. 1C; *P < 0.05 sham and §P < 0.05 versus post-MI HF with βIIV5-3). βIIV5-3 treatment improved left ventricular end systolic diameter (LVESd) significantly (Table 1) (§P < 0.05 versus post-MI HF with βIIV5-3). Although βIIV5-3 treatment slightly improved LVEDd and posterior wall thickness (PWT), it did not attain statistical significance (Table 1). There were no significant differences among groups in terms of HR, BP and BW. However, Post-MI HF rats treated with TAT displayed increased mortality (46%) compared to HF rats treated with βIIV5-3 (10%).
Table 1.
Body-weight and cardiovascular measurements in sham and heart failure rats
| Parameters | SHAM | MI-HFTAT | MI-HFβIIV5-3 |
|---|---|---|---|
| n | 8 | 7 | 9 |
| HR (bpm) | 371 ± 8 | 358 ± 11 | 381 ± 9 |
| BP (mmHg) | 122 ± 2.4 | 119 ± 3.3 | 117 ± 3.7 |
| BW (g) | 404 ± 20 | 438 ± 18 | 410 ± 25 |
| LVW/BW (mg/g) | 2.7 ± 0.1 | 3.3 ± 0.2* | 2.8 ± 0.1§ |
| PWT (mm) | 1.6 ± 0.1 | 2.4 ± 0.1* | 1.8 ± 0.1 |
| LVEDd (mm) | 7.5 ± 0.3 | 10.0 ± 0.5* | 8.8 ± 0.5 |
| LVESd (mm) | 3.7 ± 0.2 | 8.3 ± 0.5* | 5.7 ± 0.4*§ |
HR: heart rate; BP: blood pressure; BW: body-weight; LVWBW: left ventricular weight/body-weight ratio; PWT: posterior wall thickness; LVEDd: left ventricular end-diastolic diameter; LVESd: left ventricular end-systolic diameter. *P < 0.05 versus sham and §P < 0.05 versus post-MI-HF with TAT. anova with post hoc testing by Bonferron was used in those analyses.
Morphological analysis of the transverse heart sections showed reduced wall thickness and reduced left and right ventricle diameter in the βIIV5-3 treated group as compared with the post-MI TAT-treated rats (Fig. 1D). We found that PKCβII inhibition decreased the LVW/BW ratio relative to TAT-treated rats (2.8 ± 0.1 versus 3.3 ± 0.2, §P < 0.05). LVW/BW was 2.7 ± 0.1 in sham (§P < 0.05 versus TAT-treated rats; Table 1). Cardiomyocyte width in post-MI rats increased as compared with sham-operated rats and this increase was blunted in rats treated with βIIV5-3 (Fig. 1E). Quantification of cardiomyocyte width showed an increase from 27 ± 0.3 to 32 ± 0.2 μm in sham- and post-MI TAT control-treated rats (P < 0.05); and to 28 ± 0.6 μm in post-MI βIIV5-3-treated rats versus post-MI TAT-treated rats, respectively (P < 0.05; n = 3–6 for each group) (Fig. 1F).
PKCβII inhibition by βIIV5-3 suppresses myocardial fibrosis in post-MI-induced end-stage HF
On week 22, 10 weeks after infarction, collagen deposition, an index of myocardial fibrosis, was more pronounced in the peri-infarcted area, as compared with the remote area (Fig. 2A). No differences in infarcted area were observed between post-MI HF groups (Fig. 2B). When examining the remote area, collagen deposition was significantly elevated in the TAT-treated post-MI rats, as compared with TAT-treated sham-operated rats (Fig. 2C–E and F). Treatment with βIIV5-3 resulted in a significant decrease in collagen deposition in the remote area.
Fig 2.
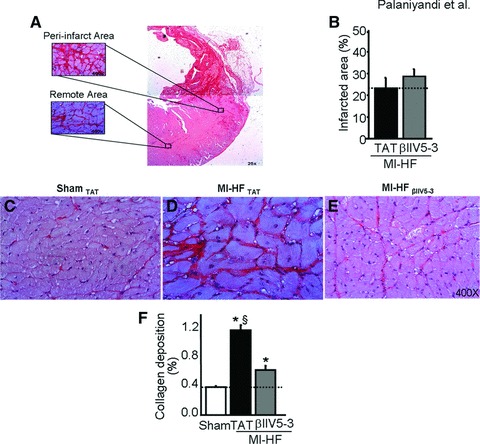
(A) Collagen deposition was found in both remote and peri-infarcted areas of post-myocardial infarction (MI) hearts as measured by picrosirius staining on week 22, 10 weeks after the myocardial infarction and 6 weeks after peptide treatment. (B) Quantification of cardiac infarcted area in post-MI heart failure (HF) rats treated with TAT or βIIV5-3. Note that no differences were observed between groups. (C–E) Representative photomicrographs (400×) from sham operated age-matched rats, post-MI HF rats treated with TAT or with βIIV5-3 showing collagen deposition in the cardiac remote area (red staining). (F) Quantitative analyses of collagen deposition in the cardiac remote area, n = 6 per group; *P < 0.05 sham; §P < 0.05 versus post-MI HF with βIIV5-3. Student’s t-test (one-tailed distribution/two-sample equal variance) was used to compare the effect of PKCβII on infarcted area. Cardiac collagen deposition was analysed by the anova with a post hoc testing by Bonferroni.
Increased myocardial fibrosis is due, at least in part, to an increase in TGFβ1 levels and a subsequent activation of SMAD-signalling cascades [23]. We found that myocardial levels of active TGFβ1 and phospho SMAD2/3 increased in post-MI TAT-treated hearts as compared with sham-operated rats (Fig. 3A–D). A 6-week treatment of rats with HF with βIIV5-3 significantly attenuated the increase in both TGFβ1 and phospho SMAD2/3 level in the remote area of the left ventricle (Fig. 3A–D). Upon quantification, we found that the percentage of phospho SMAD2/3-positive nuclei in post-MI TAT-treated hearts increased as compared with sham-operated rats (222 ± 8 versus 100 ± 2), whereas treatment with βIIV5-3 resulted in a significant decrease in phospho SMAD2/3-positive nuclei (89 ± 2) (not shown in figure).
Fig 3.
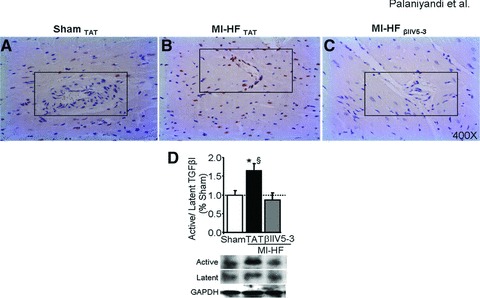
(A–C) Phospho SMAD2/3 immunostaining of hearts from sham-operated rats, or from post-myocardial infarction (MI) heart failure (HF) rats treated with TAT or βIIV5-3. A difference in immunostaining (brown) is noted in the peri-vascular region (indicated by box). n = 3 per group. At least 10 high power fields were counted from each slide. (D) Cardiac active and latent TGFβ1 levels were determined by immunoblot. Data are presented as percentage of control (sham) group, n = 6 per group. *P < 0.05 sham; §P < 0.05 versus post-MI HF with βIIV5-3. Data were analysed by the anova with a post hoc testing by Bonferroni.
PKCβII inhibition by βIIV5-3 reduces myocardial inflammation in post-MI-induced end-stage HF
Infiltrating inflammatory cells in the myocardium contribute to cardiac remodelling through secretion of cytokines [24, 25]. Histochemical analyses demonstrated peri-vascular inflammation throughout the myocardium, predominantly in the peri-infarct region. Our analysis suggested that apart from the peri-vascular region, myocardial inflammation was increased in the epicardium in rats with HF (Fig. 4B and B1) as compared with sham controls (Fig. 4A and A1). However, after 6 weeks of βIIV5-3 treatment, inflammatory cell infiltration was significantly inhibited in rats with post-MI-induced end-stage HF (Fig. 4C and C1).
Fig 4.
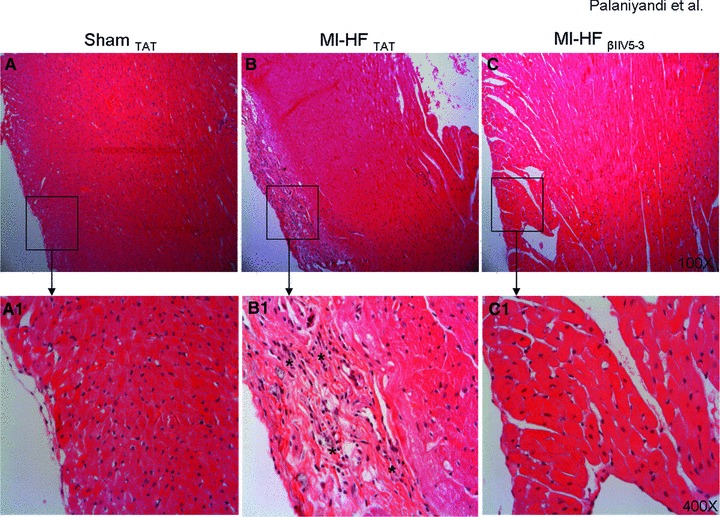
(A–C) Infiltration of inflammatory cells in the epicardial region is shown by haematoxylin and eosin stained myocardial sections of post-myocardial infarction heart failure rats treated with TAT and βIIV5-3, as well as sham-operated normal rats. 100×. Respective representative photomicrographs are presented, n = 3–5 per group. (A1–C1) Higher magnification of these photomicrographs is shown in the lower panels. 400×. Infiltrated inflammatory cells in the epicardial region are indicated by asterisks. The haematoxylin and eosin was used to identify and to quantify the infiltration of polymorphonuclear cells, based upon the localization of the cell and morphology of the nucleus of the cell.
PKCβII inhibition by βIIV5-3 decreases cardiac mast cell density and mast cell degranulation in post-MI-induced end-stage HF
Mast cells reside close to blood vessels in the myocardium and are important contributors to cardiac pathology as they release pro-fibrogenic, pro-inflammatory and pro-hypertrophic factors upon stimulation [26]. A two-fold increase in mast cell density (Fig. 5B and D) and a two-fold increase in mast cell degranulation (Fig. 5F and H) were observed in the failed myocardium of rats 10 weeks after the induction of MI, as compared with sham-operated rats (Fig. 5A and D and E and H). βIIV5-3 treatment from week 4 to week 10 after MI decreased mast cell density by 30% relative to TAT-treated rats (P < 0.05; Fig. 5C and D) and decreased mast cell degranulation by 20% (P < 0.05; Fig. 5G and H) in post-MI animals. Mast cell degranulation was more apparent in the epicardial region (Fig. 5F) whereas fewer degranulated mast cells were found in the endocardial regions in any of the three groups (Fig. 5A–C).
Fig 5.
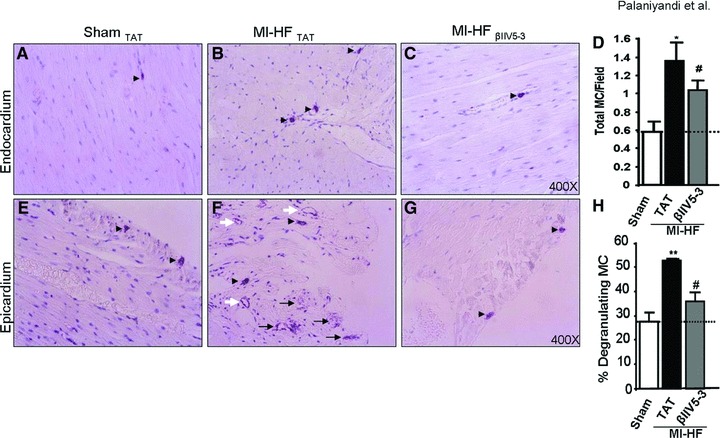
(A–C) Toluidine blue-positive mast cells are shown (as indicated by black arrowheads) in the endocardium (upper panels) of sham and post-myocardial infarction (MI) rats with heart failure treated with TAT or βIIV5-3. (D) Quantification of total mast cell (MC) numbers including both degranulating and non-degranulating mast cells, n = 3 per group. (E–G) Mast cell degranulation is greater in the epicardial region (lower panels) of post-MI heart failure (HF) rats with TAT treatment as compared with sham-operated rats and post-MI HF rats treated with βIIV5-3. Non-degranulated mast cells, partially degranulated mast cells and completely degranulated mast cells are indicated by black arrowheads, black and white arrows, respectively. The completely degranulated emptied mast cells have only outer cell membrane staining (white arrows). (H) Quantification of degranulated mast cells is presented as percent of total mast cell number, n = 3 per group. *P < 0.05 and **P < 0.01 versus sham; #P < 0.05 versus post-MI HF with TAT. Data were analysed by the anova with a post hoc testing by Bonferroni.
Discussion
In this study, we analysed myocyte hypertrophy, myocardial fibrosis, cardiac inflammation and mast cell degranulation in a region remote from the infarcted area. We hypothesized that remodelling of the remote area as induced by MI contributed to cardiac dysfunction. Indeed, with the use of the PKCβII-selective inhibitor, βIIV5-3, remodelling in the remote area was blunted and cardiac function was improved in a rat model of post-MI HF.
In this model, rats were subjected to MI and developed symptoms of end-stage HF. A 6-week treatment with βIIV5-3, initiated 4 weeks after MI induction, attenuated cardiac dysfunction, myocyte hypertrophy, collagen deposition, myocardial inflammation and mast cell density and degranulation. In addition, PKCβII inhibition normalized both right and left ventricles wall thickness (Fig. 1D and Table 1).
In hearts of patients with HF, both PKCβI and II levels are higher relative to age-matched normal hearts [8]. A more recent study also reported increased expression of PKCβII in end-stage human HF (dilated cardiomyopathy) [10]. Similarly, we also found a 50% increase in PKCβII activity in the myocardium of rats with end-stage HF (10 weeks after MI) as compared with sham-operated rats; treatment with βIIV5-3 for 6 weeks selectively decreased PKCβII levels close to the levels in the sham rats (Fig. 1B). As expected, after 6-week treatment, the PKCβII-specific inhibitor βIIV5-3 did not affect the levels or activity of PKCβI, whereas it completely blunted the activation of PKCβII (Fig. 1B).
The role of PKCβI in HF in mice is presently controversial. Because PKCβI and βII are alternatively spliced forms of the same gene product, some of the studies did not distinguish between the two isozymes. Although it was found that adult mice express little PKCβ in the myocardium [9], several reports using a variety of tools demonstrate that PKCβ is an important isozyme in cardiac disease. In fact, targeted overexpression of PKCβII in mice resulted in cardiac hypertrophy with myocardial dysfunction, fibrosis, inflammation and foetal gene expression [11]. Moreover, activated overexpression of PKCβII was lethal in neonatal mice, whereas in adult mice it induced hypertrophy and affected cardiac contractility [27]. These studies show the importance of PKCβII in cardiac hypertrophy in mice. In contrast, in one study, PKCβ KO mice showed no cardiac symptoms [28]. This may reflect compensatory effects by other PKC isozymes or other signalling enzymes. Further, a number of studies suggest that mice are not ideally suitable as models of human diseases. Similar to HF in humas, in a hypertension-induced HF model in rats, PKCβII levels increased in the late HF stage and not during the early hypertrophy stage [5]. Here we show an increase in fractional shortening following a 6-week βIIV5-3 treatment in rats that developed end-stage pathological hypertrophy 4 weeks after MI. We also found that a reduction in cardiomyocyte width and heart/BW ratio. These data indicate that PKCβII activation contributes to the development of cardiac hypertrophy and dysfunction in rats. Because PKCβII levels and activity are elevated in failed human hearts, we suggest that PKCβII inhibition is a potential target for the treatment of HF.
Supporting this suggestion is the study of Boyle et al., who reported that a 4-week treatment with ruboxistaurin (LY333531), an inhibitor of PKCβ, beginning a week after induction of MI, inhibited cardiac fibrosis in rats [29]. A similar reduction in myocardial fibrosis (collagen deposition) following treatment with a specific PKCβII inhibitor has been observed by us in the MI-induced end-stage HF in rats. Moreover, βIIV5-3 treatment resulted in decreased TGFβ1 levels and SMAD2/3 phosphorylation in the hearts of post-MI HF rats. TGFβ1 ligation to its receptor leads to SMAD2/3 phosphorylation in the cytoplasm and translocation of phospho SMAD2/3 into the nucleus to stimulate proliferation through increased transcription and protein synthesis of select genes [23]. We found that fibroblast stimulation and collagen release was inhibited by chronic treatment with βIIV5-3 (Fig. 2), presumably by limiting the TGF–SMAD signalling pathway.
A 6-week βIIV5-3 treatment also caused a reduction in infiltration of inflammatory cells into post-MI myocardium. Previous studies reported that PKCβII activation is involved in a variety of inflammatory processes. PKCβII activation by interferon-γ leads to translocation of PKCβII into the nucleus and activation of the transcription factors that regulate the expression of critical inflammatory genes such as major histocompatability complex II in microglial cells [30]. High glucose-induced interleukin (IL)-6 release from monocytes was attenuated by PKCβII inhibition [31]. We also found a reduction in mast cell number and degranulation after treating rats with βIIV5-3. A role for PKCβII in different stages of the MC degranulation process has been previously suggested by in vitro studies. Aggregation of the FCRI on mast cells elicits a PKCβ-dependent fos binding of DNA and resulting gene transcription in mast cell culture [32]. PKC-βII regulates Akt activity by directly phosphorylating it on Ser-473 in antigen-IgE-stimulated mast cells [33] and PKCβII-deficient mast cells exhibit reduced IL-6 production [34]. Stimulation by antigen or PMA and a calcium ionophore induces Ser (1917) phosphorylation of myosin heavy chain II in mast cells, which coincides with the exocytosis process in these cells, an effect that is mediated by PKCβII [35]. These reports are consistent with our findings here on the role of PKCβII in mast cell activation in vivo. However, the molecular mechanism leading to the beneficial effects of PKCβII inhibition remains to be elucidated.
In conclusion, we show that sustained selective inhibition of PKCβII by βIIV5-3 attenuated adverse cardiac remodelling and improved myocardial function in an end-stage HF model induced by MI in rats. If proven to be safe, such a specific inhibitor of PKCβII may benefit human patients with MI-induced HF.
Acknowledgments
We thank Dr. Adrienne Gordon for critical review and helpful discussions and Katt C. Mattos for technical assistance. This study was supported by National Institute of Health Grant HL076675 to DM-R. S.S.P was supported by a seed grant from Stanford Cardiovascular Institute. J.C.B.F. holds a doctoral fellowship from Fundação de Amparo a Pesquisa do Estado de São Paulo – Brasil (FAPESP 06/56321-6) and CAPES PDEE program (2177-07-2).
Conflict of Interest
DM-R is the founder of KAI Pharmaceuticals, Inc. However, none of the work in her academic laboratory is supported by the company. Other authors have no disclosure.
References
- 1.Churchill E, Budas G, Vallentin A, et al. PKC isozymes in chronic cardiac disease: possible therapeutic targets. Annu Rev Pharmacol Toxicol. 2008;48:569–99. doi: 10.1146/annurev.pharmtox.48.121806.154902. [DOI] [PubMed] [Google Scholar]
- 2.Palaniyandi SS, Sun L, Ferreira JC, et al. Protein kinase C in heart failure: a therapeutic target. Cardiovasc Res. 2009;82:229–39. doi: 10.1093/cvr/cvp001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hambleton M, Hahn H, Pleger ST, et al. Pharmacological- and gene therapy-based inhibition of protein kinase Calpha/beta enhances cardiac contractility and attenuates heart failure. Circulation. 2006;114:574–82. doi: 10.1161/CIRCULATIONAHA.105.592550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Inagaki K, Chen L, Ikeno F, et al. Inhibition of delta-protein kinase C protects against reperfusion injury of the ischemic heart in vivo. Circulation. 2003;108:2304–7. doi: 10.1161/01.CIR.0000101682.24138.36. [DOI] [PubMed] [Google Scholar]
- 5.Inagaki K, Iwanaga Y, Sarai N, et al. Tissue angiotensin II during progression or ventricular hypertrophy to heart failure in hypertensive rats; differential effects on PKC epsilon and PKC beta. J Mol Cell Cardiol. 2002;34:1377–85. doi: 10.1006/jmcc.2002.2089. [DOI] [PubMed] [Google Scholar]
- 6.Inagaki K, Koyanagi T, Berry NC, et al. Pharmacological inhibition of epsilon-protein kinase C attenuates cardiac fibrosis and dysfunction in hypertension-induced heart failure. Hypertension. 2008;51:1565–9. doi: 10.1161/HYPERTENSIONAHA.107.109637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Braun MU, Mochly-Rosen D. Opposing effects of delta- and zeta-protein kinase C isozymes on cardiac fibroblast proliferation: use of isozyme-selective inhibitors. J Mol Cell Cardiol. 2003;35:895–903. doi: 10.1016/s0022-2828(03)00142-1. [DOI] [PubMed] [Google Scholar]
- 8.Bowling N, Walsh RA, Song G, et al. Increased protein kinase C activity and expression of Ca2+-sensitive isoforms in the failing human heart. Circulation. 1999;99:384–91. doi: 10.1161/01.cir.99.3.384. [DOI] [PubMed] [Google Scholar]
- 9.Sabri A, Steinberg SF. Protein kinase C isoform-selective signals that lead to cardiac hypertrophy and the progression of heart failure. Mol Cell Biochem. 2003;251:97–101. [PubMed] [Google Scholar]
- 10.Simonis G, Briem SK, Schoen SP, et al. Protein kinase C in the human heart: differential regulation of the isoforms in aortic stenosis or dilated cardiomyopathy. Mol Cell Biochem. 2007;305:103–11. doi: 10.1007/s11010-007-9533-3. [DOI] [PubMed] [Google Scholar]
- 11.Wakasaki H, Koya D, Schoen FJ, et al. Targeted overexpression of protein kinase C beta2 isoform in myocardium causes cardiomyopathy. Proc Natl Acad Sci U S A. 1997;94:9320–5. doi: 10.1073/pnas.94.17.9320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Piacentini L, Gray M, Honbo NY, et al. Endothelin-1 stimulates cardiac fibroblast proliferation through activation of protein kinase C. J Mol Cell Cardiol. 2000;32:565–76. doi: 10.1006/jmcc.2000.1109. [DOI] [PubMed] [Google Scholar]
- 13.Baines CP, Zhang J, Wang GW, et al. Mitochondrial PKCepsilon and MAPK form signaling modules in the murine heart: enhanced mitochondrial PKCepsilon-MAPK interactions and differential MAPK activation in PKCepsilon-induced cardioprotection. Circ Res. 2002;90:390–7. doi: 10.1161/01.res.0000012702.90501.8d. [DOI] [PubMed] [Google Scholar]
- 14.Sentex E, Wang X, Liu X, et al. Expression of protein kinase C isoforms in cardiac hypertrophy and heart failure due to volume overload. Can J Physiol Pharmacol. 2006;84:227–38. doi: 10.1139/y05-120. [DOI] [PubMed] [Google Scholar]
- 15.Bates E, Bode C, Costa M, et al. Intracoronary KAI-9803 as an adjunct to primary percutaneous coronary intervention for acute ST-segment elevation myocardial infarction. Circulation. 2008;117:886–96. doi: 10.1161/CIRCULATIONAHA.107.759167. [DOI] [PubMed] [Google Scholar]
- 16.Takano H, Hasegawa H, Nagai T, et al. Implication of cardiac remodeling in heart failure: mechanisms and therapeutic strategies. Intern Med. 2003;42:465–9. doi: 10.2169/internalmedicine.42.465. [DOI] [PubMed] [Google Scholar]
- 17.Cohn JN. Structural basis for heart failure. Ventricular remodeling and its pharmacological inhibition. Circulation. 1995;91:2504–7. doi: 10.1161/01.cir.91.10.2504. [DOI] [PubMed] [Google Scholar]
- 18.Pfeffer MA, Pfeffer JM, Fishbein MC, et al. Myocardial infarct size and ventricular function in rats. Circ Res. 1979;44:503–12. doi: 10.1161/01.res.44.4.503. [DOI] [PubMed] [Google Scholar]
- 19.Oliveira RS, Ferreira JC, Gomes ER, et al. Cardiac anti-remodelling effect of aerobic training is associated with a reduction in the calcineurin/NFAT signalling pathway in heart failure mice. J Physiol. 2009;587:3899–910. doi: 10.1113/jphysiol.2009.173948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pereira MG, Ferreira JC, Bueno CR, Jr, et al. Exercise training reduces cardiac angiotensin II levels and prevents cardiac dysfunction in a genetic model of sympathetic hyperactivity-induced heart failure in mice. Eur J Appl Physiol. 2009;105:843–50. doi: 10.1007/s00421-008-0967-4. [DOI] [PubMed] [Google Scholar]
- 21.Palaniyandi SS, Watanabe K, Ma M, et al. Inhibition of mast cells by interleukin-10 gene transfer contributes to protection against acute myocarditis in rats. Eur J Immunol. 2004;34:3508–15. doi: 10.1002/eji.200425147. [DOI] [PubMed] [Google Scholar]
- 22.Stebbins EG, Mochly-Rosen D. Binding specificity for RACK1 resides in the V5 region of beta II protein kinase C. J Biol Chem. 2001;276:29644–50. doi: 10.1074/jbc.M101044200. [DOI] [PubMed] [Google Scholar]
- 23.Moustakas A, Souchelnytskyi S, Heldin CH. Smad regulation in TGF-beta signal transduction. J Cell Sci. 2001;114:4359–69. doi: 10.1242/jcs.114.24.4359. [DOI] [PubMed] [Google Scholar]
- 24.Chen D, Assad-Kottner C, Orrego C, et al. Cytokines and acute heart failure. Crit Care Med. 2008;36:S9–16. doi: 10.1097/01.CCM.0000297160.48694.90. [DOI] [PubMed] [Google Scholar]
- 25.Niebauer J. Inflammatory mediators in heart failure. Int J Cardiol. 2000;72:209–13. doi: 10.1016/s0167-5273(99)00190-4. [DOI] [PubMed] [Google Scholar]
- 26.Palaniyandi Selvaraj S, Watanabe K, et al. Involvement of mast cells in the development of fibrosis in rats with postmyocarditis dilated cardiomyopathy. Biol Pharm Bull. 2005;28:2128–32. doi: 10.1248/bpb.28.2128. [DOI] [PubMed] [Google Scholar]
- 27.Bowman JC, Steinberg SF, Jiang T, et al. Expression of protein kinase C beta in the heart causes hypertrophy in adult mice and sudden death in neonates. J Clin Invest. 1997;100:2189–95. doi: 10.1172/JCI119755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roman BB, Geenen DL, Leitges M, et al. PKC-beta is not necessary for cardiac hypertrophy. Am J Physiol Heart Circ Physiol. 2001;280:H2264–70. doi: 10.1152/ajpheart.2001.280.5.H2264. [DOI] [PubMed] [Google Scholar]
- 29.Boyle AJ, Kelly DJ, Zhang Y, et al. Inhibition of protein kinase C reduces left ventricular fibrosis and dysfunction following myocardial infarction. J Mol Cell Cardiol. 2005;39:213–21. doi: 10.1016/j.yjmcc.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 30.Nikodemova M, Watters JJ, Jackson SJ, et al. Minocycline down-regulates MHC II expression in microglia and macrophages through inhibition of IRF-1 and protein kinase C (PKC)alpha/betaII. J Biol Chem. 2007;282:15208–16. doi: 10.1074/jbc.M611907200. [DOI] [PubMed] [Google Scholar]
- 31.Devaraj S, Venugopal SK, Singh U, et al. Hyperglycemia induces monocytic release of interleukin-6 via induction of protein kinase c-{alpha} and -{beta} Diabetes. 2005;54:85–91. doi: 10.2337/diabetes.54.1.85. [DOI] [PubMed] [Google Scholar]
- 32.Lewin I, Jacob-Hirsch J, Zang ZC, et al. Aggregation of the Fc epsilon RI in mast cells induces the synthesis of Fos-interacting protein and increases its DNA binding-activity: the dependence on protein kinase C-beta. J Biol Chem. 1996;271:1514–9. doi: 10.1074/jbc.271.3.1514. [DOI] [PubMed] [Google Scholar]
- 33.Kawakami Y, Nishimoto H, Kitaura J, et al. Protein kinase C betaII regulates Akt phosphorylation on Ser-473 in a cell type- and stimulus-specific fashion. J Biol Chem. 2004;279:47720–5. doi: 10.1074/jbc.M408797200. [DOI] [PubMed] [Google Scholar]
- 34.Nechushtan H, Leitges M, Cohen C, et al. Inhibition of degranulation and interleukin-6 production in mast cells derived from mice deficient in protein kinase Cbeta. Blood. 2000;95:1752–7. [PubMed] [Google Scholar]
- 35.Ludowyke RI, Elgundi Z, Kranenburg T, et al. Phosphorylation of nonmuscle myosin heavy chain IIA on Ser1917 is mediated by protein kinase C beta II and coincides with the onset of stimulated degranulation of RBL-2H3 mast cells. J Immunol. 2006;177:1492–9. doi: 10.4049/jimmunol.177.3.1492. [DOI] [PubMed] [Google Scholar]
- 36.Kraft AS, Anderson WB. Phorbol esters increase the amount of Ca2+, phospholipid-dependent protein kinase associated with plasma membrane. Nature. 1983;301:621–3. doi: 10.1038/301621a0. [DOI] [PubMed] [Google Scholar]