

Abstract
Obesity has emerged as one of the principle worldwide health concerns of the modern era, and there exists a tremendous unmet clinical need for safe and effective therapies to combat this global pandemic. The prevalence of obesity and its associated co-morbidities, including cardiovascular and metabolic diseases, has focused drug discovery and development on generating effective modalities for the treatment and prevention of obesity. Early efforts in the field of obesity pharmacotherapy centered on agents with indeterminate mechanisms of action producing treatment paradigms characterized by significant off-target effects. During the past two decades, new insights have been made into the physiologic regulation of energy balance and the subordinate central and peripheral circuits coordinating appetite, metabolism, and lipogenesis. These studies have revealed previously unrecognized molecular targets for controlling appetite and managing weight from which has emerged a new wave of targeted pharmacotherapies to prevent and control obesity.
INTRODUCTION
Obesity rates have escalated in the past few decades, expanding from a condition affecting only a minor proportion of the adult populations of industrialized nations, into a global pandemic. It is estimated that >1 billion adults worldwide are overweight (BMI >25 kg/m2), 300 million of whom are clinically obese (BMI >30 kg/m2).1 The number of US adults classified as obese has doubled over the past 20 years and more than tripled in developing countries, coinciding with the adoption of a Western lifestyle. Importantly, the rates of childhood obesity have also reached epidemic levels in developed countries. The dramatic rise in obesity rates reflects a combination of genetic susceptibility, increased availability of high-energy foods, and decreased requirement for physical activity in modern society. Due to the wide array of chronic diseases for which obesity is a risk factor, escalating obesity rates have serious health and economic consequences. Indeed, the negative consequences of chronic obesity exceed those of either smoking or alcohol abuse. In turn, this has established a health and economic imperative to develop therapeutic strategies to prevent and treat the growing obesity problem.
HISTORY OF ANTI-OBESITY PHARMACOTHERAPY
The history of anti-obesity pharmacotherapy is filled with disappointments and cases of unsafe practices, abuse, and drugs with limited efficacy and serious adverse effects. The beginning of modern weight loss drug therapy began in the late 19th century with the discovery that sheep thyroid extract increased metabolic rate and induced significant weight loss. However, the use of thyroid hormone treatment in euthyroid patients led to an increased risk of cardiac arrhythmias and cardiac arrest.2 In the 1930s, 2,4-dinitrophenol (DNP), an uncoupling agent which increases metabolic rate, became a popular weight loss drug, but it was discontinued due to adverse effects including agranulocytosis, dermatitis, cataracts, and fatal hyperthermia.3
From the 1940s through the 1960s, there was widespread use of amphetamines which culminated with the popularization of “rainbow pill” regimens, which included combinations of amphetamines, digitalis, thyroid hormone, diuretics, and laxatives, with barbiturates to counteract the nervousness and hyperactivity of the stimulants. These diet pill regimens induced mortality due to myocardial toxicity and sudden death.4
Aminorex, an anorectic drug with similar properties to amphetamine, was put into clinical use in Europe in the late 1960s with catastrophic results. An epidemic of chronic pulmonary hypertension struck users of the drug, with a mortality rate of ~50% among affected individuals.5 Fenfluramine, a drug which stimulates serotonin release and inhibits presynaptic serotonin reuptake, was combined with the sympathomimetic drug, phentermine, in 1992 to create the popular weight loss drug, Fen-Phen, which showed promising results in promoting weight loss. However, much like with aminorex, it was soon observed that patients being treated with fenfluramine were at risk for developing pulmonary hypertension and, of even greater concern, valvular heart disease, causing both fenfluramine and its derivative dexfenfluramine to be withdrawn from the market in 1997.6
From the 1970s through the 1990s, the sympathomimetic amine, phenylpropanolamine (PPA), was commonly found in many cough and cold remedies, as well as appetite suppressants. However, studies showed the association between PPA use and the occurrence of intracranial bleeding and hemorrhagic stroke.7
More recently, dietary supplements containing ephedra alkaloids, sometimes combined with caffeine and/or gauarana, were marketed as weight loss agents and became very popular, but case reports and studies showed the dangers of these agents due to reported adverse effects of palpitations, hypertension, heart attack, stroke, and sudden death,8 prompting the FDA to ban the sale of ephedra-containing supplements in 2004. However, several other over-the-counter weight loss products currently fuel a billion dollar supplement industry which includes products purported to: modulate carbohydrate metabolism (e.g. chromium, ginseng), increase satiety (e.g. soluble fiber-containing supplements - guar gum, glucomannan, psyllium), enhance fat oxidation (hydroxycitric acid, conjugated linoleic acid, green tea, licorice, pyruvate, vitamin B6, L-carnitine), and block lipid absorption (chitosan).9 While these products appear to be tolerated by users, no evidence exists for their long-term safety, and none of these supplements have been proven in clinical trials to induce significant weight loss in overweight and obese patients.
Finally, rimonabant, a potent, selective endocannabinoid (CB1-receptor) antagonist recently showed great promise in clinical trials for inducing weight loss and was expected to gain FDA-approval for anti-obesity therapy. However, the psychiatric side effects of rimonabant therapy, including an increased risk for depression and suicide, caused an FDA-appointed advisory panel to recommend the drug not be approved, and caused the European Medicines Agency (EMEA) to recommend its suspension in October 2008. In response, Sanofi-Aventis, which developed and marketed rimonabant, terminated studies of that agent, triggering the discontinuation of the clinical development of other CB1 antagonists.10
TARGETED PHARMACOTHERAPY OF OBESITY
There has been a dramatic shift in the clinical, drug development, and regulatory communities concerning obesity as a legitimate health problem and a primary etiologic agent of prevalent chronic diseases, including cardiovascular disease and type II diabetes. Currently, the most effective means of achieving long-term weight loss in obese patients is through bariatric surgery. However, due to its inherent risks, this procedure is reserved for morbidly obese patients and those with serious co-morbidities. Therefore, physicians often enroll overweight and obese patients in counseling and lifestyle modification programs to combat their weight problems. However, while patients enrolled in these programs enjoy some initial success, most regain the majority of their lost weight within 5 years.
The solution to this apparent inability to safely achieve long-term weight reduction may be the introduction of anti-obesity pharmacotherapeutics into mainstream clinical practice. The only two drugs currently FDA-approved for the long-term treatment of obesity, sibutramine and orlistat, enhance and maintain weight loss in overweight and obese patients when combined with lifestyle modification. However, sibutramine and orlistat are associated with cardiovascular and GI side effects, respectively, which make them undesirable to both patients and physicians.
With the global rise in obesity over the past few decades much research has focused on the endogenous regulation of appetite and energy balance, and these studies have yielded several central and peripheral anti-obesity molecular targets. The development of anti-obesity drugs is tainted by a history of morbidity and mortality, but with a greater understanding of the physiology of energy balance, and the pathophysiology of its dysregulation, the next generation of molecularly targeted agents should be effective, safe, and well-tolerated.
Neuropeptide Signaling Modulators
The arcuate nucleus of the hypothalamus, the primary neural signaling site for peripheral appetite-regulating hormones, is mainly composed of two types of appetite-regulating neurons: 1), those expressing the anorexigenic (appetite-suppressing) neuropeptides pro-opiomelanocortin (POMC) and cocaine- and amphetamine-regulated transcript (CART) and 2) those expressing the orexigenic (appetite-stimulating) neuropeptides agouti-related peptide (AgRP) and neuropeptide Y (NPY). The primary effect of most circulating satiety hormones on appetite and energy balance is mediated by the modulation of the signaling of these neurons. POMC/CART and NPY/AgRP neurons project to several hypothalamic nuclei, including the dorsomedial nucleus, paraventricular nucleus, lateral hypothalamus, and prefornical area, each containing secondary neurons which process and integrate the information. These second-order neurons express neuropeptides (melanin concentrating hormone, orexin A/B, thyrotropin-releasing hormone, corticotropin-releasing hormone, brain-derived neurotrophic factor) which are the downstream mediators of energy homeostasis.
Signaling at neuronal Y1- and Y5-receptors produces the orexigenic effects of NPY, and pharmacologic blockade or genetic deletion of the Y1- and Y5-receptors reduces food intake and weight in mice, with Y1-receptor signaling appearing to be the major mediator of the orexigenic effects of NPY. An orally active Y5-receptor antagonist (MK-0557) (Merck) was unable to induce clinically meaningful weight loss in a 1-year clinical trial.11 However, another selective Y5-receptor antagonist (S-2367) (Shionogi USA, Inc.) induced a mean placebo-adjusted weight loss of 3.0% baseline weight (p<0.0001) over 54 weeks of therapy.12 While the efficacy of this drug is limited, pharmacologic Y1-receptor antagonism or a combination Y1/Y5-receptor antagonism may prove more effective.
While Y1- and Y5-receptor signaling produce appetite-stimulating effects, Y2- and Y4-receptor signaling produce an anorexic phenotype due to their pre-synaptic inhibition of NPY release. The Y2- and Y4-receptors are the targets of the satiety hormones, PYY and pancreatic polypeptide, respectively, and currently two drugs: obinepitide (7TM Pharma), a Y2/Y4-receptor agonist, and TM30339 (7TM Pharma), a selective Y4-receptor agonist, are in phase I/II clinical trials. Obinepitide significantly reduced food intake for up to 9 hrs following dosing in clinical testing.13
Additionally, a specific AgRP inhibitor (TTT-435) (TransTech Pharma) is in phase II clinical trials14, and BMS-830216 (Bristol-Myers Squibb), a pharmacological antagonist of signaling of the orexigenic neuropeptide, melanin-concentrating hormone, which is expressed in second-order neurons in the lateral hypothalamus, is in phase I/II clinical testing.15
Endogenous Hormones
Leptin
Leptin, an adipose tissue-derived hormone, has been shown to act as both a short- and long-term regulator of energy balance and to signal in the arcuate nucleus where it inhibits NPY/AgRP signaling and stimulates POMC/CART signaling. However, early clinical trials of leptin therapy for the treatment of obesity failed because the majority of obese patients exhibit leptin resistance. Recently, though, leptin has reemerged as a targeted anti-obesity therapeutic. The administration of chemical chaperones which resolve ER stress, including 4-phenyl butyric acid (PBA) and tauroursodeoxycholic acid (TUDCA), increases leptin sensitivity and its ability to induce hypophagia and weight loss in mice.16 Further, pramlintide, a synthetic analogue of pancreatic amylin, sensitizes mice to the effects of leptin, and pramlintide/metreleptin combination therapy induced a weight loss of 12.7 ± 0.9% (11.5 ± 0.9 kg) without plateau in obese patients during a 20-week trial period.17
Glucagon-like peptide-1 (GLP-1)
GLP-1, a peptide derived from the preproglucagon gene, is an incretin secreted in response to the ingestion of a meal by intestinal L-cells, and it has been observed to have central anorectic effects. Two proteolysis-resistant GLP-1 analogues (exenatide, liraglutide) are FDA-approved for adjuvant treatment of type II diabetes and have been observed to induce weight loss in clinical trials. However, the requirement of daily injection for their delivery is a major barrier to weight loss therapy. In that context, a long-acting release formulation of exenatide (exenatide-LAR), injected once weekly, induced reductions in HbA1c (−1.7%), fasting plasma glucose (−40 mg/dL), and body weight (−5.8 lbs).18 Further, nasal and transdermal formulations of exenatide are in early clinical development.19 Moreover, phase I clinical testing of a long-acting oral GLP-1 analogue (NN9924), which utilizes sodium N-[8-(2-hydroxybenzoyl) amino] caprylate (SNAC) carrier technology was initiated in January 2010.20
Oxyntomodulin
Oxyntomodulin (OXM), another derivative of the preproglucagon gene, is secreted post-prandially along with GLP-1 by intestinal L-cells, but it has more anorectic properties and minimal incretin function. While GLP-1 has been proposed to primarily signal in the nucleus tractus solitarius of the brainstem, OXM signals directly in the arcuate nucleus, and it has been observed to stimulate POMC neurons.21 Repeated injections of OXM significantly reduced caloric intake and increased energy expenditure in overweight and obese subjects22, but the utility of OXM is also limited by the necessity of injection for delivery. A long-acting OXM analogue, TKS1225, has been developed and was acquired by Wyeth Pharmaceuticals in 2008.23
Peptide YY (PYY)
PYY is another gut-derived satiety hormone primarily secreted by L-cells of the terminal ileum and colon in response to the ingestion of food. It produces anorexigenic signals through the activation of presynaptic Y2-receptors which inhibit the release of NPY by neurons in the arcuate nucleus.24 Clinical trials have revealed that obese patients are sensitive to the anorectic effects of PYY administration, with continuous infusion inhibiting feeding in a dose-dependent manner (maximal inhibition, 35%; p < .001 vs. control).25 However, as continuous infusion of PYY is not a useful therapeutic strategy, intranasal delivery of PYY (3–36) (Nastech/Merck) has been investigated, and, over 12-weeks of therapy, PYY (200 µg, 600 µg) has been shown to induce placebo-adjusted mean weight losses of −1.4 kg and −2.3 kg, respectively. Unfortunately, >50% of the subjects receiving 600 µg PYY (3–36) withdrew from the study due to nausea and vomiting,26 indicating that further improvements to the formulation and/or delivery method of PYY remain. Thus, along with GLP-1, oral delivery of PYY is also being developed utilizing SNAC carrier technology.20
Pancreatic Polypeptide (PP)
Pancreatic polypeptide (PP), a satiety peptide which is structurally similar to PYY, is primarily produced in the pancreas and reduces appetite primarily through signaling in the dorsal vagal complex, particularly the area postrema, which communicates with the hypothalamus. Its circulating levels rise rapidly following meal ingestion, rising proportionally to caloric intake and remaining elevated for up to 6 hrs. In a trial of PP use in healthy non-obese individuals, IV infusion (10 pmol/kg/min) reduced appetite and caloric intake by 21.8 ± 5.7% (p< 0.01) at a buffet meal 2 hrs following infusion. PP infusion also inhibited food consumption over 24 hrs, with a reduction in food intake of 25.3 ± 5.8%.27 IV infusion, however, remains a limitation, and new delivery methods could advance this therapeutic into clinical practice.
Amylin
Amylin, or islet amyloid polypeptide (IAPP), is synthesized and secreted along with insulin by pancreatic β-cells, and patients with type I diabetes exhibit a deficiency of both hormones. Furthermore, both insulin and amylin have low fasting levels, and their circulating levels rise in response to meal ingestion. In addition to working synergistically with insulin to regulate post-prandial glucose levels, amylin has central anorectic functions through signaling at the area postrema within the dorsal vagal complex. Pramlintide, a synthetic amylin analogue which was developed by Amylin Pharmaceuticals, has a similar pharmacodynamic and pharmacokinetic profile to endogenous amylin.28 Similar to the GLP-1 analogues discussed previously, pramlintide is approved for the treatment of diabetes and induces weight loss in diabetic patients, unlike traditional diabetic medications. Clinical trials have demonstrated the utility of pramlintide in aiding glycemic control and inducing modest weight loss in type I and type II diabetic patients. Further, a pooled post-hoc analysis of weight loss in pramlintide-treated type II diabetic subjects, revealed that, overall, pramlintide therapy (120 µg, 3× daily, or 150µg q.i.d.) induced an average weight loss of 2.6 kg (p < .0001) over 52 weeks of therapy.29 Thus, pramlintide appears to be an option for the treatment of diabetic patients, since most other diabetic medications induce weight gain. Moreover, as discussed earlier, pramlintide improves leptin sensitivity, and it may have greater utility inducing weight loss in diabetic and non-diabetic patients when combined with leptin therapy.
Ghrelin
Ghrelin, which is predominantly expressed in the stomach, is the only known circulating orexigenic hormone and signals both on vagal afferents and in the arcuate nucleus where it enhances NPY orexigenic signaling.30 An experimental ghrelin vaccine, CYT009-GhrQb, was discontinued in 2006, but a novel vaccine of ghrelin conjugated to the hapten, keyhole limpet hemocyanin (KLH), decreased feeding and induced weight loss in rodent models.31 Further, in 2006, NOXXON Pharma AG granted Pfizer an exclusive worldwide license to NOX-B11, a ghrelin-neutralizing RNA spiegelmer, which is an aptamer that binds to and inactivates ghrelin and has been demonstrated to block the orexigenic activity of exogenous ghrelin administration in rats.32 Specific ghrelin antagonists (Elixir Pharmaceuticals) are also in preclinical testing. Moreover, a new molecular target, ghrelin O-acyltransferase (GOAT), which adds an eight-carbon octanoate moiety to a ghrelin serine residue required for receptor binding, has been identified.33
Modulators of Monoamine Neurotransmission
Bupropion
Bupropion is structurally similar to the amphetamine derivative, diethylpropion, and functions as a norepinephrine and dopamine reuptake inhibitor. Bupropion has been marketed as an anti-depressant and for smoking cessation, but recently it has shown utility in the treatment of obesity. Rats injected intra-peritoneally (IP) with bupropion exhibited feeding cessation in a dose-dependent manner.34 Additionally, mice treated with either a selective norepinephrine or dopamine reuptake inhibitor reduced acute food intake, with combined treatment of both inhibitors showing an additive effect.35 This suggests that the dual role of bupropion as a norepinephrine and dopamine reuptake inhibitor is central to its appetite-suppressing ability, and the effect of bupropion on appetite has been specifically linked to the regulation of hypothalamic POMC neuronal signaling.
Three long-term clinical trials evaluated the effectiveness of bupropion as a weight loss drug, two of which investigated the drug’s effects in depressed patients. These studies revealed that bupropion is an effective, long-term (1-year) treatment for weight loss when combined with lifestyle modification, and it appears to be more effective in non-depressed patients (see Supplementary Information). However, bupropion could serve as a valuable adjunctive therapy to elevate mood in depressed patients in whom weight gain secondary to anti-depressant therapy is an issue. In the trial of non-depressed obese patients (BMI 30–44 kg/m2) participating in a lifestyle intervention program, including counseling on energy-restricted diets, meal replacements, and exercise, subjects receiving bupropion-SR at doses of 300 mg/d and 400 mg/d lost 7.2% and 10.1% of initial body weight, respectively, compared to 5.0% loss for placebo following 24 weeks of therapy. A loss of ≥5% of body weight was observed in 46%, 59%, and 83% of placebo-, bupropion-SR 300-, and bupropion-SR 400-treated subjects, respectively (p<0.0001 vs. placebo), while a loss of ≥10% of body weight was observed in 20%, 33%, and 46% of those subjects, respectively (p=0.0008 vs. placebo). Finally, subjects switched from placebo to bupropion-SR 300 or bupropion-SR 400 at 24 weeks lost an additional 1.2% and 2.7% baseline weight, respectively, by week 48 (total weight loss = 6.4%, 7.2%, respectively), while those subjects continuing bupropion-SR 300 or bupropion-SR 400 therapy maintained weight losses of 7.5% and 8.6%, respectively, following 48 weeks of therapy (see Supplementary Information).
5HT2C Agonists (Lorcaserin)
Central serotonin levels play a role in feeding behavior and energy balance, and two selective serotonin reuptake inhibitors (SSRIs), fluoxetine and sertraline, induce weight loss in obese subjects (see Supplementary Information). The proposed mechanism by which serotonergic drugs produce anorectic effects is through stimulation of central 5-HT2C receptors, and the development of selective 5-HT2C agonists may be safer and have greater utility in anti-obesity therapy compared to the non-selective serotonergic drugs currently available. While several selective 5-HT2C agonists are effective in suppressing food intake and inducing weight loss in rodents, only lorcaserin hydrochloride (ADP-356) has moved into clinical testing. In a 12-week trial, obese subjects treated with lorcaserin (10 mg q.d., 15 mg q.d., or 10 mg b.i.d.) experienced placebo-adjusted weight losses of 1.5 kg, 2.3 kg, and 3.3 kg (p < 0.001 for each group), respectively, and the percentage of patients completing the trial who lost ≥5% of their initial body weight was 2.3%, 12.8%, 19.5%, and 31.2% for the placebo, 10 mg q.d., 15 mg q.d., and 10 mg b.i.d. groups, respectively (see Supplementary Information). A phase III program, consisting of the BLOOM (Behavioral modification and Lorcaserin for Overweight and Obesity Management) and BLOSSOM (Behavioral modification and LOrcaserin Second Study for Obesity Management) trials, evaluated >7,000 patients for up to two years. Significant weight loss associated with lorcaserin use was observed in both trials, prompting Arena Pharmaceuticals to submit a New Drug Application (NDA), which was accepted for filing by the FDA in February 2010.36
H1-Receptor Agonists & H3-Receptor Antagonists
Beyond serotonergic signaling, central histamine signaling has been implicated in appetite regulation, and the weight gain associated with antipsychotic medication may reflect the inhibition of H1-histamine receptor signaling. Histamine signaling is autoregulated by the pre-synaptic H3-histamine receptor, which inhibits histamine synthesis and release upon activation. Thus, H1-receptor agonists and H3-receptor antagonists are both in development as anti-obesity therapeutics, although more attention has been focused on the development of H3-receptor antagonists, since they are expressed almost exclusively in the central nervous system and their modulation will have fewer peripheral adverse effects. In preclinical testing, a selective H3-receptor antagonist, A-331440, at oral doses of 5 mg/kg and 15 mg/kg b.i.d, induced significant weight loss in mice raised on a high-fat diet, with the highest dose reducing weight to levels comparable to mice raised on a low-calorie diet.37 Schering-Plough recently completed a phase II clinical trial for the use of an H3-receptor, antagonist, SCH-497079, in treating obesity.38 Also, OBEcure, Ltd. has recently had positive results in phase II trials investigating the use of Histalean, a drug formulation based on the H1-receptor agonist/H3-receptor antagonist, betahistine, in causing weight loss.39
Peripheral Modulators of Metabolism and Lipogenesis
Cetilistat
Due to the undesired gastrointestinal side effects produced by orlistat, a lipase inhibitor which reduces intestinal fat absorption, improved lipase inhibitors which have equal or greater efficacy while generating fewer adverse effects are being developed. Cetilistat is a novel lipase inhibitor which is currently in clinical development. A recent trial compared the efficacy and safety of cetilistat and orlistat relative to placebo in obese patients with type II diabetes on metformin. Subjects receiving cetilistat (80 or 120 mg t.i.d.) experienced similar significant reductions in weight relative to placebo compared to the orlistat (120 mg t.i.d) group (3.85 kg, p=0.01; 4.32 kg, p=0.0002; 3.78 kg, p=0.008, respectively). Further, similar significant reductions in waist circumference relative to placebo were observed in the cetilistat 80 mg and 120 mg groups (4.3 cm, p=0.033; 4.5 cm, p = 0.037 vs. 3.2 cm) and in the orlistat group (4.4 cm; p=0.019). Significant reductions in HbA1c levels also were observed in the cetilistat 80 mg and 120 mg, as well as the orlistat group. Thus, cetilistat (80 mg or 120 mg t.i.d.) was comparable in efficacy to orlistat (120 mg t.i.d.). However, while the proportion of patients in the orlistat group reporting adverse events (93%) was similar to that in the cetilistat groups, patients in the orlistat group reported more events (541 AEs vs. 428 AEs in the 120 mg cetilistat group, p=0.0148), with more events being severe (55 vs. 31 in the 120 mg cetilistat group, p=0.0546) (see Supplemental Information). Since phase I trials showed no correlation between cetilistat dose/fecal fat content and the incidence of oily stools, a common GI side effect experienced with orlistat use, the author of this study proposes that the GI side effects associated with orlistat are not a class effect common to all drugs targeting gastric/pancreatic lipase. However, long-term studies of the safety and efficacy of cetilistat are still required in order to determine if this agent is indeed a better tolerated and equally efficacious therapeutic option to orlistat.
Growth Hormone (GH)
Typically, obese individuals display blunted secretion of GH, as well as reduced GH half-life, frequency of GH secretion, and daily GH production. Similarly, GH secretion in response to exogenous and endogenous stimuli is reduced in obese individuals, and weight loss restores normal spontaneous and stimulated GH secretion. GH-replacement therapy has been tested to reduce body fat and improve obesity-related co-morbidities. However, a review of 16 clinical trials on GH administration in obesity demonstrated that when administered in combination with a hypocaloric diet, GH does not induce more weight loss than diet alone. When administered with an isoenergetic diet, GH only induced a modest reduction in weight compared to placebo. Furthermore, several studies reported worsening insulin resistance in subjects treated with GH.40
More recently, an Australia-based biotechnology company, Metabolic Pharmaceuticals Limited, developed AOD-9604, which is a modified fragment of amino acids 177–191 of GH that mimics the lipolytic effects of GH without producing growth effects. In a 12-week randomized clinical trial, subjects receiving AOD-9604 (1 mg/d) lost an average of 2.6 kg, compared to 0.8 kg in the placebo group.41 AOD-9604 did not produce weight loss in a dose-dependent manner as subjects receiving AOD-9604 (10 mg/d) showed a smaller reduction in weight. Development of this drug was terminated in 2007 because the drug failed to induce significant weight loss in a 24-week trial of 536 subjects.42 However, in 2009 Phosphagenics Limited signed a collaborative research and option agreement with Metabolic Pharmaceuticals Limited to license AOD9604 for use as a cosmeceutical product aimed at reducing cellulite and subcutaneous fat using their proprietary transdermal delivery platform (TPM).43
Diazoxide
Individuals who are most insulin-sensitive display accelerated weight gain, while insulin resistance is associated with a decreased risk of further weight gain. Therefore, the insulin resistance observed in obese individuals may be an adaptive mechanism to prevent further fat accumulation. Diazoxide, a potassium channel activator which opens the K+-ATP channel in pancreatic β-cells thereby inhibiting insulin secretion, is useful in the treatment of hypoglycemia but also has potential to treat obesity (see Supplementary Information). However, hyperglycemia is a concern, particularly in obese patients with poor glycemic control, and larger studies of longer duration are required to evaluate the efficacy and safety of diazoxide in obese hyperinsulinemic patients.
β3-Adrenoreceptor Agonists
The β3-adrenergic receptor is expressed by adipocytes and activation by cognate β-agonists induces lipolysis and increased fatty acid oxidation as well as insulin activity in adipose tissue. However, a selective human β3-agonist, L-796568 (350 mg/d), failed to reduce weight or increase 24-hour energy expenditure in overweight and obese non-diabetic men over 28 days (see Supplementary Information). The absence of efficacy of this targeted agent may reflect the low numbers of β3-adrenoreceptors on human, compared to murine, adipocytes, and the weak effect of β3 activation on lipolysis in human subcutaneous adipose tissue, compared to β1- and β2-adrenoreceptor activation (see Supplementary Information). Eli Lilly is currently sponsoring a phase II trial to study the effectiveness of the β3-agonist, LY-377604, combined with sibutramine, in inducing weight loss in overweight males and females.44
11β-Hydroxysteroid Dehydrogenase Type 1 Inhibitors
11β-Hydroxysteroid dehydrogenase type 1 (11β-HSD1) resides in the endoplasmic reticulum of most tissues and converts inactive cortisone to active cortisol. Thus, 11β-HSD1 potentiates the activity of cortisol at the tissue level by increasing its local concentration. Patients with increased circulating glucocorticoids often develop features of the metabolic syndrome, including central obesity, insulin resistance, hypertension, and dyslipidemia. Typical obese patients exhibit these same pathologies in the absence of abnormal circulating glucocorticoid levels, suggesting that changes in cortisol metabolism at the tissue level may be responsible. Pharmacologic inhibition of 11β-HSD1 in humans with the nonselective 11β-HSD inhibitor, carbenoxolone improved whole body insulin sensitivity in seven healthy male volunteers (see Supplementary Information). In a trial of lean and obese individuals, obese subjects exhibited more rapid conversion of cortisone to cortisol in adipose tissue, but carbenoxolone treatment did not inhibit adipose 11β-HSD1 activity or improve insulin sensitivity (see Supplementary Information). Therefore, selective 11β-HSD1 inhibitors which are effective in adipose tissue are in development by several pharmaceutical companies, as these agents may be efficacious in improving insulin sensitivity and reducing weight.
Angiogenesis Inhibitors
Expansion of the adipose capillary bed in obesity suggests that anti-angiogenesis may be a therapeutic strategy for managing weight. Activated adipocytes produce an array of vascular growth factors, including vascular endothelial growth factor (VEGF), VEGF-C, VEGF-D, soluble VEGF receptor-2 (sVEGFr2), hepatocyte growth factor (HGF), angiopoietin-2, and angiogenin, and serum concentrations of several of these growth factors are higher in overweight and obese individuals.45 The excess of vascular growth factors in overweight and obese patients, in addition to allowing for expansion of adipose tissue, also may underlie the increased incidence of several types of metastatic cancers in these individuals. In addition to secreting vascular growth factors, adipocytes also release several matrix metalloproteases (MMPs) which modulate the extracellular matrix and allow matrix-bound vascular growth factors to induce angiogenesis. Thus, angiogenesis, as well as MMP, inhibitors are in development as targeted anti-obesity therapeutics.
Treatment of leptin-deficient ob/ob mice with the angiogenesis inhibitor, TNP-470, induced a dose-dependent reduction in weight, while control ob/ob mice gained weight during the same period. Furthermore, aged, weight-stable ob/ob mice lost weight with TNP-470 treatment, indicating that the adipose vasculature is susceptible to inhibition even when the tissue is not growing. Moreover, mice treated with TNP-470 immediately began regaining weight upon cessation of treatment.46 In addition to decreasing adipose vascularity, TNP-470 also decreased food intake, which is partly responsible for the weight loss. TNP-470 also prevented obesity in diet-induced obesity mouse models, and angiogenesis inhibition suppressed adipose tissue growth without inducing a significant reduction in lean tissue mass.46 Currently, the South Korea-based biotechnology company, AngioLab Inc., is conducting phase II trials for the use of the anti-angiogenic/anti-MMP drug, ALS-L1023, in treating obesity.47
Sirtuin 1 (SIRT1) Activators
For decades it has been recognized that calorie restriction of rodents extends both mean and maximum survival time, as well as decreasing and delaying the incidence of several diseases associated with aging, such as cancer, atherosclerosis, and diabetes. Calorie restriction has been associated with an upregulation of SIRT1, an NAD-dependent deacetylase, in a variety of rodent tissues. SIRT1 enhances fat mobilization and lipolysis by binding to peroxisome proliferator-activated receptor-γ (PPAR-γ) and repressing the expression of PPAR-γ-regulated genes, including those mediating fat storage. SIRT1 also interacts with PGC-1α, inducing the expression of mitochondrial genes involved in oxidative metabolism and fatty acid oxidation.
Resveratrol (RSV), a potent allosteric SIRT1 activator, protects mice from diet-induced obesity and insulin resistance, enhances mitochondrial activity in brown adipose tissue and skeletal muscle, and increases the aerobic capacity of skeletal muscle. Further, mice treated with SRT1720, a potent, selective synthetic activator of SIRT1, were resistant to diet-induced obesity and insulin resistance due to enhanced oxidative metabolism in skeletal muscle, liver, and brown adipose tissue (see Supplementary Information). In 2004, GlaxoSmithKline founded Sirtris Pharmaceuticals, a company focused on the development of drugs which target sirtuins, and currently they are investigating several SIRT1 activators in phase I and phase II trials for the treatment of type II diabetes and obesity.48 Other pharmaceutical companies are also developing specific SIRT1 activators, which are being investigated to define their utility in the treatment of obesity and metabolic diseases. Furthermore, as new insights are made into the transcriptional regulation of appetite-regulating neuropeptides as well as the genes involved in cellular metabolism, new drugs targeting these pathways may have potential as anti-obesity pharmacotherapeutics.
Combination Therapeutics
Contrave (Bupropion + Naltrexone)
Combination therapy has proven effective in treating a variety of conditions, from hypertension, to heart disease, to infectious diseases. Therefore, combination therapy may prove similarly effective in combating obesity. Bupropion (BUP) is a norepinephrine- and dopamine-reuptake inhibitor and, as previously noted, its anorectic potential is linked to activation of POMC-expressing hypothalamic neurons. Naltrexone (NTX) is an opioid receptor antagonist which alone is ineffective in producing meaningful weight loss. However, opioid agonists inhibit POMC-neuronal signaling, an effect which may be repressed by NTX. Together, these considerations suggest a paradigm in which BUP induction of POMC-neuronal signaling is potentiated by NTX antagonism of the normal inhibitory mechanism that limits sustained POMC activation.49
In July 2009, the Contrave trial sponsor, Orexigen Therapeutics, Inc., announced the results of three 56-week phase III trials. In one trial, obese patients receiving Contrave-32 [BUP (360 mg/d) + NTX (32 mg/d)], along with a diet and exercise regimen, experienced a mean weight loss of 8.1%, compared to only 1.8% in the placebo group. Another trial of 1,496 obese subjects yielded similar results with no significant difference in patients treated with Contrave-32 vs. Contrave-48 [BUP (360 mg/d) + NTX (48 mg/d)]. Finally, in a trial of 505 obese patients with type II diabetes, subjects treated with Contrave-32, along with a diet and exercise regimen, experienced a mean weight loss of 5.9% vs. 2.2% in the placebo group (see Supplementary Information). On the basis of these results, Orexigen will submit a New Drug Application (NDA) with the FDA in 2010.
Empatic (Bupropion + Zonisamide)
Bupropion, an antidepressant, and zonisamide, an anti-epileptic, each induce modest weight loss in obese individuals. In a 24-week trial, obese subjects receiving zonisamide (400 mg/d) + bupropion (300 mg/d) achieved more weight loss than zonisamide (400 mg), bupropion (300 mg), or placebo (−9.2, −6.6, −3.6, and −0.4%, respectively). A Phase IIb trial explored the efficacy of different ratios of bupropion and zonisamide in the combination, and all combinations elicited significantly more weight loss than placebo, with bupropion (360 mg) + zonisamide (360 mg) having the greatest efficacy (8.6% weight loss vs. 1.1% for placebo). Another Phase IIb trial investigated the efficacy of Empatic-360 (bupropion 360 mg/zonisamide 360 mg) and Empatic-120 (bupropion 360 mg/zonisamide 120 mg) vs. placebo. In this trial of 729 obese subjects, those receiving either Empatic-360 or Empatic-120 experienced significantly greater weight loss compared to the placebo group (9.9%, 7.7% vs. 1.7%, respectively (p<0.001)). Also, significantly more subjects lost ≥5% and ≥10% of their baseline weight in the Empatic-360 and Empatic-120 groups compared to the placebo group (82.6%, 59.3% vs. 18.9%, respectively (p<0.001) and 47.7%, 35.2% vs. 5.7%, respectively (p<0.001)). Thus, both drugs appear effective, with Empatic-360 having greater efficacy (see Supplementary Information). Adverse events were similar to those reported for each of the individual drugs, with headache, nausea, insomnia, anxiety and dry mouth being most common. Phase III clinical trials for Empatic are currently being planned.
Qnexa (Topiramate + Phentermine)
Phentermine is an amphetamine derivative which has central anorectic effects and is still prescribed in the US for the short-term treatment of obesity. Topiramate is an anti-epileptic drug with multiple mechanisms of action which induced weight loss in clinical trials but is associated with several neuropsychiatric effects, especially when administered at high doses (192 mg/d). Therefore, the two drugs were combined (Qnexa) to increase the efficacy and safety of these drugs by lowering the individual daily doses. Two 1-year phase III trials of Qnexa for the treatment of obesity, sponsored by Vivus, Inc., were completed in 2009. In the first study, 1,267 morbidly obese patients (mean BMI = 42.1 kg/m2) were randomized to receive mid-dose Qnexa, full-dose Qnexa, or placebo along with a hypocaloric diet (500 kcal deficit) and advised to implement a lifestyle modification program. For those who completed the 52-week study, subjects receiving mid-dose and full-dose Qnexa experienced mean weight losses of 7.0% and 14.7%, respectively, compared to 2.5% in the placebo group (p<0.0001). In the second study, 2,487 overweight and obese patients with high blood pressure, high cholesterol, or type II diabetes were placed on the same regimen, and mid-dose and full-dose Qnexa subjects who completed the study experienced mean weight losses of 10.5% and 13.2%, respectively, compared to 2.4% in the placebo group (p<0.0001). Qnexa therapy also was associated with significant improvements in blood pressure, triglycerides, and HbA1c levels (see Supplementary Information). While topiramate has been associated with adverse effects including suicidality, metabolic acidosis, acute myopia, secondary angle closure glaucoma, and congenital malformations, to date, Qnexa has not been associated with those risks.
CONCLUSION
Escalating rates of obesity have serious global health and economic consequences, and currently there is a critical unmet clinical need for safe and effective therapies to combat this condition. In that context, an increased understanding of the molecular circuits regulating appetite, cellular metabolism, and lipogenesis have revealed new molecular targets to treat obesity. However, we have only a rudimentary understanding of the molecular mechanisms controlling energy balance, and it is anticipated that deeper insights into these mechanisms will produce novel molecular targets for therapy. Beyond the discovery of anti-obesity drug targets, new combination therapies and improved delivery methods, including oral, intranasal, and transdermal formulations, will enhance the efficacy of current drugs and their utility for patients and physicians.
While greater insights provided by the new biology are providing more effective molecular targets for obesity, there are considerable hurdles that must be considered in the development of drugs for clinical application of the worldwide populations afflicted by this pandemic. Indeed, in the context of the availability of safe and effective drugs for the co-morbidities of obesity, new anti-obesity therapies must demonstrate long-term advantages in safety, tolerability and efficacy to be translated into clinical practice. The FDA has established stringent requirements for the development of anti-obesity pharmacotherapeutics, with strict safety and efficacy standards requiring trials of ≥1 year duration that include >4,500 subjects.50 Also, these agents cannot be approved for the treatment of an obesity-related co-morbidity, such as type II diabetes or cardiovascular disease, without demonstrating their efficacy for treating those conditions independently of their effect on weight loss.50 Finally, in part due to these considerations, insurance companies often do not provide coverage for these agents because obesity is not uniformly recognized as a unique disease entity. Thus, as discovery efforts yield novel molecularly targeted agents to treat obesity, regulatory and reimbursement policies must evolve to facilitate their rapid integration into clinical practice algorithms to improve the care of overweight and obese patients.
Supplementary Material
Figure 1. Endogenous signaling of appetite-regulating hormones, neuropeptides, & neurotransmitters and the drugs targeting these pathways.
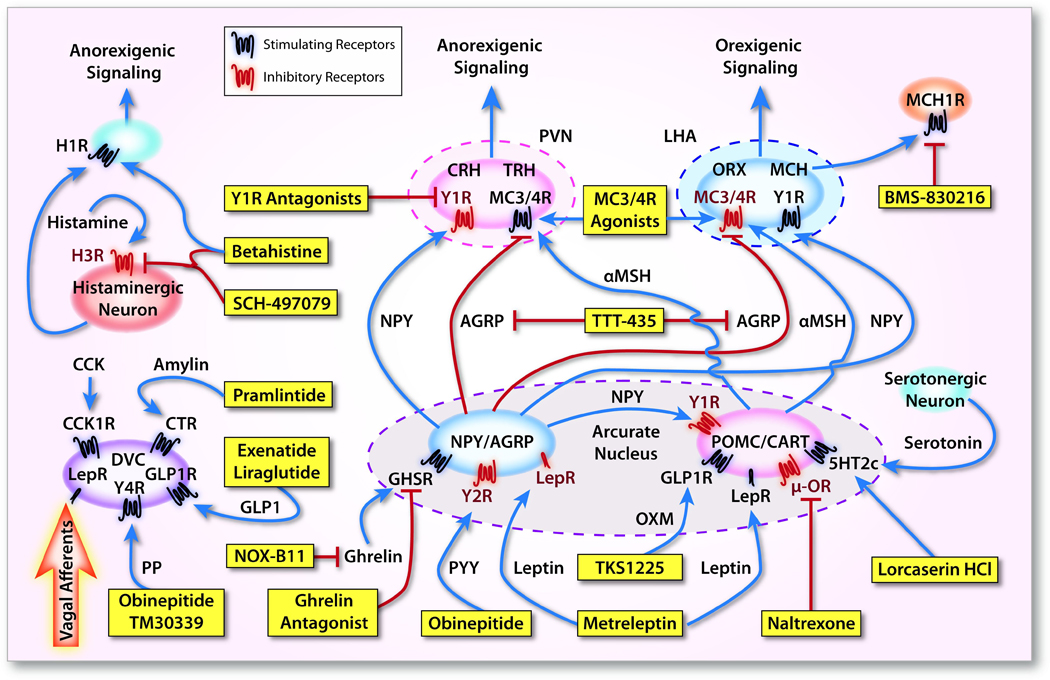
The arcuate nucleus of the hypothalamus is the primary neural signaling site for peripheral satiety hormones. It is composed of two primary types of appetite-regulating neurons: 1) those expressing the anorexigenic neuropeptides pro-opiomelanocortin (POMC) and cocaine- and amphetamine-regulated transcript (CART) and 2) those expressing the orexigenic neuropeptides agouti-related peptide (AgRP) and neuropeptide Y (NPY). There is communication between the hypothalamus and the brainstem, particularly the dorsal vagal complex (DVC) which is targeted by several satiety hormones and also receives satiety signals from vagal afferents from the GI tract. NPY/AgRP and POMC/CART neurons act on second-order neurons of the hypothalamus, including the corticotropin-releasing hormone (CRH)- and thyrotropin-releasing hormone (TRH)-expressing neurons of the paraventricular nucleus (PVN) and the orexin- and melanin-concentrating hormone (MCH)-expressing neurons of the lateral hypothalamic area (LHA). These second-order neurons target higher brain centers to induce appetite suppression or stimulation. Monoamine neurotransmission is also important in appetite regulation, as central histamine and serotonin signaling suppress appetite. Central noradrenergic and dopaminergic signaling (not shown) also reduce appetite. Blue line – activating; Red line – inhibiting; AgRP, agouti-related peptide; CART, cocaine- and amphetamine-regulated transcript; CCK, cholecystokinin; CCK1R, cholecystokinin receptor-1; CTR, calcitonin receptor; CRH, corticotropin-releasing hormone; DVC, dorsal vagal complex; GHSR, growth hormone secretagogue receptor; GLP-1, glucagon-like peptide-1; GLP1R, glucagon-like peptide-1 receptor; H1R, histamine receptor-1; H3R, histamine receptor-3; 5HT2c, 5-hydroxytryptamine receptor-2C; LepR, leptin receptor; LHA, lateral hypothalamic area; MC3/4R, melanocortin 3/4 receptor; MCH, melanin concentrating hormone; MCH1R, melanin concentrating hormone receptor-1; α-MSH, α-melanocyte stimulating hormone; NPY, neuropeptide Y; μ-OR, μ-opioid receptor; ORX, orexin; POMC, pro-opiomelanocortin; PP, pancreatic polypeptide; PVN, paraventricular nucleus; TRH, thyrotropin-releasing hormone; Y1R, Y1-receptor; Y2R, Y2-receptor; Y4R, Y4-receptor.
Figure 2. Molecular targets for anti-obesity pharmacotherapeutics outside the CNS.
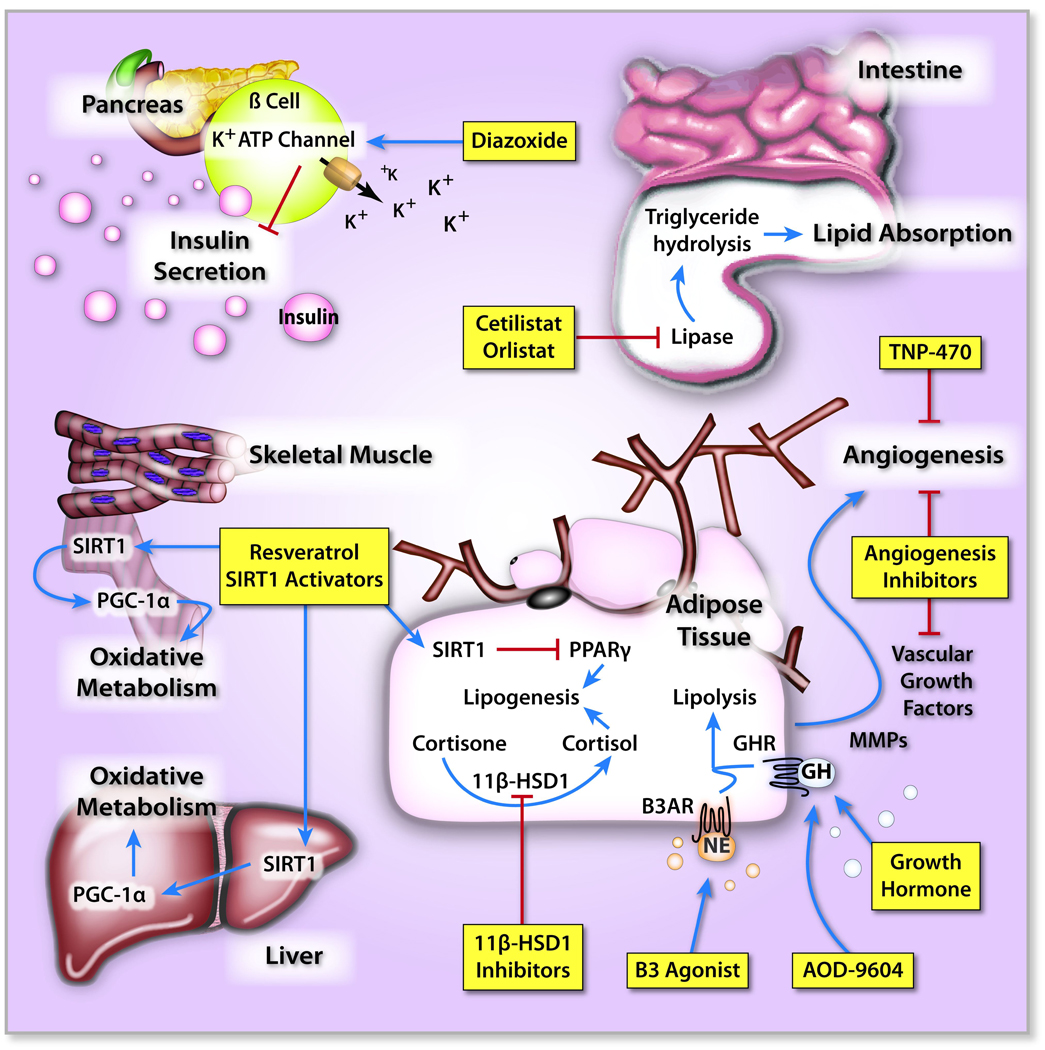
In the intestine, the inhibition of gastric/pancreatic lipase reduces triglyceride hydrolysis and lipid absorption. Within the pancreas, the opening of K+-ATP channels reduces insulin secretion, which leads to enhanced nutrient catabolism and reduced nutrient storage. In adipose tissue, both β3-adrenergic receptor (β3AR) and growth hormone receptor (GHR) activation induce lipolysis. Also, inhibition of 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1), which converts inactive cortisone to active cortisol, may induce weight loss since high cortisol levels are associated with increased fat accumulation. Activated adipocytes also produce an array of vascular growth factors and matrix metalloproteases (MMPs) that induce an expansion of the capillary bed which aids the growth of adipose tissue. Inhibition of angiogenesis within the adipose tissue, therefore, may be effective in reducing adiposity. Finally, weight loss may be achieved by modulation of cellular metabolism through the activation of sirtuin 1 (SIRT1), which 1) enhances fat mobilization and lipolysis by binding to peroxisome proliferator-activated receptor-γ (PPAR-γ) in adipocytes, thereby repressing the expression of PPAR-γ-regulated genes, including those mediating fat storage, and 2) induces the expression of mitochondrial genes involved in oxidative metabolism and fatty acid oxidation within the liver and skeletal muscle through the activation of peroxisome proliferator-activated receptor-γ coactivator (PGC-1α). Blue arrow – activating; Red arrow – inhibiting; β3AR, β3-adrenergic receptor; 11β-HSD1, 11β-hydroxysteroid dehydrogenase type 1; GH, growth hormone; GHR, growth hormone receptor; MMP, matrix metalloprotease; NE, norepinephrine; PGC-1α, peroxisome proliferator-activated receptor-γ coactivator; PPAR-γ, peroxisome proliferator-activated receptor-γ; SIRT1, sirtuin 1.
Table 1.
Historical Milestones in the Pharmacology of Weight Loss
| Drug | Time of Popular Use |
Mechanism of Action |
Adverse Effects |
|---|---|---|---|
| Thyroid Hormone | Early 1900s –1980s | Activation of thyroid hormone receptor | Reduced lean body mass, cardiac arrhythmias, cardiac arrest |
| Dinitrophenol (DNP) | 1930s | Uncoupling agent – dissipation of mitochondrial chemiosmotic gradient | Agranulocytosis, dermatitis, cataracts, fatal hyperthermia |
| “Rainbow pills” | 1940s–1960s | Multiple mechanisms – combination of amphetamines, digitalis, thyroid hormone, diuretics, laxatives, and barbiturates | Myocardial toxicity, sudden death |
| Aminorex | 1960s | Norepinephrine releasing agent | Pulmonary hypertension |
| Fenfluramine/dexfenfluramine | 1990s | Serotonin releasing agent/serotonin reuptake inhibitor | Pulmonary hypertension, heart valve disease |
| Phenylpropanolamine (PPA) | 1970s–1990s | Norepinephrine releasing agent | Intracranial bleeding, hemorrhagic stroke |
| Ephedra | 1990s–2000s | Adrenergic receptor activation | Palpitations, hypertension, heart attack, stroke, sudden death |
| Rimonabant | 2000s | CB1-receptor antagonist | Depression, suicide ideation |
Table 2.
Centrally-Acting Anti-Obesity Drugs
| Target | Drug | Mechanism of Action |
Company | Status |
|---|---|---|---|---|
| NPY | MK-0557 | Y5 receptor antagonist | Merck | Phase II |
| S-2367 | Y5 receptor antagonist | Shionogi USA, Inc. | Phase II | |
| AgRP | TTT-435 | AgRP inhibitor | TransTech Pharma | Phase II |
| MCH | BMS-830216 | MCH-1 receptor antagonist | Bristol-Myers Squibb | Phase I/II |
| CCK | GI 181771X | Selective CCK-A agonist | Glaxo Smith Kline | Terminated |
| GLP-1 | Vildagliptin | DDP-IV inhibitor | Novartis | Phase III |
| Sitagliptin | DDP-IV inhibitor | Merck | Approved for Diabetes | |
| Liraglutide | Proteolysis resistant GLP-1 analogue | Novo Nordisk | Approved for Diabetes | |
| Exenatide | Proteolysis resistant GLP-1 analogue | Amylin/Eli Lilly | Approved for Diabetes | |
| Exenatide-LAR | Long-acting proteolysis resistant GLP-1 analogue | Amylin/Eli Lilly | FDA Regulatory Review | |
| NN9924 | Long-acting oral GLP-1 analogue | Novo Nordisk | Phase I | |
| Oxyntomodulin (OXM) | TKS1225 | Long-acting OXM analogue | Thiakis, Ltd./Wyeth/Pfizer | Pre-Clinical |
| PYY | PYY (3–36) | Intranasal active PYY | Nastech/Merck | Phase I/II |
| Ghrelin | NOX-B11 | Ghrelin inactivator | NOXXON Pharma Ag/Pfizer | Pre-Clinical |
| Oral Ghrelin Antagonist | Ghrelin antagonist | Elixir Pharmaceuticals | Pre-Clinical | |
| Pancreatic polypeptide (PP) | Obinepitide | Y2/Y4 receptor agonist | 7TM Pharma | Phase I/II |
| TM30339 | Selective Y4 receptor agonist | 7TM Pharma | Phase I/II | |
| Amylin | Pramlintide | Synthetic amylin analogue | Amylin Pharmaceuticals | Approved for Diabetes |
| Leptin | Metreleptin/Pramlintide | Leptin/amylin analogues | Amylin Pharmaceuticals | Phase II |
| 5HT2C Receptor | Lorcaserin HCl | 5HT2C agonist | Arena Pharmaceuticals, Inc. | FDA Review |
| H1/H3 Receptor | SCH-497079 | H3 receptor antagonist | Schering-Plough | Phase II |
| Histalean | H1-receptor agonist/H3 receptor antagonist | OBEcure, Ltd. | Phase II | |
| Noradrenergic, Dopaminergic, & Opioid Signaling | Contrave (Bupropion-SR + Naltrexone) | Norepinephrine- and dopamine-reuptake inhibitor/μ-opioid receptor antagonist | Orexigen Therapeutics, Inc. | Phase III |
| Noradrenergic, Dopaminergic, & GABA Signaling | Empatic (Bupropion-SR + Zonisamide) | Norepinephrine- and dopamine-reuptake inhibitor/GABA receptor activator | Orexigen Therapeutics, Inc. | Phase III |
| Noradrenergic & GABA Signaling | Qnexa (Topiramate + Phentermine) | Norepinephrine releasing agent/GABA receptor activator | Vivus, Inc. | Phase III |
Table 3.
Peripherally-Acting Anti-Obesity Drugs
| Target | Drug | Mechanism of Action |
Company | Status |
|---|---|---|---|---|
| Gastric/Pancreatic Lipase | Cetilistat | Lipase inhibitor | Norgine/Takeda | Phase III |
| β3-Adrenergic Receptor | LY377604 | β3-AR agonist | Eli Lilly | Phase II |
| K+-ATP Channel | Diazoxide | Opens K+-ATP channel | Essentialis, Inc. | Phase II/III |
| Angiogenesis | ALS-L1023 | Angiogenesis/MMP inhibitor | AngioLab, Inc. | Phase II |
| SIRT1 | SRT2104 | SIRT1 activator | Sirtris Pharmaceuticals | Phase II |
| SRT2379 | SIRT1 activator | Sirtris Pharmaceuticals | Phase I | |
| SRT501 | SIRT1 activator | Sirtris Pharmaceuticals | Phase II |
ACKNOWLEDGEMENTS
Support was provided by NIH grants R01 CA75123, R01 CA95026, RC1 CA146033, and P30 CA56036 and from Targeted Diagnostic and Therapeutics, Inc (SAW). SAW is the Samuel M.V. Hamilton Professor of Medicine.
Abbreviations
- AgRP
agouti-related peptide
- BUP
bupropion
- bupropion-SR
bupropion sustained release
- CART
cocaine- and amphetamine-regulated transcript
- DNP
dinitrophenol
- exenatide-LAR
exenatide long-acting release
- GH
growth hormone
- GLP-1
glucagon-like peptide-1
- GOAT
ghrelin O-acyltransferase
- HbA1c
Hemoglobin A1c
- HGF
hepatocyte growth factor
- 11β-HSD1
11β-hydroxysteroid dehydrogenase type 1
- IAPP
islet amyloid polypeptide
- KLH
heyhole limpet hemocyanin
- MMP
matrix metalloprotease
- NAD
nicotinamide adenine dinucleotide
- NPY
neuropeptide tyrosine
- NTX
naltrexone
- OXM
oxyntomdulin
- PP
pancreatic polypeptide
- PYY
peptide tyrosine tyrosine
- PBA
4-phenyl butyric acid
- POMC
pro-opiomelanocortin
- PPAR-γ
peroxisome proliferator-activated receptor-γ
- PGC-1α
peroxisome proliferator-activated receptor-γ coactivator
- PPA
phenylpropanolamine
- RSV
resveratrol
- SSRI
selective serotonin reuptake inhibitor
- SIRT1
sirtuin-1
- SNAC
sodium N-[8-(2-hydroxybenzoyl) amino] caprylate
- TUDCA
tauroursodeoxycholic acid
- VEGF
vascular endothelial growth factor
Footnotes
FINANCIAL AND COMPETING INTERESTS DISCLOSURE
Michael A. Valentino is the recipient of a pre-doctoral fellowship from the Pharmaceutical Research and Manufacturers of America (PhRMA) Foundation. SAW is a consultant to Merck and CombiMab, Inc., the Chair of the Data Safety Monitoring Board for the C-Cure Trial™ sponsored by Cardio3 Biosciences; and the Chair (uncompensated) of the Scientific Advisory Board to Targeted Diagnostics and Therapeutics, Inc.
REFERENCES
- 1.Rodgers A, Vaughan P, Prentice T, Edejer TT-T, Evans D, Lowe J. Geneva, Switzerland: World Health Organization; 2002. Reducing Risks, Promoting Healthy Life. 2002. [Google Scholar]
- 2.Bhasin S, Wallace W, Lawrence JB, Lesch M. Sudden death associated with thyroid hormone abuse. American Journal of Medicine. 1981;71:887–890. doi: 10.1016/0002-9343(81)90392-2. [DOI] [PubMed] [Google Scholar]
- 3.Dinitrophenol not acceptable for N.N.R. JAMA. 1935;105:31–33. [Google Scholar]
- 4.Asher WL. Mortality rate in patients receiving "diet pills". Current Therapeutic Research - Clinical and Experimental. 1972;14:525–539. [PubMed] [Google Scholar]
- 5.Gurtner HP. Aminorex and pulmonary hypertension. A review. Cor Vasa. 1985;27:160–171. [PubMed] [Google Scholar]
- 6.Connolly HM, Crary JL, McGoon MD, Hensrud DD, Edwards BS, Edwards WD, Schaff HV. Valvular heart disease associated with fenfluramine phentermine. N. Engl. J. Med. 1997;337:581–588. doi: 10.1056/NEJM199708283370901. [DOI] [PubMed] [Google Scholar]
- 7.Kernan WN, Viscoli CM, Brass LM, Broderick JP, Brott T, Feldmann E, Morgenstern LB, Wilterdink JL, Horwitz RI. Phenylpropanolamine and the risk of hemorrhagic stroke. N. Engl. J. Med. 2000;343:1826–1832. doi: 10.1056/NEJM200012213432501. [DOI] [PubMed] [Google Scholar]
- 8.Haller CA, Benowitz NL. Adverse cardiovascular and central nervous system events associated with dietary supplements containing ephedra alkaloids. N. Engl. J. Med. 2000;343:1833–1838. doi: 10.1056/NEJM200012213432502. [DOI] [PubMed] [Google Scholar]
- 9.Saper RB, Eisenberg DM, Phillips RS. Common dietary supplements for weight loss. American Family Physician. 2004;70:1731–1738. [PubMed] [Google Scholar]
- 10.Heal DJ, Gosden J, Smith SL. Regulatory challenges for new drugs to treat obesity and comorbid metabolic disorders. Br. J. Clin. Pharmacol. 2009;68:861–874. doi: 10.1111/j.1365-2125.2009.03549.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Erondu N, Gantz I, Musser B, Suryawanshi S, Mallick M, Addy C, Cote J, Bray G, Fujioka K, Bays H, Hollander P, Sanabria-Bohórquez SM, Eng W, Långström B, Hargreaves RJ, Burns HD, Kanatani A, Fukami T, MacNeil DJ, Gottesdiener KM, Amatruda JM, Kaufman KD, Heymsfield SB. Neuropeptide Y5 receptor antagonism does not induce clinically meaningful weight loss in overweight and obese adults. Cell Metabolism. 2006;4:275–282. doi: 10.1016/j.cmet.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 12.Sargent BJ, Moore NA. New central targets for the treatment of obesity. Br. J. Clin. Pharmacol. 2009;68:852–860. doi: 10.1111/j.1365-2125.2009.03550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.7TM Pharma research pipeline. Available from: http://www.7tm.com/Pipeline.aspx.
- 14.TransTech Pharma obesity product pipeline. Available from: http://www.ttpharma.com/TherapeuticAreas/MetabolicDiseases/Obesity/TTP435/tabid/126/Default.aspx.
- 15.Bristol-Myers Squibb metabolics clinical trial registry. Available from: http://ctr.bms.com/OneBmsCtd/InitTrialAction.do?linkname=Metabolics&type=pharma&sortby=default.
- 16.Ozcan L, Ergin AS, Lu A, Chung J, Sarkar S, Nie D, Myers MG, Jr, Ozcan U. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metabolism. 2009;9:35–51. doi: 10.1016/j.cmet.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 17.Roth JD, Roland BL, Cole RL, Trevaskis JL, Weyer C, Koda JE, Anderson CM, Parkes DG, Baron AD. Leptin responsiveness restored by amylin agonism in diet-induced obesity: Evidence from nonclinical and clinical studies. Proc. Natl. Acad. Sci. U. S. A. 2008;105:7257–7262. doi: 10.1073/pnas.0706473105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amylin Pharmaceuticals, Inc. and Eli Lilly and Company: Exenatide once weekly provided sustained improvements in glycemic control with weight loss over two years: DURATION-1 interim long-term data presented at ADA 2009. Available from: http://www.amylin.com/assets/001/5097.pdf.
- 19.Amylin Pharmaceuticals, Inc. research pipeline. Available from: http://www.amylin.com/research/pipeline/exenatide-once-weekly.htm.
- 20.Novo Nordisk starts phase 1 trial with long-acting oral GLP-1 analogue (13 Jan 2010) Available from: http://www.novonordisk.com/press/news/news.asp?sNewsTypeGUID=&lMonth=&lYear=&sLanguageCode=&sSearchText=NN9924&sShowNewsItemGUID=acc555cc-2124-4ebf-ad6d-7f923e2e2a78&sShowLanguageCode=en-GB.
- 21.Dakin CL, Small CJ, Batterham RL, Neary NM, Cohen MA, Patterson M, Ghatei MA, Bloom SR. Peripheral oxyntomodulin reduces food intake and body weight gain in rats. Endocrinology. 2004;145:2687–2695. doi: 10.1210/en.2003-1338. [DOI] [PubMed] [Google Scholar]
- 22.Wynne K, Park AJ, Small CJ, Meeran K, Ghatei MA, Frost GS, Bloom SR. Oxyntomodulin increases energy expenditure in addition to decreasing energy intake in overweight and obese humans: a randomised controlled trial. Int. J. Obes. 2006;30:1729–1736. doi: 10.1038/sj.ijo.0803344. [DOI] [PubMed] [Google Scholar]
- 23.Wyeth Pharmaceuticals acquires Thiakis Limited. Available from: http://www.drugs.com/news/wyeth-pharmaceuticals-acquires-thiakis-limited-15289.html.
- 24.Larhammar D. Structural diversity of receptors for neuropeptide Y, peptide YY and pancreatic polypeptide. Regul. Pept. 1996;65:165–174. doi: 10.1016/0167-0115(96)00110-3. [DOI] [PubMed] [Google Scholar]
- 25.Degen L, Oesch S, Casanova M, Graf S, Ketterer S, Drewe J, Beglinger C. Effect of peptide YY3-36 on food intake in humans. Gastroenterology. 2005;129:1430–1436. doi: 10.1053/j.gastro.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 26.Gantz I, Erondu N, Mallick M, Musser B, Krishna R, Tanaka WK, Snyder K, Stevens C, Stroh MA, Zhu H, Wagner JA, MacNeil DJ, Heymsfield SB, Amatruda JM. Efficacy and safety of intranasal peptide YY3-36 for weight reduction in obese adults. J. Clin. Endocrinol. Metab. 2007;92:1754–1757. doi: 10.1210/jc.2006-1806. [DOI] [PubMed] [Google Scholar]
- 27.Batterham RL, Le Roux CW, Cohen MA, Park AJ, Ellis SM, Patterson M, Frost GS, Ghatei MA, Bloom SR. Pancreatic polypeptide reduces appetite and food intake in humans. J. Clin. Endocrinol. Metab. 2003;88:3989–3992. doi: 10.1210/jc.2003-030630. [DOI] [PubMed] [Google Scholar]
- 28.Young AA, Vine W, Gedulin BR, Pittner R, Janes S, Gaeta LSL, Percy A, Moore CX, Koda JE, Rink TJ, Beaumont K. Preclinical pharmacology of pramlintide in the rat: Comparisons with human and rat amylin. Drug Development Research. 1996;37:231–248. [Google Scholar]
- 29.Maggs D, Shen L, Strobel S, Brown D, Kolterman O, Weyer C. Effect of pramlintide on A1C and body weight in insulin-treated African Americans and Hispanics with type 2 diabetes: a pooled post hoc analysis. Metabolism. 2003;52:1638–1642. doi: 10.1016/j.metabol.2003.06.003. [DOI] [PubMed] [Google Scholar]
- 30.Date Y, Murakami N, Toshinai K, Matsukura S, Niijima A, Matsuo H, Kangawa K, Nakazato M. The role of the gastric afferent vagal nerve in ghrelin-induced feeding and growth hormone secretion in rats. Gastroenterology. 2002;123:1120–1128. doi: 10.1053/gast.2002.35954. [DOI] [PubMed] [Google Scholar]
- 31.Zorrilla EP, Iwasaki S, Moss JA, Chang J, Otsuji J, Inoue K, Meijler MM, Janda KD. Vaccination against weight gain. Proc. Natl. Acad. Sci. U. S. A. 2006;103:13226–13231. doi: 10.1073/pnas.0605376103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moran TH, Dailey MJ. Gut peptides: targets for antiobesity drug development? Endocrinology. 2009;150:2526–2530. doi: 10.1210/en.2009-0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang J, Zhao T-J, Goldstein JL, Brown MS. Inhibition of ghrelin O-acyltransferase (GOAT) by octanoylated pentapeptides. Proceedings of the National Academy of Sciences. 2008;105:10750–10755. doi: 10.1073/pnas.0805353105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zarrindast MR, Hosseini-Nia T. Anorectic and behavioral effects of bupropion. Gen. Pharmacol. 1988;19:201–204. doi: 10.1016/0306-3623(88)90061-4. [DOI] [PubMed] [Google Scholar]
- 35.Billes SK, Cowley MA. Inhibition of dopamine and norepinephrine reuptake produces additive effects on energy balance in lean and obese mice. Neuropsychopharmacology. 2007;32:822–834. doi: 10.1038/sj.npp.1301155. [DOI] [PubMed] [Google Scholar]
- 36.Arena Pharmaceuticals announces FDA acceptance of lorcaserin NDA for filing. Available from: http://invest.arenapharm.com/releasedetail.cfm?ReleaseID=446755.
- 37.Hancock AA, Bennani YL, Bush EN, Esbenshade TA, Faghih R, Fox GB, Jacobson P, Knourek-Segel V, Krueger KM, Nuss ME, Pan JB, Shapiro R, Witte DG, Yao BB. Antiobesity effects of A-331440, a novel non-imidazole histamine H 3 receptor antagonist. Eur. J. Pharmacol. 2004;487:183–197. doi: 10.1016/j.ejphar.2004.01.015. [DOI] [PubMed] [Google Scholar]
- 38.Gemkow MJ, Davenport AJ, Harich S, Ellenbroek BA, Cesura A, Hallett D. The histamine H3 receptor as a therapeutic drug target for CNS disorders. Drug Discovery Today. 2009;14:509–515. doi: 10.1016/j.drudis.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 39.Obecure Phase II trial shows positive results for Histalean™ in obese women under 50. Available from: http://www.obecure.com/astatic%20news/51584.php.
- 40.Shadid S, Jensen MD. Effects of growth hormone administration in human obesity. Obesity. 2003;11:170–175. doi: 10.1038/oby.2003.27. [DOI] [PubMed] [Google Scholar]
- 41.Bray GA, Greenway FL. Pharmacological treatment of the overweight patient. Pharmacol. Rev. 2007;59:151–184. doi: 10.1124/pr.59.2.2. [DOI] [PubMed] [Google Scholar]
- 42.The Phase 2B trial results for Metabolic's drug, AOD9604, do not support the commercial viability of the drug as a treatment for obesity. Development of the drug for this condition is terminated. Available from: http://www.metabolic.com.au/files/POQ26IMG5C/ASX_OPTIONSStudyResults_21Feb2007.pdf.
- 43.Phosphagenics Limited signs agreement with Metabolic Pharmaceuticals Limited. Available from: http://www.phosphagenics.com/site/DefaultSite/filesystem/documents/20090813%20Met abolic%20Agreement.pdf.
- 44.A weight loss study in overweight men and women. Available from: http://clinicaltrials.gov/ct2/show/NCT00993421.
- 45.Silha JV, Krsek M, Sucharda P, Murphy LJ. Angiogenic factors are elevated in overweight and obese individuals. Int. J. Obes. Relat. Metab. Disord. 2005;29:1308–1314. doi: 10.1038/sj.ijo.0802987. [DOI] [PubMed] [Google Scholar]
- 46.Rupnick MA, Panigrahy D, Zhang CY, Dallabrida SM, Lowell BB, Langer R, Folkman MJ. Adipose tissue mass can be regulated through the vasculature. Proc. Natl. Acad. Sci. U. S. A. 2002;99:10730–10735. doi: 10.1073/pnas.162349799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.AngioLab’s candidates. Available from: http://angiolab.co.kr/
- 48.Sirtris pipeline. Available from: http://www.sirtrispharma.com/pipeline.html.
- 49.Greenway FL, Whitehouse MJ, Guttadauria M, Anderson JW, Atkinson RL, Fujioka K, Gadde KM, Gupta AK, O'Neil P, Schumacher D, Smith D, Dunayevich E, Tollefson GD, Weber E, Cowley MA. Rational design of a combination medication for the treatment of obesity. Obesity. 2009;17:30–39. doi: 10.1038/oby.2008.461. [DOI] [PubMed] [Google Scholar]
- 50.FDA: Guidance for industry developing products for weight management. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm071612.pdf.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.