

Abstract
Missense mutations, which replace one Gly with a larger residue in the repeating sequence of the type I collagen triple helix, lead to the hereditary bone disorder osteogenesis imperfecta (OI). Previous studies suggest that these mutations may interfere with triple-helix folding. NMR was used to investigate triple-helix formation in a series of model peptides where the residue replacing Gly, as well as the local sequence environment, was varied. NMR measurement of translational diffusion coefficients allowed the identification of partially folded species. When Gly was replaced by Ala, the Ala residue was incorporated into a fully folded triple helix, whereas replacement of Gly by Ser or Arg resulted in the presence of some partially folded species, suggesting a folding barrier. Increasing the triple-helix stability of the sequence N-terminal to a Gly-to-Ser replacement allowed complete triple-helix folding, whereas with the substitution of Arg, with its large side chain, the peptide achieved full folding only after flexible residues were introduced N-terminal to the mutation site. These studies shed light on the factors important for accommodation of Gly mutations within the triple helix and may relate to the varying severity of OI.
Introduction
Mutations in Type I collagen, the major structural component of skin, bone, tendon, and ligament, lead to osteogenesis imperfecta (OI), or brittle bone disease, a hereditary disorder distinguished by fragile bones (1–4). Type I collagen is a heterotrimer, consisting of two α1 chains and one α2 chain. Like all collagens, type I adopts a characteristic triple-helix conformation consisting of three extended polyproline-II-like chains supercoiled about a common axis with one-residue staggering (5,6). The close packing of the three chains can only accommodate Gly as every third residue, generating the repetitive (Gly-Xaa-Yaa)n sequence pattern (5,6). The repeating tripeptide sequence is perfectly maintained throughout the 1014-residue-long triple-helix domain of type I collagen (7,8), and even a single Gly mutation at any site results in OI. More than 600 cases of OI have been reported to result from missense mutations that replace a Gly with another bulkier amino acid in the α1 chain, and >400 cases exist for the α2 chain of type I collagen (3,9). Defective folding of collagen has been implicated in the etiology of OI. A delay in folding was first suggested based on the observation that OI collagens contain increased posttranslational modifications, which can only take place on unfolded chains (10). More recently, abnormal folding has been supported by reports of endoplasmic reticulum stress and unfolded protein response in an OI mouse model (11,12). The phenotype of OI is highly variable, ranging from mild cases with multiple fractures to perinatal lethal cases (2,3). Factors thought to be related to this variable severity include the identity of the residue replacing Gly, the neighboring sequences around the Gly mutation site, and the proximity of the mutation to interaction sites or salt bridges (13–21), but it is not clear how these factors impact triple-helix conformation and dynamics.
Triple-helical peptides with OI mutations are useful for evaluating the effects of mutations in a systematic, well controlled environment and have proved amenable to biophysical characterization (14,22–26). Introduction of mutations within a host-guest (Gly-Pro-Hyp)8 peptide system showed that the degree of triple-helix destabilization depended on the identity of the residue replacing Gly: the order of destabilization is Ala, Ser < Cys < Arg < Val < Glu, Asp (21). Although recent studies on OI collagens suggest that their thermal stability is not strongly dependent on the residue replacing Gly (15), the order of destabilization in peptides does show a correlation with the severity and lethality of OI mutations (20,21), which could relate to their effect on folding. NMR studies on triple-helix peptides are challenging, since the characteristic rodlike shape and repeating Gly-Xaa-Yaa sequences result in resonance broadening and overlapping (27). Despite these challenges, useful information has been gained from NMR studies of peptide models for native and mutant collagens using selectively 15N-labeled residues, distances derived from nuclear Overhauser effects, dynamics using hydrogen exchange, temperature-dependent amide proton chemical shifts, relaxation times, and, recently, NMR diffusion experiments (17,25,27–31).
NMR studies of peptide models with selectively labeled residues have allowed characterization of the perturbations at an OI mutation site as well as at sites N-terminal or C-terminal to it (14,32–34). For instance, peptide T1-892, which contains six triplets from the α1(I) chain (residues 892–909, GPAGPAGPVGPAGARGPA) at the N-terminus and a stabilizing (GPO)4 sequence at the C-terminus, forms a stable triple helix, and introduction of a Gly-to-Ala or Gly-to-Ser substitution at residue 901, modeling an OI mutation, maintains the stable C-terminal triple helix but has a disordered region N-terminal to the mutation (32). The presence of a more stabilizing N-terminal sequence, GPO(GAO)3 (creating a peptide denoted as T1-898), allows complete folding in the presence of the Gly-to-Ala mutation (33), showing the importance of the surrounding sequence. Recent folding studies suggest that when Gly is replaced by Ala or Ser in T1-898, a less stable, nonnative structure is formed initially before the final folded state (35).
NMR diffusion experiments have been developed in our laboratory to explore the low population of intermediate species that are in equilibrium with the unfolded and fully triple-helical states for collagen-like peptides (25). If there are partially folded molecules in which the labeled residue is disordered within a trimer species, then the translational diffusion coefficients of the disordered peaks will be reduced, reflecting their presence in a larger trimer species (25). Such diffusion experiments are applied here to a set of peptides that model Gly mutations to clarify the sequence constraints that lead partially folded species to accumulate in the presence of a mutation. Our data suggest that the ability of the peptide to fully form a triple helix depends on the identity of the residue replacing Gly, as well as on the stability of the sequence N-terminal to the mutation. Interestingly, it appears that replacement of Gly by Arg, which has a very large side chain, requires a nearby flexible sequence to fold completely.
Materials and Methods
Sample preparation
Peptides were synthesized by the Tufts University Core Facility (Boston, MA) and were purified on a reverse-phase high-pressure liquid chromatography system (Shimadzu, Columbia, MD) with a C-18 column. The identities of all peptides were confirmed by mass spectrometry using matrix-assisted laser desorption ionization time of flight.
Circular dichroism spectroscopy
Circular dichroism (CD) spectra were recorded on an Aviv model 62DS spectrophotometer (Aviv Biomedical, Lakewood, NJ). Cells of path length 0.1 cm were used, and the temperature of the cells was controlled using a Peltier thermoelectric temperature controller (Hewlett-Packard, Palo Alto, CA). Unless explicitly stated otherwise, samples were prepared with a concentration of 1 mg/mL in 20 mM phosphate-buffered saline (PBS) (10 mM NaH2PO4, 10 mM Na2HPO4, and 150 mM NaCl) at pH 7. Peptide concentrations were determined by tyrosine absorbance at 275 nm using ɛ275 = 1400 M−1cm−1. CD was used to track melting transitions by monitoring the ellipticity at 225 nm as a function of temperature from 0°C to 60°C. Standard procedure of a rate of 0.3°C/min and a 2-min equilibration time was employed (36). Peptides were equilibrated at 0°C for at least 40 h before all melting experiments.
NMR experiments
T1-898 peptide sets were synthesized with 15N-labeled amino acids at selective positions for NMR characterization (Table S1 in the Supporting Material). Peptide T1-898 was selectively 15N-labeled at positions G7, A18, and G28, and peptide T1-898(G16A) was labeled at positions G7, A16, and G28. Peptides T1-898(G16S) and GPOT1-898(G16S) were labeled at positions G7, V15, A18, and G28; and G7, V15, A18, and G22, respectively. Peptides T1-898(G16R), GPOT1-898(G16R), and GAAT1-898(G16R) were labeled at positions G7, G13, and G28; G7, V15, A18, and G22; and G7, A15, and G22, respectively (Table S1). Samples were prepared in 10% D2O/90% H2O with a concentration of 6 mM at pH adjusted to ∼2.5 by addition of hydrochloric acid.
NMR experiments were performed on an INOVA 500 or 600 MHz spectrometer (Varian, Palo Alto, CA). 1H-15N heteronuclear single quantum coherence (HSQC) was performed at 0°C (5°C for peptide GPOT1-898(G16S)). For peptide GAAT1-898(G16R), a hydrogen-exchange experiment was performed at 0°C to solve the overlapping problem of G22. The peptide was lyophilized and re-dissolved in 100% D2O to exchange out the protons of the disordered state. As the NH of Gly in the disordered state exchanged much faster than those in the triple helix, after several hours in D2O, the intensities of disordered resonances were significantly decreased compared with trimer resonances, giving us an opportunity to observe the trimer resonances of G22.
Residue-specific translational diffusion measurements were performed as described before (25). Experiments employing a convection-compensated light-emitting diode followed by HSQC (CCLED-HSQC) were used to measure the residue-specific diffusion coefficients of the selectively labeled peptides at 0°C (5°C for peptide GPOT1-898(G16S)) and 40°C. All the samples were equilibrated for >24 h before the diffusion experiments. Peptide GPOT1-898(G16S) has weak disordered peaks at low temperature, and satisfactory data could be achieved only at 5°C. The gradient strength was calibrated on a standard doped 1% H2O in 99% D2O sample using the value 1.90 × 10−9 m2 s−1 for the diffusion coefficient of HDO at 25°C (37,38).
All data were processed using the FELIX 2004 software package (MSI, San Diego, CA). Data analysis of translational diffusion measurements was conducted as described previously (25). The diffusion coefficients at temperature T (0°C or 5°C) were normalized to values at 40°C by using the Stokes-Einstein equation, Dnormalized,T = DT × 313.15 × ηT/(T × η40°C), where DT is the directly measured diffusion coefficient at T, and ηT is the viscosity calculated for the solution of 90% H2O and 10% D2O at a specific temperature (39). Then, a ratio of Dnormalized,T to D40°C was calculated as the normalized diffusion coefficient for each residue, denoted as DM,T (DM0°C in the case of all peptides except GPOT1-898(G16S), where it was DM5°C). The error of DM,T was calculated as
| (1) |
The relative populations of partially folded species and pure monomer for each labeled residue were derived from a two-parameter fitting, assuming slow exchange rate between partially folded species and pure monomer on the diffusion timescale (25).
Some potential problems, such as cross-relaxation and exchange of the amide protons with water, could be associated with the diffusion measurements (40–46). To reduce the oscillatory modulation and signal attenuation arising from cross-relaxation and chemical exchange, the bipolar pulse pairs in the CCLED-HSQC were used (43–46). The experiments were performed at low pH (2.5) to suppress the exchange process of the amide protons with water. At pH 2.5, even at 40°C, the water-amide proton exchange rate was very slow relative to the diffusion timescale based on theoretical calculations and NMR CLEANEX-PM experiments, suggesting little potential effect (47,48). Sample volumes were minimized by using shigemi tubes to decrease artifacts due to nonlinearity of the pulsed field gradients. A control experiment with a short diffusion delay (Δ) of 50 ms was done at 10°C and 40°C, and revealed essentially the same diffusion coefficients as those measured with a 280-ms delay, indicating that the diffusion measurements were not affected by the potential artifacts.
Results
Residue-specific NMR diffusion experiments were performed to investigate the structural disruption caused by replacing a Gly in the repeating sequence of peptide T1-898 with Ala, Ser, Arg, or Asp, and to characterize the nature of the molecular species that arise from different substitutions (Table S1). Previous CD studies of these peptides have shown that the Ser replacement causes greater destabilization of the triple helix than the Ala replacement, whereas Arg and Asp do not allow complete folding of the triple helix (35).
Equilibrium conformations of T1-898 and T1-898(G16A)
The HSQC spectrum of peptide T1-898 at 0°C contains features typical of a triple-helical conformation, consistent with previous results (33). 15N-labeled residues at three different positions (G7, A18, and G28) show one or more triple-helical resonances in addition to resonances due to a disordered form (Fig. 1 A). Triple-helical resonances are distinguished from disordered resonances by their much faster 15N transverse relaxation rates (R2). Unlike disordered resonances, triple-helical resonances disappear at 40°C, a temperature higher than the melting temperatures of the peptides (data not shown). Disordered resonances, which were referred to simply as monomer resonances in our earlier studies, are used here to clarify and emphasize that these resonances may represent unfolded monomer as well as unfolded regions in a partially folded trimer. Other minor resonances observed in the HSQC spectrum arise due to cis-trans isomerization of Gly-Pro and Pro-Hyp bonds in the peptide in the unfolded states (49). The disordered resonances with the highest intensities have been assigned, and they represent the major component of cis-trans conformers: the all-trans form. The observation of distinct disordered and triple-helical resonances for all residues supports the concept of a native triple-helical-to-disordered equilibrium in solution.
Figure 1.
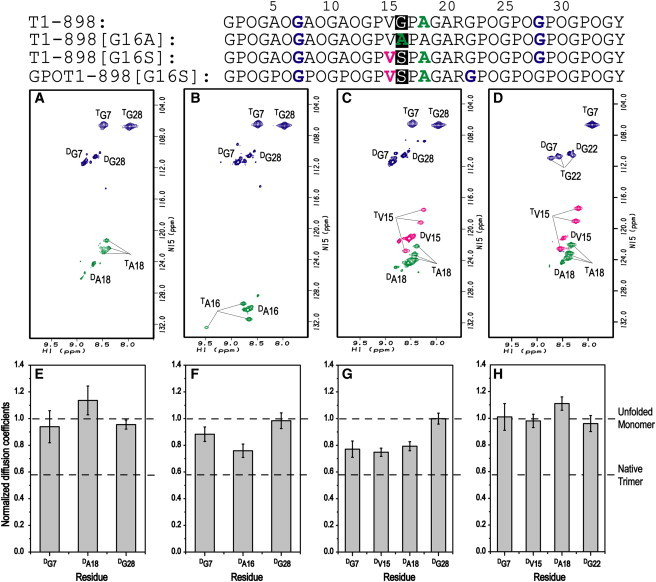
Effect of Gly-to-Ala/Ser substitution on equilibrium conformations of peptide T1-898. (A–D) 1H-15N HSQC spectra of T1-898 (A), T1-898(G16A) (B), T1-898(G16S) (C), and GPOT1-898(G16S) (D) at 0°C. Peaks corresponding to the disordered and triple-helical states are denoted by superscripts D and T, respectively. Disordered resonances, referred to simply as monomer resonances in our earlier studies, are used here to clarify and emphasize that these resonances may represent M, as well as unfolded regions in a PFT. Minor disordered resonances arise due to cis-trans isomerization in the unfolded state of the Pro/Hyp-rich chains (49). The assigned disordered resonances represent the major all-trans isomer. (E–H) Histograms of the residue-specific translational diffusion coefficients of 15N-labeled residues in disordered states for peptide T1-898 (E), T1-898(G16A) (F), T1-898(G16S) (G), and GPOT1-898(G16S) (H). Diffusion coefficients at 0°C (5°C for peptide GPOT1-898(G16S)) are normalized to those at 40°C. Peptide GPOT1-898(G16S) has weak disordered peaks at low temperature and satisfactory data could be achieved only at 5°C. The black dashed lines represent the normalized diffusion coefficients for M (theoretically with a value of 1) and native trimer (with a value of 0.58 for G28 in peptide T1-898(G16A)).
NMR translational diffusion coefficients are used to detect equilibrium intermediates in the self-associating triple-helical peptides. If there is a simple two-state equilibrium between disordered monomer species and fully folded triple-helical trimer species, then the diffusion constants of all disordered peaks will reflect the monomer state. If, however, a given residue is in a disordered state within a partially folded trimer, the diffusion constant will be smaller than that of the monomer, since the trimer species has very different hydrodynamic properties from the monomer. This model of diffusion data analysis is based on the premise that the disordered resonances originate from either the completely unfolded monomer or unfolded parts of the partially folded trimer, and this assumption is supported by previous studies (25,32,34,50). NMR relaxation studies of a peptide with a Gly-to-Ser mutation indicated that peaks due to disordered residues arise primarily from the disordered region in a partially folded species (32). NMR studies have also shown that the nonuniform diffusion coefficients observed for disordered resonances are best explained by the overlapping chemical shifts of residues in unfolded monomers and in disordered regions of partially folded trimers (25).
Residue-specific translational diffusion measurements were performed on the disordered peaks of the T1-898 peptide at 0°C and 40°C to characterize equilibrium species in solution, according to the methodology developed by Li et al. (25). We are not able to measure translational diffusion coefficients of triple-helical resonances due to low sensitivity. At 40°C, when the protein is unfolded, the disordered resonances represent the unfolded monomeric protein and serve as a control for the disordered diffusion coefficient. The diffusion coefficients are uniform at these high temperatures for all labeled positions within the T1-898 peptide, consistent with sampling a similar averaged denatured shape across the protein (Table S2). However, at a lower temperature such as 0°C, disordered resonances may represent a combination of pure monomers as well as unfolded regions within partially folded species, resulting in reduced diffusion coefficients. Therefore, comparing the values of the diffusion coefficient of disordered residues at 0°C with those at 40°C allows us to detect the existence of additional species relative to pure monomer based on the diffusion model. Moreover, this model, although qualitative, allows us to make structural predictions about which region of the peptide is disordered based on information about which part of the protein has smaller diffusion coefficients. Peptide T1-898 contains no Gly mutations, and all labeled residues show normalized diffusion coefficients at 0°C that are close to 1, which is consistent with sampling a pure monomer state (Fig. 1 E). Thus, the HSQC and diffusion experiments support a two-state unfolded monomer (M) to fully folded trimer (FT) equilibrium model.
The HSQC spectrum of peptide T1-898(G16A) shows distinct triple-helical resonances for all labeled residues at positions G7, A16, and G28 (Fig. 1 B), which is consistent with previous studies showing that the replacement of the central Gly with Ala results in a peptide that contains the features of a fully folded triple helix (33). However, normalized diffusion coefficients at 0°C (DM0°C) indicate that this peptide contains more than a simple monomer/trimer equilibrium. The DM0°C values for G7 and G28 are ∼1, indicating that the two terminal residues are not in partially folded states and suggesting that the two ends of the peptide are folded (Fig. 1 F). The much lower DM0°C of A16 suggests a triple-helical conformation with a looser middle region. Hence, the peptide T1-898(G16A) appears to have an intermediate partially folded trimer species (PFT) in addition to the FT and M forms, and the M/PFT ratio is quantified to be 1:1.2, indicating that about half of the disordered chemical shift for residue A16 is arising from a partially folded trimer.
Effects of a GPO-rich renucleation sequence on a Gly-to-Ser substitution
A peptide with a central Gly replaced by Ser (T1-898(G16S)) models a mild case of OI resulting from a Gly-to-Ser mutation at position 901 in the α1 chain of type I collagen (3). The HSQC spectrum (Fig. 1 C) of T1-898(G16S) at 0°C shows distinct triple-helical resonances for all four labeled residues, G7, V15, A18, and G28, indicating that when a Ser replaces the Gly, the Ser can be incorporated into a triple-helical conformation. The residue-specific diffusion coefficients of the resonances corresponding to the disordered state (Fig. 1 G) indicate that DM0°C for G7, V15, and A18 are similar and <1, whereas DM0°C for G28 is ∼1. The structural implications are that T1-898(G16S) is a partially folded species whose conformation contains a folded C-terminus, and an unfolded N-terminus and mutation site. Peptide T1-898(G16S) contains some PFT, as well as M and FT, and the M/PFT ratio is calculated as 1:1.8. One explanation for the difference in the conformations of the two terminal ends is that the C-terminus has the highly stable and stronger nucleation sequence (GPO)4, whereas the N-terminus has the less stable GPO(GAO)3 sequence (33).
Previous studies have suggested that triple-helix folding starts from the C-terminal (GPO)4 sequence, whereas the N-terminal sequence may constitute a renucleation domain, which could be a crucial factor in the completion of folding of triple helix N-terminal to a mutation (33,51). Studies have also suggested that (GPO)4 is a more efficient nucleation domain than GPO(GAO)3 (33). To create a peptide with a Gly-to-Ser substitution that would be less prone to fraying at the N-terminus, a new peptide was designed with a more stabilizing (GPO)4 sequence to replace the GPO(GAO)3. As expected, the thermal stability of peptide GPOT1-898(G16S) increased significantly to 28°C compared to the Tm of 14.5°C for T1-898(G16S). The HSQC spectrum of GPOT1-898(G16S) shows triple-helical resonances for the four labeled residues G7, V15, A18, and G22, indicating the presence of FT (Fig. 1 D). The diffusion experiments were carried out at 5°C and 40°C to study the disordered resonances (Fig. 1 H), and they show that the values of DM5°C for G7, V15, A18, and G22 are all close to 1, suggesting that the peptide has no partially folded species, only M in equilibrium with FT.
Effects of renucleation sequence and nearest neighboring sequence on a Gly-to-Arg substitution
Peptide T1-898(G16R), modeling a Gly-to-Arg mutation, shows a low thermal stability (Tm ∼ 7°C) compared to the control T1-898 (Tm ∼ 35°C) and other mutant peptides, and it was suggested that this peptide did not fold completely even at low temperature (35). In the HSQC spectrum at 0°C, G13 shows no triple-helical resonance, indicating that peptide T1-898(G16R) does not adopt a triple-helical conformation in the middle, near the substitution site (Fig. 2 A). Both triple-helical and disordered resonances are present for the residues at both ends of this peptide, G7 and G28, and these resonances have the same chemical shifts as G7 and G28 in other T1-898 peptide variants. Even though triple-helical resonances are observed for both end residues, it is not clear whether some molecules are folded at only one end (C- or N-terminus) or whether there are molecular species with both folded ends but a disordered or distorted middle region. Peptide T1-898(G16D) shows a similar low Tm value, and an HSQC spectrum similar to that of T1-898(G16R), with the absence of a trimer resonance for G13 (Fig. S1 A). Reduced diffusion coefficients of peptides T1-898(G16R) and T1-898(G16D) at 0°C also indicated the presence of PFTs, although the measurements may be less accurate due to overlapping of the resonances corresponding to the disordered state (Fig. 3 A and Fig. S1 B).
Figure 2.
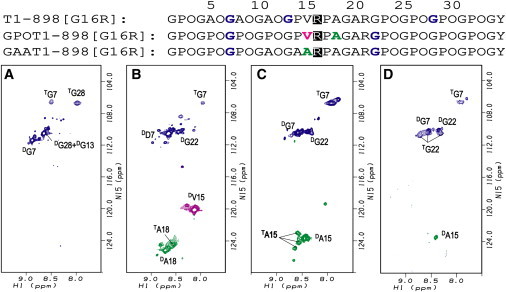
Effect of Gly-to-Arg substitutions in different neighboring-sequence environments. Shown are 1H-15N HSQC spectra of T1-898(G16R) (A), GPOT1-898(G16R) (B), GAAT1-898(G16R) (C), and GAAT1-898(G16R) after ∼20 h in 100% D2O (D) at 0°C. The peaks corresponding to the disordered and triple helical states are denoted by superscripts D and T, respectively. Minor disordered resonances arise due to cis-trans isomerization in the unfolded state of the Pro/Hyp-rich chains (49). After ∼20 h in D2O, the disordered peaks of Gly in GAAT1-898(G16R) have decreased intensities compared with the triple-helical peaks of Gly, as the NH of Gly in the disordered state exchanges much faster than those in the triple helix conformation.
Figure 3.
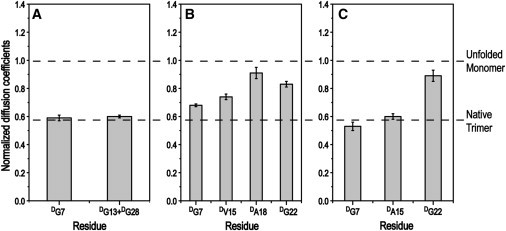
NMR diffusion measurements of T1-898(G16R), GPOT1-898(G16R), and GAAT1-898(G16R). Histograms of the residue-specific translational diffusion coefficients of 15N-labeled residues in disordered states for peptide T1-898(G16R) (A), GPOT1-898(G16R) (B), and GAAT1-898(G16R) (C). Diffusion coefficients at 0°C are normalized to those at 40°C (20°C for GPOT1-898(G16R)). The black dashed lines represent the normalized diffusion coefficients for M (theoretically with a value of 1) and native trimer (with a value of 0.58 for G28 in peptide T1-898(G16A)).
To promote folding through the Gly-to-Arg mutation site, the design that was used to successfully complete triple-helix formation in the presence of a Gly-to-Ser substitution was applied to the T1-898(G16R) peptide. We were surprised to find that replacing GPO(GAO)3 with (GPO)4 at the N-terminus reduced the stability of the new peptide, GPOT1-898(G16R), to 5°C (Table S1). This lowering of stability is consistent with the HSQC spectrum at 0°C, which does not show clear triple-helical peaks for V15 and G22 (Fig. 2 B). It suggests that the peptide could not fold through the mutation site and that only partially folded species could exist, which is also indicated by the smaller diffusion coefficients of disordered resonances of G7 and V15 (Fig. 3 B). In contrast to the Gly-to-Ser mutation, the rigid GPO triplets destabilize rather than stabilize the triple-helix formation for this Gly-to-Arg mutation, suggesting that more flexibility may be needed to incorporate the large Arg residues within a triple helix.
Since the introduction of rigid sequences was unfavorable, it is possible that flexible local sequence environments are needed to incorporate an Arg into the triple helix (19). Thus, a peptide was designed with more flexible residues introduced N-terminal to the Gly-to-Arg mutation. Peptide GAAT1-898(G16R) was designed by modifying the triplet next to the mutation site from GPV to a more flexible GAA, and the N-terminal renucleation region was modified from (GPO)4 to (GPO)3GAO. The thermal stability of the new peptide (Tm ∼ 13°C) is increased significantly compared to that of the other two Gly-to-Arg peptides (Table S1). The HSQC spectrum of the peptide at 0°C reveals a single strong triple-helical resonance for G7 and three well dispersed triple-helical resonances for A15 (Fig. 2 C). Compared with GPOT1-898(G16R), the triple-helical resonance for G7 in GAAT1-898(G16R) becomes much stronger. In GPOT1-898(G16R), the A18 near the mutation site shows only a poorly defined triple-helical resonance, whereas the A15 in GAAT1-898(G16R), just beside the mutation site, shows three triple-helical resonances. Similar to the case for GPOT1-898(G16R), the triple-helical resonances for G22 in GAAT1-898(G16R) are not observed, probably due to overlapping with disordered resonances. To solve the overlap problem, the peptide is lyophilized and redissolved in 100% D2O to exchange out the protons of the disordered state, allowing the triple-helical resonances to be observed without overlap. The HSQC spectrum of the peptide at 0°C after ∼20 h in D2O shows three triple-helical resonances of G22 (Fig. 2 D), which, together with triple-helical resonances for G7 and A15, suggests that the peptide GAAT1-898(G16R) forms a fully folded triple-helical conformation at 0°C. However, PFT may still exist for the peptide, as indicated by the small diffusion coefficients of G7 and V15, where measurements may be hindered by overlapped resonances (Fig. 3).
Role of charge in folding for substitution of Gly by charged residues
The effects of electrostatic interactions when a Gly is replaced with a charged residue are studied on T1-898(G16R) (Table S1). Experiments were performed in 2 M NaCl to determine whether the charge repulsion of the side chain of the Arg substitution destabilizes the peptides. The pKa of the side-chain amino group of Arg is 12.48, and studies at pH >12.5 are not desirable, as alkaline treatment of collagen has been shown to result in the loss of inter- and intramolecular bonds (52,53). The ellipticity value of NaCl-treated T1-898(G16R) decreased compared to the peptide in PBS, but there is no change in Tm (Fig. 4). It is clear that masking the charge did not help the folding of T1-898(G16R). CD experiments of T1-898(G16D) at pH 3.5 and pH 7 also indicated that charges do not affect the molecular features of the peptide significantly (Fig. S1 C).
Figure 4.
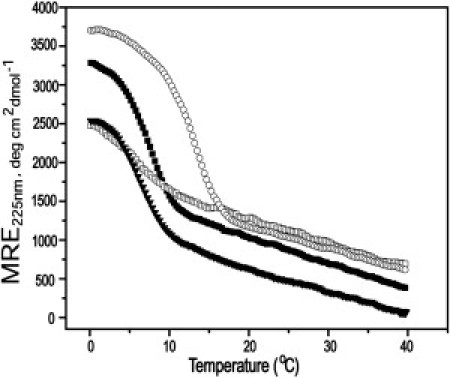
Effect of charge on peptide T1-898(G16R). CD melting transition of peptides GAAT1-898(G16R) (open circles), T1-898(G16R) (solid squares), and GPOT1-898(G16R) (open squares) at 1 mg/mL in 20 mM PBS buffer, pH 7, and T1-898(G16R) (inverted triangles) at 2 M NaCl.
Discussion
Missense mutations leading to substitution of a Gly in the triple helix with a bulkier residue (Ala, Ser, Cys, Val, Arg, Asp, or Glu) result in various OI phenotypes (1–3). Studies have shown that the identity of the amino acid replacing Gly is correlated with the lethality of OI (3,18,21). Gly→Ala replacements are very underrepresented and those found are largely nonlethal; Gly→Ser replacements are mixtures of lethal and nonlethal; and Gly→Arg replacements at positions >178 are predominantly lethal in the α1(I) chain of type I collagen (3,9). Computational analysis also indicates a relationship between the degree of conformational perturbation in terms of hydrogen bonding and the identity of the residues near the mutation site (18,19,54). However, the molecular mechanism for this correlation is still poorly understood. It is known that OI mutations in collagen delay triple-helix folding (55,56). X-ray and NMR studies have shown that Gly-to-Ala and Gly-to-Ser mutations locally perturb the triple-helix structure and register in collagen model peptides (17,24,32).
Here, NMR diffusion techniques were utilized to investigate low-level populations of partially folded species in a set of triple-helical peptides with different Gly substitutions. Interpretation of the diffusion data is based on the model assumption that partially folded intermediate may exist in equilibrium with M and FT, and the chemical shifts of residues in the unfolded region of the PFT intermediate are totally overlapped with those in the M state. This model assumes slow exchange rates between PFT, M, and FT. Although this is largely a qualitative model, it allows us to investigate how the sequence environment affects incorporation of the residue replacing Gly within the triple helix.
The peptide set based on T1-898 models a Gly substituted by Ala, Ser, Asp, and Arg in a defined sequence environment (Table S1). NMR HSQC and diffusion studies on this set of peptides show that an Ala can be incorporated into the triple helix at 0°C but that, at least in some molecular species, the triple helix is partially unfolded in the middle at the mutation site (Fig. 5). The Ser could also be incorporated into the T1-898 triple helix but a relatively larger proportion of species with a folded C-terminus but disordered N-terminus and central region was found at 0°C (Fig. 5). It seems that the small residues Ala and Ser can be tolerated within a triple-helix conformation only at the expense of generating some species with central or terminal disorder in equilibrium with the fully folded species. However, introduction of Arg or Asp as a Gly replacement does not allow complete triple-helix formation and results in partially folded molecules (Fig. 5). The more severe OI phenotype caused by Arg/Asp replacements compared with Ala/Ser replacements of Gly in the Gly-Xaa-Yaa repeating sequence could relate to the folding difficulty seen in these peptide models, where the short length leads to exaggerated structural consequences.
Figure 5.
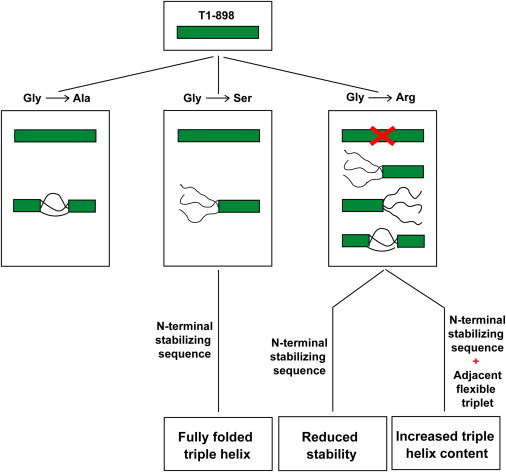
Effect of different Gly substitutions on the equilibrium conformations. Peptide T1-898 forms a fully folded triple-helix conformation. When Gly is replaced by Ala, the Ala residue is incorporated into a fully folded triple helix; but in some molecular species, the trimer is partially unfolded in the middle at the mutation site. A Gly-to-Ser substitution results in the presence of some partially folded species with unfolding at the N-terminus, but replacing the N-terminal region by a stabilizing/stronger renucleation sequence, (GPO)4, leads to fully folded triple helix. Introduction of Arg prevents complete triple-helix formation, leaving only partially folded species. A stabilizing/stronger renucleating sequence, (GPO)4, at the N-terminal alone does not help the peptide to fold through the mutation site, whereas addition of flexible residues near the mutation site in addition to a stabilizing N-terminal sequence can incorporate the Arg residue into a fully folded triple helix.
A relationship is seen between the requirement for a good nucleation sequence at both ends of the peptide and successful triple-helix folding when a Gly is replaced by another residue near the center. With a C-terminal (GPO)4 end, the presence of an N-terminal GPO(GAO)3 sequence allowed successful incorporation of Ala, whereas a Ser could only be incorporated with the more stabilizing (GPO)4 N-terminus. However, we were surprised to find that addition of the N-terminal (GPO)4 does not promote complete triple-helix formation when an Arg is present, suggesting the need for strong renucleation and, in the case of large residues, an additional requirement of local flexibility (Fig. 5). Modification of the triplet next to the mutation site from GPV to GAA and of the N-terminal renucleation sequence from (GPO)4 to (GPO)3GAO to make a more flexible local environment, led to some fully folded triple helix in the presence of a Gly→Arg mutation (Fig. 5). The presence of the bulky V15 residue next to Arg may hinder the modifications needed to incorporate the Arg, which may be aided when a more flexible environment is introduced. Thus, it appears that a balance between stabilizing sequences, strong renucleation, and local flexibility needs to be achieved for accommodation of bulky residues within triple-helical peptides. Our data suggest that how far the structural perturbation extends along the chain in the mutant collagen depends on the residue replacing Gly, and that this could affect the interactions of mutant molecules within fibrils with proteoglycans. The larger substituting residues lead to an increased population of PFTs, with larger unfolded regions for Arg > Ser > Ala. The presence of partially folded species in model peptides is likely to reflect a difficulty in triple-helix folding around a mutation, and this could impact recognition and degradation, triggering the unfolded protein response in the endoplasmic reticulum.
The destabilizing effects of Gly mutations are magnified in peptide models compared to the consequences of these mutations in OI collagens. The shorter length of the peptides (∼10 triplets vs. 338 triplets in type I collagen) and their imino-acid-rich nature (∼40–66% imino acid vs. ∼20% imino acids in type I collagen) are both likely to be factors in the sensitivity of peptides to Gly substitutions. This is consistent with results showing that the degree of destabilization of (Gly-Pro-Hyp)8 peptides strongly depends on the identity of the residue replacing Gly, whereas Makareeva et al. found no relation between the decrease in thermal stability of different OI collagens and the identity of the residue replacing Gly (15,21). The ability to characterize the molecular consequences of a Gly substitution in peptides, together with the amplification of structural effects, can contribute insights about OI mutations. In collagen, the long stretches of Gly-Xaa-Yaa sequences on both sides of a mutation must facilitate complete triple-helix formation, although difficulties may arise when the mutation near the C-terminus or the N-terminus is affected by factors seen in these studies. In addition, all peptides studied here are homotrimers, with Gly substitutions in all three chains, whereas type I collagen from OI patients represents a mixture of molecular species containing mutations in no chains, one chain, or two chains. However, studies of heterotrimer peptides containing different numbers of mutant chains (57), and recent studies on bacterial collagen models with mutations in all three chains (58), suggest that the consequences of having collagen molecules with three mutant chains is not dramatically different from the case with just one or two mutant chains. The advantage of characterizing the well-defined, homogeneous peptide molecules containing mutations in all three chains is important and likely to be relevant to collagen diseases.
Acknowledgments
This work was supported by grants from the National Institutes of Health (GM45302 to J.B. and GM60048 to B.B.), the National Science Foundation (DBI-0403062 and DBI-0320746 to J.B.), and a BOYSCAST fellowship (Department of Science and Technology, Government of India) to B.M.
Footnotes
Balaraman Madhan's present address is Central Leather Research Institute, Chennai, India.
Yingjie Li's present address is OSI Pharmaceuticals, Farmingdale, NY.
Barbara Brodsky's present address is Department of Biomedical Engineering, Tufts University, Medford, MA.
Contributor Information
Barbara Brodsky, Email: brodsky@umdnj.edu.
Jean Baum, Email: jean.baum@rutgers.edu.
Supporting Material
References
- 1.Myllyharju J., Kivirikko K.I. Collagens and collagen-related diseases. Ann. Med. 2001;33:7–21. doi: 10.3109/07853890109002055. [DOI] [PubMed] [Google Scholar]
- 2.Byers P.H., Cole W.G. Osteogenesis imperfecta. In: Royce P.M., Steinmann B., editors. Connective Tissue and Its Hereditable Disorders: Molecular, Genetic, and Medical Aspects. Wiley-Liss; New York: 2002. pp. 385–430. [Google Scholar]
- 3.Marini J.C., Forlino A., Byers P.H. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum. Mutat. 2007;28:209–221. doi: 10.1002/humu.20429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shoulders M.D., Raines R.T. Collagen structure and stability. Annu. Rev. Biochem. 2009;78:929–958. doi: 10.1146/annurev.biochem.77.032207.120833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramachandran G.N., Kartha G. Structure of collagen. Nature. 1955;176:593–595. doi: 10.1038/176593a0. [DOI] [PubMed] [Google Scholar]
- 6.Rich A., Crick F.H. The structure of collagen. Nature. 1955;176:915–916. doi: 10.1038/176915a0. [DOI] [PubMed] [Google Scholar]
- 7.Kielty C.M., Grant M.E. The collagen family: strucutre, assembly, and organization in the extracellular matrix. In: Royes P.M., Steinmann B., editors. Connective Tissue and Its Heritable Disorders: Molecular, Genetic, and Medical Aspects. Wiley-Liss; New York: 2002. pp. 159–222. [Google Scholar]
- 8.van der Rest M., Aubert-Foucher E., Goldschmidt D. Structure and function of the fibril-associated collagens. Biochem. Soc. Trans. 1991;19:820–824. doi: 10.1042/bst0190820. [DOI] [PubMed] [Google Scholar]
- 9.Dalgleish, R. Osteogenesis imperfecta and Ehlers-Danlos syndrome variant databases. Leiden University Medical Center, Leiden, The Netherlands. http://www.le.ac.uk/genetics/collagen/.
- 10.Bonadio J., Byers P.H. Subtle structural alterations in the chains of type I procollagen produce osteogenesis imperfecta type II. Nature. 1985;316:363–366. doi: 10.1038/316363a0. [DOI] [PubMed] [Google Scholar]
- 11.Forlino A., Kuznetsova N.V., Leikin S. Selective retention and degradation of molecules with a single mutant α1(I) chain in the Brtl IV mouse model of OI. Matrix Biol. 2007;26:604–614. doi: 10.1016/j.matbio.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 12.Makareeva E., Aviles N.A., Leikin S. Chaperoning osteogenesis: new protein-folding disease paradigms. Trends Cell Biol. 2011;21:168–176. doi: 10.1016/j.tcb.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Di Lullo G.A., Sweeney S.M., San Antonio J.D. Mapping the ligand-binding sites and disease-associated mutations on the most abundant protein in the human, type I collagen. J. Biol. Chem. 2002;277:4223–4231. doi: 10.1074/jbc.M110709200. [DOI] [PubMed] [Google Scholar]
- 14.Baum J., Brodsky B. Folding of peptide models of collagen and misfolding in disease. Curr. Opin. Struct. Biol. 1999;9:122–128. doi: 10.1016/s0959-440x(99)80016-5. [DOI] [PubMed] [Google Scholar]
- 15.Makareeva E., Mertz E.L., Leikin S. Structural heterogeneity of type I collagen triple helix and its role in osteogenesis imperfecta. J. Biol. Chem. 2008;283:4787–4798. doi: 10.1074/jbc.M705773200. [DOI] [PubMed] [Google Scholar]
- 16.Xu K., Nowak I., Xu Y. Recombinant collagen studies link the severe conformational changes induced by osteogenesis imperfecta mutations to the disruption of a set of interchain salt bridges. J. Biol. Chem. 2008;283:34337–34344. doi: 10.1074/jbc.M805485200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y., Brodsky B., Baum J. NMR conformational and dynamic consequences of a gly to ser substitution in an osteogenesis imperfecta collagen model peptide. J. Biol. Chem. 2009;284:20660–20667. doi: 10.1074/jbc.M109.018077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bodian D.L., Madhan B., Klein T.E. Predicting the clinical lethality of osteogenesis imperfecta from collagen glycine mutations. Biochemistry. 2008;47:5424–5432. doi: 10.1021/bi800026k. [DOI] [PubMed] [Google Scholar]
- 19.Radmer R.J., Klein T.E. Severity of osteogenesis imperfecta and structure of a collagen-like peptide modeling a lethal mutation site. Biochemistry. 2004;43:5314–5323. doi: 10.1021/bi035676w. [DOI] [PubMed] [Google Scholar]
- 20.Persikov A.V., Pillitteri R.J., Brodsky B. Stability related bias in residues replacing glycines within the collagen triple helix (Gly-Xaa-Yaa) in inherited connective tissue disorders. Hum. Mutat. 2004;24:330–337. doi: 10.1002/humu.20091. [DOI] [PubMed] [Google Scholar]
- 21.Beck K., Chan V.C., Brodsky B. Destabilization of osteogenesis imperfecta collagen-like model peptides correlates with the identity of the residue replacing glycine. Proc. Natl. Acad. Sci. USA. 2000;97:4273–4278. doi: 10.1073/pnas.070050097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xiao J., Addabbo R.M., Baum J. Local conformation and dynamics of isoleucine in the collagenase cleavage site provide a recognition signal for matrix metalloproteinases. J. Biol. Chem. 2010;285:34181–34190. doi: 10.1074/jbc.M110.128355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brodsky B., Thiagarajan G., Kar K. Triple-helical peptides: an approach to collagen conformation, stability, and self-association. Biopolymers. 2008;89:345–353. doi: 10.1002/bip.20958. [DOI] [PubMed] [Google Scholar]
- 24.Bella J., Eaton M., Berman H.M. Crystal and molecular structure of a collagen-like peptide at 1.9 Å resolution. Science. 1994;266:75–81. doi: 10.1126/science.7695699. [DOI] [PubMed] [Google Scholar]
- 25.Li Y., Kim S., Baum J. Identification of partially disordered peptide intermediates through residue-specific NMR diffusion measurements. J. Am. Chem. Soc. 2005;127:10490–10491. doi: 10.1021/ja052801d. [DOI] [PubMed] [Google Scholar]
- 26.Madhan B., Xiao J., Brodsky B. NMR monitoring of chain-specific stability in heterotrimeric collagen peptides. J. Am. Chem. Soc. 2008;130:13520–13521. doi: 10.1021/ja805496v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li M.H., Fan P., Baum J. Two-dimensional NMR assignments and conformation of (Pro-Hyp-Gly)10 and a designed collagen triple-helical peptide. Biochemistry. 1993;32:7377–7387. doi: 10.1021/bi00080a007. [DOI] [PubMed] [Google Scholar]
- 28.Xiao J., Baum J. Structural insights from 15N relaxation data for an anisotropic collagen peptide. J. Am. Chem. Soc. 2009;131:18194–18195. doi: 10.1021/ja9056823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Y., Brodsky B., Baum J. NMR shows hydrophobic interactions replace glycine packing in the triple helix at a natural break in the (Gly-X-Y)n repeat. J. Biol. Chem. 2007;282:22699–22706. doi: 10.1074/jbc.M702910200. [DOI] [PubMed] [Google Scholar]
- 30.Bryan M.A., Hyde T., Brodsky B. Collagen model peptides: support for an internal nucleation site in type I collagen. Biophys. J. 2003;84:337a. (Abstr.) [Google Scholar]
- 31.Fan P., Li M.H., Baum J. Backbone dynamics of (Pro-Hyp-Gly)10 and a designed collagen-like triple-helical peptide by 15N NMR relaxation and hydrogen-exchange measurements. Biochemistry. 1993;32:13299–13309. doi: 10.1021/bi00211a043. [DOI] [PubMed] [Google Scholar]
- 32.Liu X., Kim S., Baum J. Nuclear magnetic resonance shows asymmetric loss of triple helix in peptides modeling a collagen mutation in brittle bone disease. Biochemistry. 1998;37:15528–15533. doi: 10.1021/bi981147u. [DOI] [PubMed] [Google Scholar]
- 33.Hyde T.J., Bryan M.A., Baum J. Sequence dependence of renucleation after a Gly mutation in model collagen peptides. J. Biol. Chem. 2006;281:36937–36943. doi: 10.1074/jbc.M605135200. [DOI] [PubMed] [Google Scholar]
- 34.Bhate M., Wang X., Brodsky B. Folding and conformational consequences of glycine to alanine replacements at different positions in a collagen model peptide. Biochemistry. 2002;41:6539–6547. doi: 10.1021/bi020070d. [DOI] [PubMed] [Google Scholar]
- 35.Bryan M.A., Cheng H., Brodsky B. Sequence environment of mutation affects stability and folding in collagen model peptides of osteogenesis imperfecta. Biopolymers. 2011;96:4–13. doi: 10.1002/bip.21432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Persikov A.V., Xu Y., Brodsky B. Equilibrium thermal transitions of collagen model peptides. Protein Sci. 2004;13:893–902. doi: 10.1110/ps.03501704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mills R. Self-diffusion in normal and heavy water in the range 1–45°. J. Phys. Chem. 1973;77:685–688. [Google Scholar]
- 38.Holz M., Weingartner H. Calibration in accurate spin-echo self-diffusion measurements using 1H and less-common nuclei. J. Magn. Reson. 1991;92:115–125. [Google Scholar]
- 39.Cho C.H., Urquidi J., Robinson G.W. Thermal offset viscosities of liquid H2O, D2O, and T2O. J. Phys. Chem. B. 1999;103:1991–1994. [Google Scholar]
- 40.Choy W.Y., Mulder F.A., Kay L.E. Distribution of molecular size within an unfolded state ensemble using small-angle x-ray scattering and pulse field gradient NMR techniques. J. Mol. Biol. 2002;316:101–112. doi: 10.1006/jmbi.2001.5328. [DOI] [PubMed] [Google Scholar]
- 41.Dvinskikh S.V., Furo I.I., I Cross-relaxation effects in stimulated-echo-type PGSE NMR experiments by bipolar and monopolar gradient pulses. J. Magn. Reson. 2000;146:283–289. doi: 10.1006/jmre.2000.2158. [DOI] [PubMed] [Google Scholar]
- 42.Orekhov V.Y., Korzhnev D.M., Arseniev A.S. Sampling of protein dynamics in nanosecond time scale by 15N NMR relaxation and self-diffusion measurements. J. Biomol. Struct. Dyn. 1999;17:157–174. doi: 10.1080/07391102.1999.10508348. [DOI] [PubMed] [Google Scholar]
- 43.Chen A.D., Johnson C.S., Shapiro M.J. Chemical exchange in diffusion NMR experiments. J. Am. Chem. Soc. 1998;120:9094–9095. [Google Scholar]
- 44.Derrick T.S., Lucas L.H., Larive C.K. F-19 diffusion NMR analysis of enzyme-inhibitor binding. Magn. Reson. Chem. 2002;40:S98–S105. [Google Scholar]
- 45.Tillett M.L., Lian L.Y., Norwood T.J. Practical aspects of the measurement of the diffusion of proteins in aqueous solution. J. Magn. Reson. 1998;133:379–384. doi: 10.1006/jmre.1998.1472. [DOI] [PubMed] [Google Scholar]
- 46.Chou J.J., Baber J.L., Bax A. Characterization of phospholipid mixed micelles by translational diffusion. J. Biomol. NMR. 2004;29:299–308. doi: 10.1023/B:JNMR.0000032560.43738.6a. [DOI] [PubMed] [Google Scholar]
- 47.Bai Y., Milne J.S., Englander S.W. Primary structure effects on peptide group hydrogen exchange. Proteins. 1993;17:75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hwang T.L., van Zijl P.C., Mori S. Accurate quantitation of water-amide proton exchange rates using the phase-modulated CLEAN chemical EXchange (CLEANEX-PM) approach with a Fast-HSQC (FHSQC) detection scheme. J. Biomol. NMR. 1998;11:221–226. doi: 10.1023/a:1008276004875. [DOI] [PubMed] [Google Scholar]
- 49.Buevich A.V., Dai Q.H., Baum J. Site-specific NMR monitoring of cis-trans isomerization in the folding of the proline-rich collagen triple helix. Biochemistry. 2000;39:4299–4308. doi: 10.1021/bi992584r. [DOI] [PubMed] [Google Scholar]
- 50.Buevich A.V., Baum J. Residue-specific real-time NMR diffusion experiments define the association states of proteins during folding. J. Am. Chem. Soc. 2002;124:7156–7162. doi: 10.1021/ja012699u. [DOI] [PubMed] [Google Scholar]
- 51.Buevich A.V., Silva T., Baum J. Transformation of the mechanism of triple-helix peptide folding in the absence of a C-terminal nucleation domain and its implications for mutations in collagen disorders. J. Biol. Chem. 2004;279:46890–46895. doi: 10.1074/jbc.M407061200. [DOI] [PubMed] [Google Scholar]
- 52.Courts A. Structural changes in collagen. The action of alkalis and acids in the conversion of collagen into eucollagen. Biochem. J. 1960;74:238–247. doi: 10.1042/bj0740238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bowes J.H., Kenten R.H. The effect of alkalis on collagen. Biochem. J. 1948;43:365–372. doi: 10.1042/bj0430365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mooney S.D., Huang C.C., Klein T.E. Computed free energy differences between point mutations in a collagen-like peptide. Biopolymers. 2001;58:347–353. doi: 10.1002/1097-0282(200103)58:3<347::AID-BIP1011>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 55.Engel J., Prockop D.J. The zipper-like folding of collagen triple helices and the effects of mutations that disrupt the zipper. Annu. Rev. Biophys. Biophys. Chem. 1991;20:137–152. doi: 10.1146/annurev.bb.20.060191.001033. [DOI] [PubMed] [Google Scholar]
- 56.Raghunath M., Bruckner P., Steinmann B. Delayed triple helix formation of mutant collagen from patients with osteogenesis imperfecta. J. Mol. Biol. 1994;236:940–949. doi: 10.1006/jmbi.1994.1199. [DOI] [PubMed] [Google Scholar]
- 57.Gauba V., Hartgerink J.D. Synthetic collagen heterotrimers: structural mimics of wild-type and mutant collagen type I. J. Am. Chem. Soc. 2008;130:7509–7515. doi: 10.1021/ja801670v. [DOI] [PubMed] [Google Scholar]
- 58.Cheng H., Rashid S., Brodsky B. Location of glycine mutations within a bacterial collagen protein affects degree of disruption of triple-helix folding and conformation. J. Biol. Chem. 2011;286:2041–2046. doi: 10.1074/jbc.M110.153965. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.