

Abstract
Rate-limiting millisecond motions in wild-type (WT) Ribonuclease A (RNase A) are modulated by histidine 48. Here, we incorporate an unnatural amino acid, thia-methylimidazole, at this site (H48C-4MI) to investigate the effects of a single residue on protein motions over multiple timescales and on enzyme catalytic turnover. Molecular dynamics simulations reveal that H48C-4MI retains some crucial WT-like hydrogen bonding interactions but the extent of protein-wide correlated motions in the nanosecond regime is decreased relative to WT. NMR Carr-Purcell-Meiboom-Gill relaxation dispersion experiments demonstrate that millisecond conformational motions in H48C-4MI are present over a similar pH range compared to WT. Furthermore, incorporation of this nonnatural amino acid allows retention of WT-like catalytic activity over the full pH range. These studies demonstrate that the complexity of the protein energy landscape during the catalytic cycle can be maintained using unnatural amino acids, which may prove useful in enzyme design efforts.
Introduction
Optimal enzyme activity requires that the protein possess the ability to undergo fluctuations between conformations. The important role of conformational flexibility in protein function (1) is underscored by efforts to incorporate elements of flexibility into the protein design process (2–5) to enhance or modulate the desired function. The amplitudes and mechanisms of these motions are often complex and occur over an enormous timescale range (6). The bond making/breaking steps in an enzyme-catalyzed reaction occur on the timescale of tens of femtoseconds (fs). Yet, the overall rates of catalytic reactions occur in tens to hundreds of milliseconds (ms), a range of 12 orders of magnitude. Thus, the overwhelming majority of events that occur during the course of enzyme-catalyzed reactions occur before and after the reaction transition state (7–13). The energy landscape on which enzyme reactions take place is extremely complex in which slower, barrier-crossing millisecond motions are often responsible for preparing the substrate and the enzyme active site for reaction and for facilitating the release of products once the reaction is complete, whereas faster thermal motions can contribute to the thermodynamics of ligand binding (14). The enormity of the timescales involved makes investigation of enzyme function a daunting task. Nonetheless, obtaining deeper insight into enzyme function and the role these motions play, requires that this issue be addressed. Motional events that occur with such a large diversity in timescale can be investigated, in part, through a combination of molecular dynamics (MD) simulations to obtain atomistic insight into thermal motions that are faster than a few nanoseconds, and NMR relaxation dispersion experiments that probe μs-ms motions. Here, we combine these two methods along with the introduction of an unnatural amino acid at a critical enzyme residue to address the robustness of enzyme function and motions to perturbations, which are not accessible by standard site-directed mutagenesis.
These issues are investigated in the enzyme ribonuclease A (RNase A) in which millisecond motions have been demonstrated to be of functional importance and where motions optimize the enzyme conformation at distinct stages along the enzymatic reaction coordinate (15,16). RNase A possesses the innate ability to presample the relevant ligand-bound conformations even in the absence of ligand (15). Studies on RNase A since the mid-1960s have implicated a histidine residue in the functional motion in this enzyme on the basis of temperature jump and NMR pH titration experiments (17,18). More recent NMR relaxation dispersion experiments have identified and characterized the role in which hydrogen bonding associated with histidine 48 (H48) plays in these important protein motions (19,20). In RNase A motions that are distant (20 Å) from the enzyme's active site are propagated, via H48, between loop 1 (residues 14–24) to β-strands 1 and 4, which contain active-site residues T45 and D83, respectively. These residues provide important pyrimidine-specificity interactions at the active site and ultimately modulate the enzyme's rate-limiting step, which is a conformational change associated with product release. In contrast, the chemistry step in RNase A is rapid and kcat/KM is limited by substrate association with the enzyme and approaches the diffusion limit (21). Replacement of hydrogen bonding interactions around H48 with deuterium resulted in a normal kinetic solvent isotope effect on enzyme motions from 1800 s–1 to 800 s–1 and similar reduction in the measured kcat value (20). Furthermore, studies have demonstrated the sensitivity of the region surrounding H48 to mutagenesis, which in all cases examined thus far have resulted in complete or severe disruption of these important ms motions (20,22). This observation is consistent with work on this and other enzymes that showed that mutation of single amino acids, even those far from the active site; detrimentally disrupt important protein motions (23–25). Nonetheless, in this previous work the limitations of site-directed mutagenesis to provide suitable amino acid substitutions that would allow subtle alterations and probing of the chemical and physical properties of important residues is a limitation to more detailed investigation of the mechanism of these ms motions.
Here, we incorporate 4-methylimidazole (a histidine analog, (Scheme 1)) into a fully 15N-labeled enzyme and demonstrate that this method can provide an additional means to further investigate the role of motions in enzyme function and can also provide a potential avenue toward the rational modulation of motions in proteins. The unmodified H48C mutant exhibits no ms motions in the essential regions in RNase A but the resulting thia-methylimidazole enzyme (H48C-4MI) restores ms motions in an otherwise motionally dead region of the H48C enzyme while retaining wild-type (WT) functional properties.
Scheme 1.
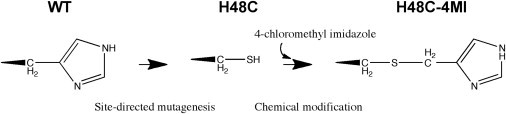
Procedure for constructing RNase A H48C-4MI.
Methods
Protein expression and purification
Plasmids for mutants H48G and H48C were produced using QuikChange (Stratagene) site-directed mutagenesis. All mutants were confirmed by DNA sequencing. Expression and purification was as described previously (22) with few modifications. Escherichia coli harboring RNase A expression plasmid were induced for 4–5 h. Refolding of the RNase A inclusion bodies was then performed at pH 5. H48C required additional modifications from previous protocol. All steps from cell lysis to column purification were strictly performed on ice or at 4°C. The urea solubilization step was shortened from overnight at room temperature to 10 min on ice. Precipitation during refolding necessitated a second round of solubilization and refolding. Soluble fractions from refolding were combined and concentrated before column purification.
Incorporation of 4-methylimidazole
Modification of cysteine residues with 4-chloromethyl-imidazole (4-CMI) was as described previously (26). Following purification, H48C was dialyzed into 400 mM borate pH 8.75. 4-CMI (Synthonix, Wake Forest, NC) was solubilized in the same buffer and added dropwise to H48C while stirring at room temperature. The reaction was performed at room temperature for two days with a 4-CMI concentration of 60 mM and protein concentration of 20 μM. 5,5′-dithiobis-(2-nitrobenzoic acid) analysis (27) confirmed the loss of 1 equivalent of protein thiol after reaction. In addition, the complete disappearance of amide resonances corresponding to H48C were observed by 1H-15N NMR HSQC spectra after reaction with 4-CMI. The resulting H48C-4MI were further purified and concentrated to 300 μM for NMR experiments.
Enzyme kinetics
The kcat/Km values for RNase A were determined as previously described using a fluorescent substrate (28). Experiments were performed as a function of pH using a combination of MES and HEPES buffers (100 mM MES, 100 mM NaCl; 100 mM HEPES, 100 mM NaCl). Assay volumes were 2.0 ml and contained 100 nM substrate (5,6-carboxyfluorescein-dArUdAdA-3′6-carboxytetramethylrhodamine) (Integrated DNA Technologies, Coralville, IA), and either 129 pM H48C-4MI or 163 pM WT RNase A. Reactions were initiated by addition of 2 μl of enzyme and fluorescence emission was monitored at 550 nm with excitation at 490 nm. Experiments were performed on a Shimadzu RF5301 fluorometer located in the Yale Richards Center for Biophysics. All experiments were performed at room temperature (295 ± 1 K). kcat/Km was calculated using the initial velocity portion of the reaction progress curve as described previously (28,29). Duplicates were performed at pH = 6.5 with both buffers to check for buffer artifacts. pH rate data were fit with Eq. 1:
| (1) |
NMR dynamics studies
Relaxation-compensated Carr-Purcell-Meiboom-Gill (rcCPMG) experiments (30) were performed on WT, mutant, and chemically modified RNase A to measure millisecond motions. All sample buffers were 5 mM MES, 7 mM NaCl, 0.01% NaN3, and 5% D2O unless otherwise stated. All spectra were collected with the proton carrier centered on the water resonance and the nitrogen carrier set to 120 ppm. WT relaxation dispersion data were collected on a 2H,15N-labeled RNase A sample for pH = 6.4 with all other experiments performed on protonated enzymes. NMR experiments with H48G (350 μM) with 53.3 mM imidazole were collected at 500 MHz with a spectral width in the direct (indirect) dimension of 9000 (2200) Hz with 1978 (128) points. H48C (100 μM) at pH 7.0 spectra were collected at 600 MHz with a spectral width in the direct (indirect) dimension of 9000 (2200) Hz with 1152 (128) points. H48C-4MI (300 μM) experiments at pH 7.0 spectra were collected at 600 MHz with a spectral width in the direct (indirect) dimension of 9056 (2200) Hz with 1160 (128) points. Fitting of fast and general equations for CPMG dispersion data were as described previously (15,31). For C84 and V43, dispersion curves gave similar fit results and therefore the data were fitted in a global manner, which is presented in the Results.
NMR lineshape analysis
Dissociation rate constants for 3′-CMP with 1H-15N-H48C-4MI and 1H-15N-H48C were determined by analysis of NMR lineshapes over the course of 3′-CMP titrations. Analyzed peaks were sliced in both the 1H and 15N dimensions using the program Sparky. One-dimensional slices were then individually and globally fit to the McConnell equations using the program LineShapeKin (http://lineshapekin.net/). Comparison of fits using Akaike's information criterion indicated that global fitting was preferred in each case by a factor of at least 700.
Modeling and MD simulations
The computational structural models for WT, H48C, and H48C-4MI enzymes are based on the crystal structure of the phosphate-free RNase A refined at 1.26 Å (Protein Data Bank code 7RSA) (32). All water molecules in the crystal structure were kept in the computational studies. All MD simulations were based on the AMBER-ff99SB (33) force field, as implemented in the NAMD2 software package (34). The generalized Amber force field (35) has been used for the nonstandard residue present in the thia-methylimidazole enzyme (H48C-4MI). The partial charges for this nonstandard residue were derived by using the RESP method (36) based on Hartree-Fock (HF)/6-31G∗ calculations performed with the GAUSSIAN 09 package (37).
Production MD runs have been performed after the following preequilibration procedure: hydrogen atoms and explicit TIP3P water (38) solvent molecules (∼8890) were added using AmberTools (version 1.4) and subsequently optimized, constraining the rest of the atoms at the crystal structure positions. A short optimization of the side chain of residue 48 has been performed only in the case of H48C and H48C-4MI mutants. The optimized solvated structures were then slowly heated to 298 K by performing MD simulations (100 ps) in the canonical NVT ensemble using Langevin dynamics. Harmonic constrains have been applied to the protein heavy atoms, with force constants set to 1 kcal/mol/Å2. During this heating procedure different parts of the system were gradually unconstrained until all atoms were set free. Fully unconstrained MD simulations were run for 4.9 ns, for a total preequilibration simulations of 5 ns.
Upon system preequilibration, 20 ns MD simulations were performed in the NPT ensemble at 298 K and 1 atm using the Langevin piston. All simulations were performed using periodic boundary conditions. Electrostatics were treated with the particle mesh Ewald method (39) and van der Waals interactions were calculated using a switching distance of 10 Å and a cutoff of 12 Å. A multiple time stepping algorithm (40) was used, where bonded, short-range nonbonded, and long-range electrostatic interactions were evaluated at every one, two, and four time steps, respectively, using a time step of integration set to 1 fs.
The Cα-Cα covariance matrix is computed according to the Pearson correlation coefficient:
where xi is the nuclear coordinates of atom i relative to the ensemble average value, where 〈 〉 denotes the ensemble average over the entire MD simulation. This is an established method typically used for quantifying correlated motion in proteins from MD simulations (41).
Results and Discussion
Site-specific chemical modifications to introduce unnatural chemical functionalities that are not available in the standard genetic code have previously been used to study enzyme mechanisms and motions. Early applications include proteases in which S195 of chymotrypsin was converted to dehydroalanine (42) and the active-site hydroxyl from subtilisin was replaced with a thiol (43,44). This early work exploited the special reactivity of these active site groups to achieve the desired modification. Coupling of chemical modification with site-directed mutagenesis allowed modulation of the chemical and physical properties at nearly any desired position. In the late 1980's, Smith et al. (45) and Kirsch and co-workers (46,47) exploited thiol reactivity to selectively incorporate aminoethylcysteine residues into ribulose bisphosphate carboxylase/oxygenase and aspartate aminotransferase, respectively, enabling this modified amino acid to carry out the normal lysine mediated enzymatic reaction. Furthermore, unnatural amino acids have been incorporated to introduce a spectroscopically active probe to interrogate protein structure and dynamics by Raman, infrared, electron paramagnetic resonance, and NMR spectroscopies (48–54). These spectroscopic studies have shed light on the functional role of protein dynamics but are limited in scope, due to the single probe being located at an isolated site in the protein structure. Characterization of the effects of an unnatural amino acid substitution on the dynamics of the entire enzyme would enable detailed insight into the energy landscape on which these motions occur.
In RNase A, histidine 48 was mutated to cysteine by standard site-directed mutagenesis protocols. Subsequently, we expressed and purified 15N-labeled H48C RNase A and reacted the resulting free thiol, introduced at position 48, with 4-chloromethylimidazole (4-CMI) (26) (Scheme 1). For WT RNase A, there are no free thiols in the enzyme, thus mutation of H48 to cysteine and subsequent chemical derivatization with 4-CMI results in incorporation of a single, 4-methyl imidazole (4-MI) group, which was verified by 5,5′-dithiobis-(2-nitrobenzoic acid) analysis (27). This single-site introduction of an unnatural amino acid results in replacement of histidine with an analog containing sulfur and an additional methylene group and is therefore two chemical bonds longer than histidine (Scheme 1 and Fig. 1 a). The larger side chain of 4-MI results in an increase in volume of this residue to 133.2 Å3 from that of a histidine side chain, which is 98.2 Å3. In addition, it is expected that the thioether linkage will alter the pKa of the methylimidazole side chain to some extent. Fig. 1 shows a comparison of 1H-15N NMR spectra for uniformly 15N-labeled WT, H48C, and H48C-4MI enzymes. The mean composite 1H and 15N chemical shift difference from WT is 0.11 ± 0.07 ppm for H48C-4MI, suggesting that chemical modification of C48 results in little perturbation of the RNase A structure. These chemical shift changes are localized to the site of mutation (Fig. S1 in the Supporting Material). The largest differences (+1.5σ) from WT for H48C-4MI are located at the end of loop 1 (Y25), β1 (K41 and T45), α3 (L51, D53, V54, and Q55), β4 (D83, C84, and E86), and β5 (T100). Small chemical shift changes from WT values were also observed in studies on the H48C and H48A RNases A (20). In addition, the H48C-4MI resonance peaks are mostly positioned between H48C and WT suggesting that the modification of C48 by 4-MI results in an enzyme structure that more closely resembles the WT than the cysteine mutant from which it is derived. This similarity with WT extends to sites of protein conformational motions as described below.
Figure 1.
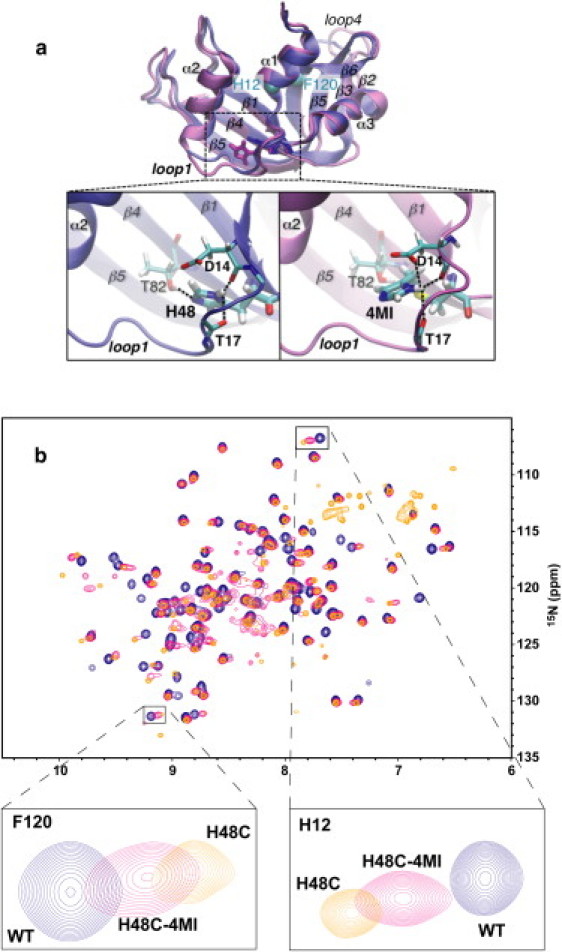
Comparison of WT, H48C, and H48C-4MI. (a) Average backbone structure WT and H48C-4MI over a 20 ns MD simulation and expansion showing H-bond interactions with loop 1 and β-strand 4 that are common to H48 and H48C-4MI. Secondary structures and relevant residues are labeled. Part (b) shows an overlay of 1H,15N HSQC spectra for WT, H48C, and H48C-4MI. Peaks are labeled to indicate the RNase A variant. The expanded region shows a close up view of F120 and H12 showing the proximity of the H48C-4MI peak to WT, relative to the H48C residues. Contour levels were adjusted to account for different [RNase A].
Millisecond motions in H48C-4MI
Conformational exchange motions can be quantitatively characterized by Carr-Purcell-Meiboom-Gill (CPMG) NMR relaxation dispersion experiments. In these experiments, the transverse relaxation rate constant (R2) is measured as a function of the CPMG pulse spacing (τcp) in a spin-echo experiment. In many cases, conformational motion manifests as dispersion in the measured R2 value as a function of τcp in which the kinetic (kex), thermodynamic (populations, (pa/b)), and structural (chemical shift differences (Δω) for the atomic sites involved in these equilibrium motions) details of the motion can be determined (55). In the fast limit in which kex > Δω, kex and the exchange contribution to the NMR resonance (Rex = papbΔω2/kex) can be determined. In other cases, conformational motion is evident in the form of elevated R2 values compared to rigid sites within the protein but shows no variation with τcp, usually indicating that motion is faster than the interval between the CPMG 180° pulses. To investigate motions in RNase A, we performed these experiments on 15N,2H-WT, 15N,1H-H48C, and 15N,1H-H48C-4MI at different solution pH values. Exchange broadening of several of the residues in loop 1 in the H48C-4MI enzyme (pH 6.4) precludes quantitation of motional parameters and suggests that motions are on the intermediate exchange timescale in H48C-4MI, unlike what is observed in H48C or H48A (Fig. S2 and Fig. S3), in which these sites are rigid. In loop 1, T17 (Fig. S3) shows elevated R2 values but no dispersion in H48C-4MI indicating that this residue retains its flexibility but its motion has likely moved to a somewhat faster timescale. A similar effect is observed for T45 in β1 and C84 in β4 (Fig. S2 and Fig. S3, Table S1). Conversion of H48C to H48C-4MI restores ms motions at pH = 6.4 as shown by Rex contributions to R2 for V43 that are >0 (3.2 ±1.8 s–1). In addition, in β1, three residues (F46, V47, and MI48) are unassigned in H48C-4MI at pH 6.4 and 7.0, due to being exchange broadened. Nonetheless, the H48C-4MI enzyme shows conformational exchange at similar sites to WT enzyme, which is not the case for the H48C enzyme (Fig. S3, Fig. S4, Fig. S5 and Table S1). Overall, relaxation rates could be quantitated for 109, 106, 78, and 98 residues, respectively, for WT (pH = 6.4 and 70) and H48C-4MI (6.4 and 7.0).
One aspect of the involvement of H48 in WT RNase A motions is that above and below the pK range for histidine, no evidence for motions is present in the region surrounding H48 such as in loop1, β1, and β4. The flat dispersion curves that are characteristic of a lack of ms motions, at these pH values, are shown in Fig. S6. In contrast, at pH = 6.4 and 7.0 the functionally important motions in WT are evident (Figs. 2 and S6). For WT residues S22, V43, and C84 the CPMG dispersion curves at pH = 6.4 and 7.0 show the upward curvature that is characteristic of ms motions (Fig. 2 and Fig. S6). These results indicate that the rate-limiting motions in WT RNase A occur within a small pH window. Motions for residues that are not part of the loop1, β1, and β4 regions, such as N71, are not affected significantly by the changes in solution pH (Fig. 2, and Fig. S3 and Fig. S6) (20). These pH insensitive regions are also not affected by mutagenesis to H48, surrounding sites, or loop 1 indicating that they are distinct from the rate-limiting motions in loop1, β1, and β4 (22,56).
Figure 2.
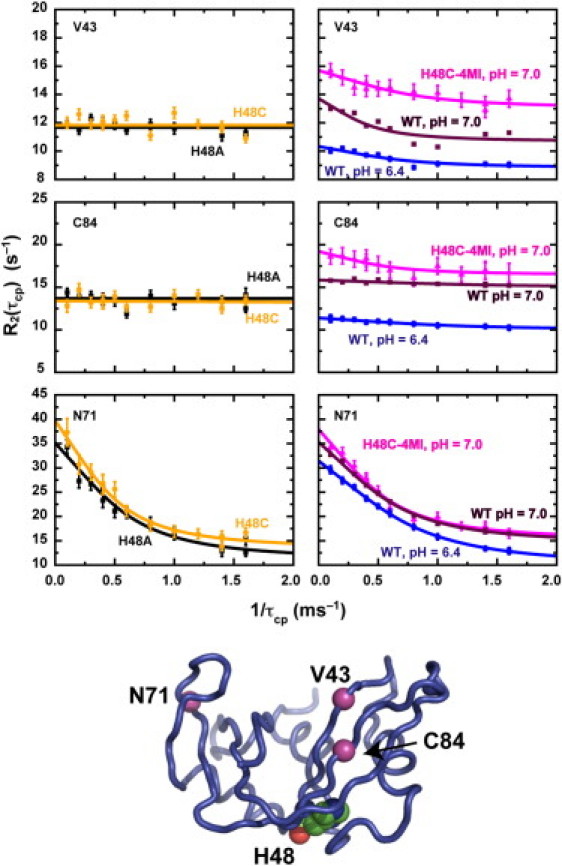
Millisecond motions in H48C-4MI. Dispersion curves for V43, C84, and N71 are displayed. On the left are dispersion curves for 1H,15N-H48A pH = 6.4 and 1H,15N-H48C, pH = 7.0. The right panel shows CPMG dispersion curves 2H,15N-WT, pH = 6.4 and 1H,15N-pH = 7.0 and 1H,15N-H48C-4MI pH = 7.0. Labels identifying the appropriate enzyme are placed closest to the relevant dispersion curve. The data are fit with the fast-limit CPMG dispersion equation. The ribbon structure of RNase A shows the location of these residues as single spheres. The side chain of thia-imidazole is shown as a sphere representation.
Mutation to H48C, like previous mutations in this region, results in the elimination of ms motions in loop1, β1, and β4 (Fig. 2). For many residues in H48C-4MI at pH = 6.4, evidence for ms motions at sites similar to WT comes from elevated R2 values and observation of exchange broadened residues (Fig. S2, Fig. S3, Fig. S4, Fig. S5, Table S1). For WT RNase A, NMR data have established that the functionally important global conformational motions in WT RNase A cluster in loop 1, β1, and β4 and move with an exchange rate constant, kex = 1800 s–1 at 298 K and pH = 6.4 and 7.0 (15,16,19,20,56). Likewise, at pH 7.0 for H48C-4MI upward sloping dispersion curves are observed for V43 and C84 on β1 and β4, respectively (Fig. 2), which are indicative of conformational exchange motion. Global fits of the fast-limit form of the CPMG dispersion equation to these two residues give a kex = 1650 ± 450 s–1, which is nearly identical to WT values (Fig. 2). Motions in loop 4 as evidenced by residue N71, indicate that it retains WT-like motions and is not affected by the thia-imidiazole substitution at residue 48 (Figs. 2 and S3) (16,20,22). Other residues in β1 and β4 remain sufficiently exchanged broadened at pH = 7.0 such that quantitation of these motions are not possible for these residues (Fig. S4). These data indicate that placing a larger imidazole group at position 48 is compatible within the WT enzyme structure and can restore ms motions that are similar to WT RNase A.
MD simulations of H48C-4MI
Molecular motions on timescales of several ns provide atomistic information on the ensemble of populated conformers that are accessible to the macromolecule. To investigate the effects of an unnatural amino acid substitution on ns timescale motions, we have modeled the effects of the 4-MI incorporation in a 20 ns MD simulation of H48C, H48C-4MI, and WT at 298 K, with emphasis on the analysis of H-bonding interactions with loop 1 and β4 that are common to H48 and H48C-4MI. Fig. 1 a shows a comparison of representative snapshot configurations for WT and H48C-4MI. The region around loop 1 (backbone root mean-square deviation 1.87 Å) is highlighted, including hydrogen bonds with side chains in loop 1 (D14 and T17) and β4 (T82). When comparing the relative stability of these fundamental interactions, we find that H48 in WT RNase A maintains an H-bond with the side chain of T82 as well as H-bonding interactions with loop 1 over the entire simulation time. Due to thermal fluctuations, however, the H-bond that is made with loop 1 is formed alternately with D14 for 75% of the time and with T17 for the rest of the simulation. Analogously, the longer and more flexible side chain of H48C4-MI stabilizes H-bonding interactions with T82 and D14 for 60% and 30% of the simulation time, respectively. The unprotonated nitrogen of the imidazole ring of H48C-4MI is alternately forming H-bonds with the polar side chains of T82 or Q101, or with the N-H group of the A20 backbone. In addition, the imidazole NH group of H48C-4MI forms H-bonds with the side chains of T82 or Y25, or with D14. Overall, the main H-bond interactions of H48 with T82 and D14 in WT are maintained in the H48C-MI enzyme, indicating that the most relevant interactions with loop 1 and β-strand 4 are present with the 4-MI single-site chemical modification of H48C (Fig. S7, Fig. S8, Fig. S9). However, the 4-MI group is more promiscuous over the 20 ns simulation, forming transient hydrogen bonding interactions with a number of other residues. These additional interactions made by the methylimidazole side chain are likely due to the additional rotational degrees of freedom relative to that of histidine.
We additionally analyzed the Cα-Cα covariance matrix over the entire 20 ns simulation for each enzyme. Shown in Fig. 3 are the results of this analysis for WT, H48C-4MI, and H48C. Qualitatively, it is clear that the extent of motional correlation in H48C-4MI more closely resembles that of WT than does H48C. In addition to correlations between loop 1 and the rest of the enzyme, correlations are decreased in the α3 and β3 regions and loop residues 85–95 (circled areas in Fig. 3). The correlations in secondary structure in WT are shown in Fig. 3 b–d. In Fig. 3 b positive correlations with α1/loop 1 with β1, 4, and 5 are indicated. In Fig. 3 d, correlations between H48 and parts of β4 and loop 1 are shown. Whereas, in Fig. 3 c, anti-correlated regions between α1/loop1 and β2, α3, and the N-terminal portion of β4 are indicated in shades of blue. Modification of C48 with methylimidazole does not fully restore WT-like ns motions or the high degree of correlated motions throughout the protein, yet the H48C-4MI is more WT-like in its fast motion behavior than is the H48C protein.
Figure 3.

Covariance analysis of ns motions in RNase A. In a, motional correlations are shown for WT, H48C-4MI, and H48C from a 20 ns MD simulation. Panels b–d show results for WT mapped onto a cartoon ribbon of RNase A. Positive correlations (b) between residues 1–22 and β1, β4, and β5 are shown. Panel c shows anticorrelations between residues 1–22 and β2, α3, and β5. Panel d shows positive correlations between H48 (stick rendering) and loop 1, β1, and β4.
4-MI restores ligand-binding interactions
Having addressed the motional properties of H48C-4MI, we next investigated the impact of this unnatural amino acid on RNase A function. Upon interaction with the product analog (3′-CMP), the NMR resonance signals in WT RNase A show linear progression until saturation is reached, which is characteristic of a two-state bimolecular binding reaction. The resulting apparent Kd for 3′-CMP for WT is 500 μM at pH 7.0 (57,58). Fig. 4 shows the chemical shift change for D121, Q11, H12, and E86 as a function of [3′-CMP] for H48C-4MI and H48C at pH = 7.0. The apparent 3′-CMP dissociation constants for these four residues, individually, are within 3% of each other and therefore all four were fit to obtain a single, apparent Kd = 300 ± 9 μM for H48C-4MI at pH = 7.0 (Fig. 4 a). In H48C, the affinity for 3′-CMP is weaker (Kd = 610 ± 32 μM, Fig. 4 b) than in H48C-4MI enzyme. The introduction of 4-methylimidazole results in slightly tighter binding for H48C-4MI compared to WT RNase A.
Figure 4.
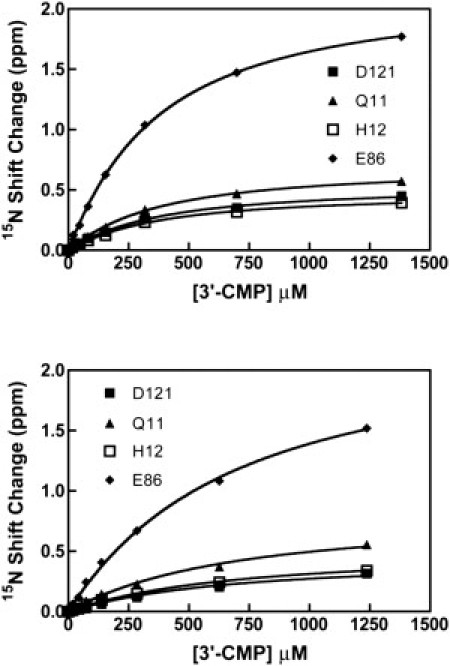
Binding of 3′-CMP to RNase A. (Top) H48C-4MI (bottom) H48C. 1H,15N chemical shift changes for four residues are plotted versus the concentration of 3′-CMP at pH = 7.0, 298 K. All data are fit with a single global one-site binding model.
The kinetics of interaction between WT RNase A and the reaction product determine the overall rate of catalytic turnover in this enzyme (21). Furthermore, the ms motions in RNase A have been demonstrated to be coupled to the release of product. The rate constant of 3′-CMP dissociation from RNase A can be determined by NMR lineshape analysis. In 1H-15N-WT RNase A under conditions in which ms motions are present, koff for 3′-CMP determined by lineshape analysis is 1700 ± 500 s–1 (16,22,56). In Fig. 5, the data used to determine koff for 1H-15N-H48C-4MI and 1H-15N-H48C are shown. For H48C-4MI, koff for 3′-CMP is 1920 ± 750 s–1 and is similar to kex measured for motions in this chemically modified enzyme. This koff value is also similar to that obtained for WT enzyme. In contrast, the data for H48C are at the upper limit for accurate measurements. The minimal line broadening at intermediate 3′-CMP titration points indicates fast dissociation. The uncertainty in fitted koff for H48C is high, koff = 10,500 ± 2700 s–1 and we estimate that koff in the H48C mutant is at least 6000 s–1 and thus ligand dissociates much faster from this enzyme than WT or H48C-MI, which have similar koff values.
Figure 5.
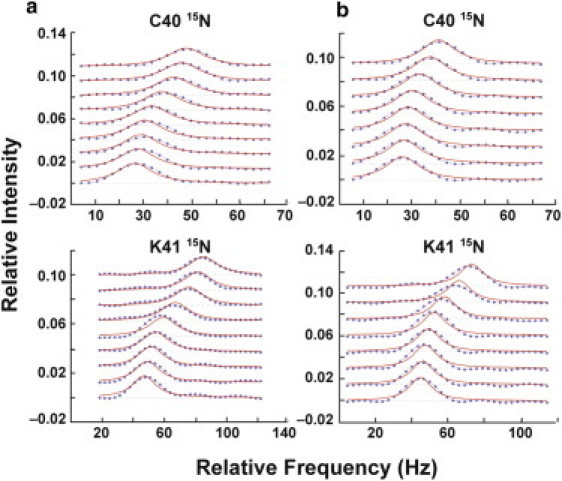
Kinetics of product, 3′-CMP interactions with RNase A. In a, global lineshape fitting to C40 and K41 in H48C-4MI. Panel (b) shows lineshape results for the same residues in H48C. All lineshape experiments were performed on protonated enzyme.
H48C-4MI is catalytically active
The kcat/KM values for WT RNase A with a fluorescent substrate (28) (6-carboxyfluorescein∼dArU(dA)2∼6-carboxytetramethylrhodamine) indicate that its cleavage is limited by the rate of enzyme encounter with substrate. For WT, at 0.1 M NaCl and pH = 7.0 kcat/KM for this substrate is 3.6 × 107 M–1 s–1 (59). We investigated the effect of solution pH on kcat/KM for WT and H48C-4MI (Fig. 6). Fitting Eq. 1. to the data gave –log of the acidic (pKa) and basic (pKb) equilibrium values of 4.1 ± 0.1 (4.7 ± 0.1) and 6.8 ± 0.1 (6.6 ± 0.1) for WT (H48C-4MI), respectively. The WT pK values obtained here are consistent with prior work on this enzyme (21). The instability of the substrate at acidic conditions precludes measurements at lower pH values. Thus, the pKa value determined from the fits are highly dependent on the data point at pH = 4.3. Therefore, we estimate the uncertainty in the pKa is slightly higher than the fitted error of 0.1 pH units. These data show similar catalytic activities over a similar pH range for WT and H48C-4MI.
Figure 6.
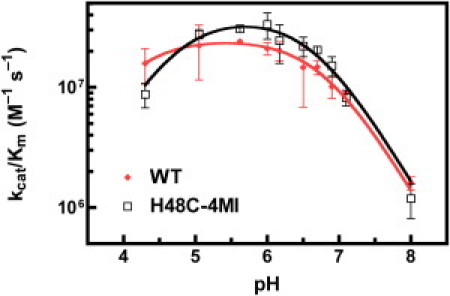
Catalytic activity in RNase A. pH dependence of kcat/KM for WT (diamonds) and H48C-4MI (squares) are shown. Equation 1 was fit to the data to determine pK values. Error bars are derived from multiple measurements.
Conclusions
Enzyme motions are essential for their function. Understanding how the structure and flexibility of the protein shapes the energy landscape over the course of the catalytic cycle is crucial for drug and protein design as well as protein engineering efforts. The complexity of this problem necessitates that multiple and unique experimental methods be used. In this work, we introduce an unnatural amino acid at a single site in uniformly 15N-labeled RNase A and subsequently characterize the effects of this amino acid substitution on protein motions, function, and structure.
The similarity between the 1H-15N HSQC spectra for WT, H48C-4MI, and H48C indicate that the structure of RNase A is not significantly altered by modification at position 48. Thus, the native fold of RNase A is robust and tolerant to single-site perturbations. This conclusion is further supported by the similarity in computationally optimized models of WT, H48C, and H48C-4MI (Fig. 1 a). In addition, almost all NMR resonances in H48C-4MI are positioned closer to the WT peak than the corresponding resonance in H48C suggesting that the 4-MI modification shifts the average solution structure to more closely resemble that of WT enzyme than does the H48C variant.
Perhaps the most interesting feature of the H48C-4MI RNase A is the similarity in pH range in which ms motions are present compared to WT. The cysteinyl methylimidazole group must be able to satisfy the main requirement that H48 does for facilitation of motion for residues in loop 1, β1, and β4. In addition, this chemical modification does not alter distant flexible regions of RNase A that have been shown to be uncoupled from residue 48. Thus, the 4-MI residue does not introduce nonnatural motions elsewhere in the enzyme. The kinetics of motion in H48C-4MI are the same, at pH = 7.0, as those in WT at pH = 6.4 and pH = 7.0. In addition, these motions occur at similar sites in the two enzymes suggesting similar motional mechanisms. Moreover, the onset of millisecond motions requires the covalent linkage to the imidazole moiety because the H48G mutant in the presence of >50 mM imidazole shows no conformational exchange motions (Fig. S10), indicating that ms motions cannot be rescued by addition of endogenous imidazole.
The amplitude of the relaxation dispersion curves, Rex = papbΔω2/kex, reflects both contributions from the equilibrium populations and chemical shift differences due to the conformational motions, which in the fast conformational exchange limit cannot be resolved into individual terms. Nonetheless, the ratio of WT and H48C-4MI dispersion curve amplitudes is the same for V43 and C84 and is equal to 0.56 ± 0.30 and 0.51 ± 0.16, respectively. Yet for N71 there is no effect with = 0.95 ± 0.11. The unnatural amino acid at position 48 likely affects the populations of the two conformations and not Δω because the HN of V43 and C84 are far from residue 48, 15.3 Å, and 9.0 Å, respectively. In addition, the isotropic chemical shifts of V43 and C84 are very close to their WT positions. Therefore effects, if any, on Δω should be rather small for these two residues. Thus, the identical increase in Rex for V43 and C84 suggests that it is the equilibrium populations (papb) that are primarily affected. This change in populations reflects about a 2 kcal/mol change in the relative differences in energy levels between the two conformations. However, the similar kex for WT and H48C-4MI suggests the barrier height between the two conformations remains the same. This conclusion will require further testing for validation.
The function of H48C-4MI is not dramatically altered relative to WT RNase A. The methylimidazole group of H48C-4MI fully restores WT binding affinity of the product analog 3′-CMP. This suggests that the H-bonding interactions made by the imidazole side chain are functionally important for ligand binding and these interactions are not satisfied by the thiol of cysteine but are sufficiently reestablished by 4-MI modification. Moreover, the pH rate data indicate that 4-methylimidazole at position 48 does not alter the ionization constants of the active site residues to an extent that it alters the catalytic activity of RNase A or its substrate association rate. The model for Michaelis complex formation in RNase A involves a diffusion-limited encounter complex followed by rearrangement to form the RNase A/substrate complex (21). Incorporation of an unnatural amino acid in RNase A does not alter this aspect of the RNase A energy landscape. This may turn out to be a clinically useful property of RNase A in which its cancer-killing ability requires catalytic activity but also the ability to evade Ribonuclease inhibitor (RI) (60). If binding to RI relies in part on the ms motions in RNase A, incorporation of unnatural amino acids may promote lower affinity interactions with RI and yet maintain high levels of catalytic activity.
In conclusion, we have exploited the versatility afforded by insertion of an unnatural amino acid into RNase A by chemical modification of a unique cysteine residue. The enzyme containing thia-methylimidazole at residue 48 possesses motions in similar regions, on similar timescales, and over a similar pH range as WT enzyme. These motions are selectively incorporated into one region of the enzyme without affecting existing motions in a separate region. These results demonstrate that conformational exchange motions may be amenable to rational engineering provided that details of motions in the WT enzyme are known in advance, such that the appropriate modification can be introduced. Furthermore, these studies indicate that chemical modification can be a route to introduce subtle alterations at amino acid sites for the study of protein motions. This should pave the way for in-depth study of the chemical and physical properties of dynamically important residues.
Acknowledgments
We thank Professors Jeff Hoch and Mark Maciejewski (University of Connecticut) for the generous use of their Varian 600 MHz NMR equipped with a cryogenically cooled probe. We thank Roger Cole and Gennady Khirich for NMR data obtained at pH = 4.5, 7.0, and 7.5.
J.P.L. acknowledges support from the National Science Foundation (NSF) MCB-0744161. E.D.W. and S.K.W. were recipients of a National Institutes of Health (NIH) biophysical training grant (5T32GM008283). V.S.B. acknowledges support for Dr. Rivalta from NIH 1R01-GM-084267-01 and GM-043278 and supercomputing time from the National Energy Research Scientific Computing Center (NERSC).
Supporting Material
References
- 1.Karplus M., Kuriyan J. Molecular dynamics and protein function. Proc. Natl. Acad. Sci. USA. 2005;102:6679–6685. doi: 10.1073/pnas.0408930102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Desjarlais J.R., Handel T.M. Side-chain and backbone flexibility in protein core design. J. Mol. Biol. 1999;290:305–318. doi: 10.1006/jmbi.1999.2866. [DOI] [PubMed] [Google Scholar]
- 3.Harbury P.B., Plecs J.J., Kim P.S. High-resolution protein design with backbone freedom. Science. 1998;282:1462–1467. doi: 10.1126/science.282.5393.1462. [DOI] [PubMed] [Google Scholar]
- 4.Nauli S., Kuhlman B., Baker D. Computer-based redesign of a protein folding pathway. Nat. Struct. Biol. 2001;8:602–605. doi: 10.1038/89638. [DOI] [PubMed] [Google Scholar]
- 5.Su A., Mayo S.L. Coupling backbone flexibility and amino acid sequence selection in protein design. Protein Sci. 1997;6:1701–1707. doi: 10.1002/pro.5560060810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frauenfelder H., Chen G., Young R.D. A unified model of protein dynamics. Proc. Natl. Acad. Sci. USA. 2009;106:5129–5134. doi: 10.1073/pnas.0900336106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hammes G.G., Porter R.W., Stark G.R. Relaxation spectra of aspartate transcarbamylase. Interaction of the catalytic subunit with carbamyl phosphate, succinate, and L-malate. Biochemistry. 1971;10:1046–1050. doi: 10.1021/bi00782a017. [DOI] [PubMed] [Google Scholar]
- 8.Hedstrom L., Szilagyi L., Rutter W.J. Converting trypsin to chymotrypsin: the role of surface loops. Science. 1992;255:1249–1253. doi: 10.1126/science.1546324. [DOI] [PubMed] [Google Scholar]
- 9.Sampson N.S., Knowles J.R. Segmental motion in catalysis: investigation of a hydrogen bond critical for loop closure in the reaction of triosephosphate isomerase. Biochemistry. 1992;31:8488–8494. doi: 10.1021/bi00151a015. [DOI] [PubMed] [Google Scholar]
- 10.Alber T., Gilbert W.A., Petsko G.A. The role of mobility in the substrate binding and catalytic machinery of enzymes. Ciba Found. Symp. 1983;93:4–24. doi: 10.1002/9780470720752.ch2. [DOI] [PubMed] [Google Scholar]
- 11.Erman J.E., Hammes G.G. Relaxation spectra of ribonuclease. IV. The interaction of ribonuclease with cytidine 2′:3′-cyclic phosphate. J. Am. Chem. Soc. 1966;88:5607–5614. doi: 10.1021/ja00975a046. [DOI] [PubMed] [Google Scholar]
- 12.Erman J.E., Hammes G.G. Relaxation spectra of ribonuclease. V. The interaction of ribonuclease with cytidylyl-3′:5′-cytidine. J. Am. Chem. Soc. 1966;88:5614–5617. doi: 10.1021/ja00975a047. [DOI] [PubMed] [Google Scholar]
- 13.Jayaraman V., Rodgers K.R., Spiro T.G. Hemoglobin allostery: resonance Raman spectroscopy of kinetic intermediates. Science. 1995;269:1843–1848. doi: 10.1126/science.7569921. [DOI] [PubMed] [Google Scholar]
- 14.Cooper A., Dryden D.T.F. Allostery without conformational change. A plausible model. Eur. Biophys. J. 1984;11:103–109. doi: 10.1007/BF00276625. [DOI] [PubMed] [Google Scholar]
- 15.Beach H., Cole R., Loria J.P. Conservation of mus-ms enzyme motions in the apo- and substrate-mimicked state. J. Am. Chem. Soc. 2005;127:9167–9176. doi: 10.1021/ja0514949. [DOI] [PubMed] [Google Scholar]
- 16.Kovrigin E.L., Loria J.P. Enzyme dynamics along the reaction coordinate: critical role of a conserved residue. Biochemistry. 2006;45:2636–2647. doi: 10.1021/bi0525066. [DOI] [PubMed] [Google Scholar]
- 17.French T.C., Hammes G.G. Relaxation spectra of ribonuclease. II. Isomerization of ribonuclease at neutral pH values. J. Am. Chem. Soc. 1965;87:4669–4673. doi: 10.1021/ja00949a002. [DOI] [PubMed] [Google Scholar]
- 18.Markley J.L. Correlation proton magnetic resonance studies at 250 MHz of bovine pancreatic ribonuclease. II. pH and inhibitor-induced conformational transitions affecting histidine-48 and one tyrosine residue of ribonuclease A. Biochemistry. 1975;14:554–561. [PubMed] [Google Scholar]
- 19.Cole R., Loria J.P. Evidence for flexibility in the function of ribonuclease A. Biochemistry. 2002;41:6072–6081. doi: 10.1021/bi025655m. [DOI] [PubMed] [Google Scholar]
- 20.Watt E.D., Shimada H., Loria J.P. The mechanism of rate-limiting motions in enzyme function. Proc. Natl. Acad. Sci. USA. 2007;104:11981–11986. doi: 10.1073/pnas.0702551104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park C., Raines R.T. Catalysis by ribonuclease A is limited by the rate of substrate association. Biochemistry. 2003;42:3509–3518. doi: 10.1021/bi026076k. [DOI] [PubMed] [Google Scholar]
- 22.Doucet N., Watt E.D., Loria J.P. The flexibility of a distant loop modulates active site motion and product release in ribonuclease A. Biochemistry. 2009;48:7160–7168. doi: 10.1021/bi900830g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Butterwick J.A., Palmer A.G., 3rd An inserted Gly residue fine tunes dynamics between mesophilic and thermophilic ribonucleases H. Protein Sci. 2006;15:2697–2707. doi: 10.1110/ps.062398606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chakravorty D.K., Hammes-Schiffer S. Impact of mutation on proton transfer reactions in ketosteroid isomerase: insights from molecular dynamics simulations. J. Am. Chem. Soc. 2010;132:7549–7555. doi: 10.1021/ja102714u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Du Z., Liu Y., Wang C. Backbone dynamics and global effects of an activating mutation in minimized Mtu RecA inteins. J. Mol. Biol. 2010;400:755–767. doi: 10.1016/j.jmb.2010.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Earnhardt J.N., Wright S.K., Silverman D.N. Introduction of histidine analogs leads to enhanced proton transfer in carbonic anhydrase V. Arch. Biochem. Biophys. 1999;361:264–270. doi: 10.1006/abbi.1998.0984. [DOI] [PubMed] [Google Scholar]
- 27.Ellman G.L. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 1959;82:70–77. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- 28.Kelemen B.R., Klink T.A., Raines R.T. Hypersensitive substrate for ribonucleases. Nucleic Acids Res. 1999;27:3696–3701. doi: 10.1093/nar/27.18.3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park C., Kelemen B.R., Raines R.T. Fast, facile, hypersensitive assays for ribonucleolytic activity. Methods Enzymol. 2001;341:81–94. doi: 10.1016/s0076-6879(01)41146-3. [DOI] [PubMed] [Google Scholar]
- 30.Loria J.P., Rance M., Palmer A.G. A relaxation-compensated Carr-Purcell-Meiboom-Gill sequence for characterizing chemical exchange by NMR spectroscopy. J. Am. Chem. Soc. 1999;121:2331–2332. [Google Scholar]
- 31.Kovrigin E.L., Kempf J.G., Loria J.P. Faithful estimation of dynamics parameters from CPMG relaxation dispersion measurements. J. Magn. Reson. 2006;180:93–104. doi: 10.1016/j.jmr.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 32.Wlodawer A., Svensson L.A., Gilliland G.L. Structure of phosphate-free ribonuclease A refined at 1.26 A. Biochemistry. 1988;27:2705–2717. doi: 10.1021/bi00408a010. [DOI] [PubMed] [Google Scholar]
- 33.Case D.A., Cheatham T.E., 3rd, Woods R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang J., Wolf R.M., Case D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 36.Bayly C.I., Cieplak P., Kollman P. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges - The RESP model. J. Phys. Chem. 1993;97:10269–10280. [Google Scholar]
- 37.Frisch M.J., Trucks G.W., Fox D.J. Gaussian; Wallingford, CT: 2009. Gaussian 09, Revision A.1. [Google Scholar]
- 38.Jorgensen W.L., Chandrasekhar J., Klein M. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 39.Darden T., York D., Pedersen L. Particle mesh Ewald: an N-log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;89:10089–10092. [Google Scholar]
- 40.Grubmüller H., Heller H., Schulten K. Generalized Verlet algorithm for efficient molecular dynamics simulations with long-range interactions. Mol. Simul. 1991;6:121–142. [Google Scholar]
- 41.Ichiye T., Karplus M. Collective motions in proteins: a covariance analysis of atomic fluctuations in molecular dynamics and normal mode simulations. Proteins. 1991;11:205–217. doi: 10.1002/prot.340110305. [DOI] [PubMed] [Google Scholar]
- 42.Weiner H., White W.N., Koshland D.E., Jr. The formation of anhydrochymotrypsin by removing the elements of water from the serine at the active site. J. Am. Chem. Soc. 1966;88:3851–3859. doi: 10.1021/ja00968a033. [DOI] [PubMed] [Google Scholar]
- 43.Neet K.E., Koshland D.E., Jr. The conversion of serine at the active site of subtilisin to cysteine: a “chemical mutation”. Proc. Natl. Acad. Sci. USA. 1966;56:1606–1611. doi: 10.1073/pnas.56.5.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Polgar L., Bender M.L. A new enzyme containing a synthetically formed active site. Thiol-subtilisin. J. Am. Chem. Soc. 1966;88:3153–3154. [Google Scholar]
- 45.Smith H.B., Hartman F.C. Restoration of activity to catalytically deficient mutants of ribulosebisphosphate carboxylase/oxygenase by aminoethylation. J. Biol. Chem. 1988;263:4921–4925. [PubMed] [Google Scholar]
- 46.Gloss L.M., Kirsch J.F. Decreasing the basicity of the active site base, Lys-258, of Escherichia coli aspartate aminotransferase by replacement with gamma-thialysine. Biochemistry. 1995;34:3990–3998. doi: 10.1021/bi00012a017. [DOI] [PubMed] [Google Scholar]
- 47.Gloss L.M., Kirsch J.F. Examining the structural and chemical flexibility of the active site base, Lys-258, of Escherichia coli aspartate aminotransferase by replacement with unnatural amino acids. Biochemistry. 1995;34:12323–12332. doi: 10.1021/bi00038a028. [DOI] [PubMed] [Google Scholar]
- 48.Waldman A.D., Birdsall B., Holbrook J.J. 13C-NMR and transient kinetic studies on lactate dehydrogenase [Cys(13CN)165]. Direct measurement of a rate-limiting rearrangement in protein structure. Biochim. Biophys. Acta. 1986;870:102–111. doi: 10.1016/0167-4838(86)90013-0. [DOI] [PubMed] [Google Scholar]
- 49.Cellitti S.E., Jones D.H., Geierstanger B.H. In vivo incorporation of unnatural amino acids to probe structure, dynamics, and ligand binding in a large protein by nuclear magnetic resonance spectroscopy. J. Am. Chem. Soc. 2008;130:9268–9281. doi: 10.1021/ja801602q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cornish V.W., Benson D.R., Schultz P.G. Site-specific incorporation of biophysical probes into proteins. Proc. Natl. Acad. Sci. USA. 1994;91:2910–2914. doi: 10.1073/pnas.91.8.2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lampe J.N., Floor S.N., Ortiz de Montellano P.R. Ligand-induced conformational heterogeneity of cytochrome P450 CYP119 identified by 2D NMR spectroscopy with the unnatural amino acid (13)C-p-methoxyphenylalanine. J. Am. Chem. Soc. 2008;130:16168–16169. doi: 10.1021/ja8071463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miyake-Stoner S.J., Refakis C.A., Mehl R.A. Generating permissive site-specific unnatural aminoacyl-tRNA synthetases. Biochemistry. 2010;49:1667–1677. doi: 10.1021/bi901947r. [DOI] [PubMed] [Google Scholar]
- 53.Weeks C.L., Polishchuk A., Spiro T.G. Investigation of an unnatural amino acid for use as a resonance Raman probe: detection limits, solvent and temperature dependence of the nuC identical withN band of 4-cyanophenylalanine. J Raman Spectrosc. 2008;39:1606–1613. doi: 10.1002/jrs.2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zheng M.L., Zheng D.C., Wang J. Non-native side chain IR probe in peptides: ab initio computation and 1D and 2D IR spectral simulation. J. Phys. Chem. B. 2010;114:2327–2336. doi: 10.1021/jp912062c. [DOI] [PubMed] [Google Scholar]
- 55.Palmer A.G., Kroenke C.D., Loria J.P. Nuclear magnetic resonance methods for quantifying microsecond-to-millisecond motions in biological macromolecules. Meth. Enzymol. 2001;339:204–238. doi: 10.1016/s0076-6879(01)39315-1. [DOI] [PubMed] [Google Scholar]
- 56.Doucet N., Khirich G., Loria J.P. Alteration of hydrogen bonding in the vicinity of histidine 48 disrupts millisecond motions in RNase A. Biochemistry. 2011;50:1723–1730. doi: 10.1021/bi1018539. [DOI] [PubMed] [Google Scholar]
- 57.Cathou R.E., Hammes G.G. Relaxation spectra of ribonuclease. I. The interaction of ribonuclease with cytidine 3′-phosphate. J. Am. Chem. Soc. 1964;86:3240–3245. doi: 10.1021/ja00949a003. [DOI] [PubMed] [Google Scholar]
- 58.Cathou R.E., Hammes G.G. Relaxation spectra of ribonuclease. 3. Further investigation of the interaction of ribonuclease and cytidine 3′-phosphate. J. Am. Chem. Soc. 1965;87:4674–4680. doi: 10.1021/ja00949a003. [DOI] [PubMed] [Google Scholar]
- 59.Park C., Raines R.T. Quantitative analysis of the effect of salt concentration on enzymatic catalysis. J. Am. Chem. Soc. 2001;123:11472–11479. doi: 10.1021/ja0164834. [DOI] [PubMed] [Google Scholar]
- 60.Rutkoski T.J., Raines R.T. Evasion of ribonuclease inhibitor as a determinant of ribonuclease cytotoxicity. Curr. Pharm. Biotechnol. 2008;9:185–189. doi: 10.2174/138920108784567344. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.