

Abstract
Context
Bilateral adrenal hyperplasias (BAHs) may be caused by mutations of genes that code for molecules that participate in cAMP signaling. Little is known about cAMP signaling in adrenal lesions associated with ACTH-independent Cushing syndrome (AICS) that do not harbor mutations in known genes.
Objective
We assessed the cAMP-signaling pathway by enzymatic and molecular studies.
Design
Samples from 27 patients (ages 5–60 years) were studied and compared with normal adrenocortical tissue (n=4) and aldosterone-producing adenomas (APA, n=5). All samples were sequenced for GNAS, PRKAR1A, PDE11A, and PDE8B sequencing defects. cAMP levels and binding, protein kinase A, and phosphodiesterase (PDE) activities were assayed. Immunohistochemistry was used for certain studies and the phosphorylation status of CREB was studied.
Patients
A total of 36 samples from patients were used.
Results
Cortisol-producing adenomas (CPAs) and other lesions that were GNAS, PRKAR1A, PDE11A, and PDE8B gene mutation-negative were compared with PRKAR1A mutation-positive lesions, normal tissue, and APAs; abnormalities of the cAMP-signaling pathway were found in both BAHs and CPAs. Interestingly, mutation-negative CPAs had significantly decreased PDE activity.
Conclusion
Lesions of the adrenal associated with AICS, independently of their GNAS, PRKAR1A, PDE11A, and PDE8B mutation status, have functional abnormalities of cAMP signaling. It is probable that epigenetic events or additional defects of genes involved in this pathway are responsible for this phenomenon.
Introduction
Cushing syndrome (CS) may be caused by ACTH-independent adrenocortical lesions (AICS) that include the relatively common cortisol-producing adenoma (CPA) and the various rare forms of bilateral adrenal hyperplasia (BAH) 1. The cAMP-signaling pathway is involved in the pathogenesis of BAHs 2, 3: GNAS-activating mutations were found in macronodular BAH in the context of McCune–Albright syndrome 4 and in massive macronodular adrenocortical disease (MMAD), also known as ACTH-independent macronodular adrenocortical hyperplasia (AIMAH) 5, and G-protein-coupled receptor (GPCR) aberrant expression has been found in both MMAD/AIMAH and sporadic adrenal lesions, including CPAs 3, 6, 7. Mutations in the PRKAR1A gene coding for the regulatory subunit 1A (R1A) of cAMP-dependent protein kinase A (PKA) are responsible for the most common form of micronodular BAH, primary pigmented nodular adrenocortical disease (PPNAD), a disease that may be isolated (iPPNAD) or part of Carney complex (CNC), a multiple endocrine neoplasia (MEN) syndrome 8, 9, 10, 11. More recently, mutations in the phosphodiesterase (PDE) genes PDE11A and PDE8B have been found in isolated micronodular adrenocortical disease (iMAD) and iPPNAD and other forms of BAHs, as well as in sporadic adrenocortical tumors (ADTs), 12, 13, 14.
However, the vast majority of benign adrenocortical lesions associated with AICS does not bear germline or somatic sequence mutations in GNAS, PRKAR1A, PDE11A, or PDE8B. A minority of these tumors demonstrate abnormal expression of one or another of these genes due to somatic allelic losses or other, possibly epigenetic, events. For example, PRKAR1A losses in sporadic ADTs and MMAD/AIMAH without coding sequence alterations 15, 16 or PDE11A under-expression in several ADTs, including CPAs, were demonstrated by both mRNA and protein studies 14, 17, 18.
The purpose of this study was to assess the overall activity of the cAMP-signaling pathway in ADTs that do not have coding sequence mutations or allelic losses for any of the above genes and genetic loci, respectively. These tumors were compared with those that had known PRKAR1A gene mutations and a small number of normal adrenal tissue samples and tissue from aldosterone-producing adenomas (APA). A relatively simple and general approach was taken; cAMP levels and cAMP-binding activity were assayed followed by total PKA and PDE activities along with the protein levels of the main molecular players. The data are suggestive of functional abnormalities of this pathway, albeit at different levels, in many of these lesions.
Materials and methods
Patients and adrenal tissue collection
All clinical studies were approved by the Eunice Kennedy Shriver National Institute of Child Health and Human Development Institutional Review Board. Thirty-two patients (21 women and 11 men), aged 5–60 years with AICS, have participated in this study (Table 1). Patients were divided in diagnostic groups according to their histopathological findings after bilateral or unilateral adrenalectomy: there were 13 patients with MMAD/AIMAH, eight with PPNAD (with or without CNC and with PRKAR1A mutations), three with iMAD, and three with CPA. All samples were analyzed simultaneously and compared with four normal adrenal glands collected from unrelated patients, as well as five APAs. The group described as ‘BAHs without PRKAR1A mutations’ includes the MMAD/AIMAH and the iMAD patients, who bear no sequence mutations in the GNAS, PRKAR1A, PDE11A, and PDE8B gene. For the CREB and immunostaining studies, an additional group of controls, 3 tissues with PDE11A mutations, were studied; these samples have been extensively described by Horvath et al. 12.
Table 1.
Patient clinical data and mutation status.
| Numbers | Patient code | Sex | Age (year) | Diagnosis | Surgery | Dimensions (cm) | Weight (g) | Diurnal cortisol | Gene | Mutations | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Rt | Lt | Rt | Lt | 12 Midnight | 8 a.m. | ||||||||
| 1A | ADT03.01 | M | 54 | MMAD/AIMAH | BADX | 9×5×3 | 6×2.5×2 | 190 | 154 | 6.2 | 12.5 | – | None |
| 2A | M1 | F | 49 | MMAD/AIMAH | BADX | 9×5×2 | 7×3×5 | 32 | 34.4 | 13.1 | 15.7 | – | None |
| 3A | ADT65.02 | F | 51 | MMAD/AIMAH | UADX-right | 6.2×4 | – | 111 | – | 7.9 | 10.2 | – | None |
| 4A | ADT55.02 | F | 42 | MMAD/AIMAH | UADX-right | 5 | – | NA | – | 3.2 | 8.2 | – | None |
| 5A | ADT36.01 | M | 46 | MMAD/AIMAH | BADX | 10×8×3.5 | 8×4.5×2.5 | 101 | 490 | 7.5 | 9.8 | – | None |
| 6A | CAR 073.01 | M | 60 | MMAD/AIMAH | BADX | 12×8×4 | 14×7×4 | 155 | 182 | 12.8 | 16.2 | – | None |
| 7A | CAR 589.01 | M | 39 | MMAD/AIMAH | BADX | 10×5.5×2.8 | 7×6×2.5 | 36 | 44 | 29.4 | 28.3 | – | None |
| 8A | ADT23.01 | F | 57 | MMAD/AIMAH | BADX | 8×8.5×3.5 | 11.5×5×2.5 | 65 | 54.3 | 22 | 31.1 | – | None |
| 9A | ADT29.03 | M | 45 | MMAD/AIMAH | BADX | 10.2×6.5×2.5 | 13×5×4 | NA | 131 | 12.6 | 19.9 | – | None |
| 10A | ADT27.03 | F | 43 | MMAD/AIMAH | BADX | 9×4×2 | 9×5×2 | 52 | 94.8 | 21.8 | 30.5 | – | None |
| 11A | ADT04.01 | F | 49 | MMAD/AIMAH | BADX | 3.5×3.5×2.8 | 2.3×1.8×1.5 | NA | NA | 32.3 | 30.6 | – | None |
| 12A | M101 | F | 46 | MMAD/AIMAH | BADX | 5×3×2.5 | 6.5×3×2.5 | 43 | 44.9 | NA | NA | – | None |
| 13A | ADT05.01 | F | 31 | MMAD/AIMAH | UADX-right | 5×2.5×2 | – | 22 | – | 22.4 | 21 | – | None |
| 1B | CAR 77.03 | M | 19 | iMAD | BADX | 5×1×0.6 | 5×1×0.5 | 13 | 25.6 | 25 | 21.8 | – | None |
| 2B | CAR545.13 | F | 16 | iMAD | UADX-left | – | 7×3.5×1.5 | – | 15.6 | 5.2 | 7.1 | – | None |
| 3B | CAR653.02 | F | 30 | iMAD | NA | NA | NA | NA | NA | NA | NA | – | None |
| 1C | CAR 021.02 | F | 23 | CNC/PPNAD | BADX | 10×2.1×1 | 7×3×1 | NA | NA | 11.4 | 11.8 | PRKAR1A | c.101_105delCTATT/p.Ser34fsX9 |
| 2C | CAR599.02 | F | 41 | CNC/PPNAD | BADX | NA | NA | NA | NA | 13 | 16 | PRKAR1A | c.682C>T/p.Arg228X |
| 3C | CAR 616.02 | F | 30 | CNC/PPNAD | BADX | 7×6.5×1.5 | 10.5×4.5×2.5 | 43 | 51 | 13.5 | 14.2 | PRKAR1A | c.491_492delTG/p.Val164fsX4 |
| 4C | CAR 047.01 | F | 35 | CNC/PPNAD | BADX | 4.5×2×1 | 5.5×4×1.5 | 17 | 12 | 8.7 | 13.7 | PRKAR1A | c.177+1G>A |
| 5C | CAR 053.03 | F | 22 | CNC/PPNAD | BADX | 5×3×1.8 | 5×2.5×2 | NA | NA | 32.2 | 31.2 | PRKAR1A | c.43_58del16/p.Leu15fs 104X |
| 6C | CAR 020.14 | M | 13 | CNC/PPNAD | BADX | 4.5×3×0.5 | 5×3×0.5 | 13 | 13.4 | 3.8 | 8.8 | PRKAR1A | c.491_492delTG/p.Val164fsX4 |
| 7C | CAR 79.03 | M | 5 | CNC/PPNAD | BADX | 3×1.3×0.8 | 3×1.5×.8 | NA | NA | 6.6 | 22 | PRKAR1A | c.496C>T/p.Gln166X |
| 8C | CAR583.03 | F | 10 | iPPNAD | BADX | NA | NA | NA | NA | NA | NA | – | None |
| 1D | ADT07.01 | F | 51 | CPA | UADX-left | – | 7×3.5×2.8 | – | 37 | 6 | 24 | – | None |
| 2D | CAR070.03 | F | 16 | CPA | UADX-left | – | 5.2×3.5×3.5 | – | NA | 19.6 | 19.5 | – | None |
| 3D | ADT54.03 | F | 18 | CPA | UADX-left | – | 5.5×4×3.5 | – | 23.7 | 22.9 | 18.4 | – | None |
| 1E | A1 | F | 44 | APA | NA | NA | NA | NA | NA | NA | 33.5 | – | None |
| 2E | ADT42.01 | M | 58 | APA | UADX-right | 8.6×4.2×3.1 | – | NA | – | 4.5 | 3.3 | – | None |
| 3E | ADT17.01 | M | 44 | APA | UADX-right | 4 | – | NA | – | 4.1 | 13.3 | – | None |
| 4E | A10 | F | 52 | APA | NA | NA | NA | NA | NA | NA | NA | – | None |
| 5E | ADT02.01 | M | 48 | APA | UADX-left | – | 6.5×4.5×5 | – | NA | 2.8 | 12.4 | – | None |
age (year), age at diagnosis; MMAD, massive macronodular adrenocortical disease; AIMAH, ACTH-independent macronodular adrenal hyperplasia; CNC, Carney complex; PPNAD, primary pigmented adrenocortical disease; iPPNAD, isolated PPNAD; iMAD, isolated micronodular adrenocortical disease; CPA, cortisol-producing adenoma; APA, aldosterone-producing adenoma; BADX, bilateral adrenalectomy; UADX, unilateral adrenalectomy; PRKAR1A, PKA regulatory subunit 1A; normal range: cortisol: 5–25μg/dl.
All samples had been collected during surgery, immediately dissected and separated from periadrenal fat and other elements, and stored in liquid nitrogen until use. DNA sequencing of the GNAS, PRKARIA, PDE11A, and PDE8B gene was completed as published elsewhere 8, 12, 19.
Assays for cAMP levels and binding activity, and PKA activity
Levels of cAMP were measured in tissue extracts using the cAMP 3H Biotrak Assay System (Amersham Biosciences). Samples were homogenized in ethanol and centrifuged at maximum 1000g for 10min. The supernatant was dried and resuspended in 50μl of assay buffer according to the manufacturer's instructions. Two samples were prepared from each surgical specimen and assayed.
The Gilman procedure was used to measure the cAMP-binding activity 20. Protein extract (see PKA activity) of 50μg in a total volume of 0.4ml, containing 50mM of Tris 7.5, 8mM of MgCl2, and 0.15μM [3H] cAMP were used as the incubation mixture. The samples were incubated for 50min at 4°C and the reaction was stopped by adding 2ml of potassium phosphate buffer (20mM). The final mixture was passed through Millipore (Billerica, MA, USA) filters followed by washing with 15ml of the same buffer (potassium phosphate buffer). The radioactivity was then assessed using liquid scintillation counting.
PKA activity was measured following the protocol described earlier by Nesterova et al. 21, 22. Tissue samples were extracted in 10mM Tris–HCl, pH 7.5, 1mM EDTA, and 0.1mM dithiothreitol (DTT) protease inhibitor cocktail I (EMD Biosciences, Darmstadt, Germany). About 10μg protein of the tissue extracts were added to the reaction mixture (50μl) containing 0.025mM [γ-32P] ATP, 5μM kemptide, 10mM MgCl2±5μM cAMP, and 5μM PKA inhibitor (PKI). The mixture was incubated for 15min at 30°C, spotted on phosphocellulose filters, and washed for three times using 0.1% phosphoric acid. The filters were left to air dry before analysis by liquid scintillation counter.
Basal levels of PKA activity represent the non-stimulated PKA activity. Total PKA activity reflects the PKA activity after the addition of cAMP; free PKA activity represents the difference in PKA activity without the addition of cAMP and that without cAMP but with PKI. Also, we calculated the PKA activity ratio; basal PKA activity/total PKA activity, to determine how much PKA is present in its active state.
PDE activity
A colorimetric method was used according to recommended protocol in order to determine the PDE activity in all our samples; BIOMOL GREENTM Reagent supplied by QuantiZymeTM Assay System, BIOMOL International, LP (Plymouth Meeting, PA, USA). This PDE assay is mainly based on the degradation of cAMP and cGMP by a cyclic nucleotide PDE and the subsequent counting of the phosphate released, using the BIOMOL GREEN Reagent. Also, a non-specific cyclic nucleotide PDE inhibitor, 3-isobutyl-1-methylxanthine, was used in order to study the degree of PDE inhibition in our samples.
Preparation of proteins, western blotting and immunohistochemistry
Tissues from patients and their tumors were processed as previously published 12, 18. Total cellular protein extracts from frozen tissues or cultured cells were prepared using RIPA buffer (20mM Hepes, 250mM NaCl, 10% glycerol, 1% NP-40, 0.5% deoxycholate, 2mM DTT, and protease inhibitor). Twenty micrograms from cell lysates (and 50μg from tissue lysates) of total protein were subjected to SDS/PAGE using a 4–20% gradient gel. The proteins were transferred to nitrocellulose membranes, and CREB and phosphor (P) CREB were detected by western blotting using polyclonal antibodies 12, 18 that are commercially available (Upstate, Waltham, MA, USA). The same antibodies were also used for immunohistochemistry (IHC). An antibody specific for PDE11A was also used for IHC as directed by the manufacturer (Abcam, Cambridge, MA, USA) at 1:500, and 1:100 dilutions, as described previously 12, 18 on paraffin-embedded tissue slides. Similar methods were used for the CREB and p-CREB immunostaining (western blot and IHC).
All slides used for IHC were then scored by at least two reviewers that were blinded to the genetic defects of these lesions or the clinical diagnosis, as we have done elsewhere 15, 19.
Statistical analysis
All assays were obtained in duplicate measurements and an average was calculated for each value. Comparisons were made using a two-sample t-test and P values were considered significant at P<0.05.
Results
Genetic studies
A total of 27 patients with AICS were studied (Table 1); both tumor and peripheral DNA were studied for GNAS, PRKARIA, PDE11A, and PDE8B gene mutations and allelic losses of the respective chromosomal loci (data not shown). Only the PPNAD patients had germline mutations of the PRKAR1A gene; these mutations are listed in Table 1. Patients with APAs 5 were operated for hyperaldosteronism; normal adrenal tissue was available from four subjects.
PKA activity
PKA activity was determined with and without the addition of cAMP and PKI in all samples (Table 2). We then used the ratio between ‘basal’ and ‘total’ activity (Fig. 1A) because of individual variability and traditionally this is the best way of assessing PKA activity 20, 21, 22. We termed ‘basal’ all PKA activities that were detected without the addition of cAMP; we called ‘free’ the difference between PKA activity without the addition of cAMP and that without cAMP but with PKI. After the addition of cAMP and PKI, total PKA activity was practically returning to activity without cAMP.
Table 2.
Protein kinase A (PKA) activity, cAMP levels, cAMP binding, and phosphodiesterase (PDE) activity in the studied tissues.
| PKA activity | ||||||||
|---|---|---|---|---|---|---|---|---|
| Basal PKA | Total PKA | (−) cAMP (+) PKI | Free PKA | PKA activity ratio | cAMP levels assay | cAMP-binding activity | PDE activity | |
| BAHs | ||||||||
| MMAD | ||||||||
| 1A | 1.91 | 10.8 | 0.26 | 1.65 | 0.18 | 6.73 | 24.83 | 7.31 |
| 2A | 8.47 | 47.05 | 0.72 | 7.75 | 0.18 | 10.44 | 29.82 | 4.69 |
| 3A | 0.95 | 11.77 | 0.24 | 0.71 | 0.08 | 8.05 | 42.21 | 7.47 |
| 4A | 5.26 | 31.64 | 0.54 | 4.71 | 0.17 | 4.51 | 28.14 | 1.18 |
| 5A | 4.22 | 26.92 | 0.1 | 4.12 | 0.16 | 7.65 | 13.72 | 5.9 |
| 6A | 1.56 | 14.75 | 0.33 | 1.23 | 0.11 | 7.64 | 24.98 | 7.88 |
| 7A | 2.17 | 9.21 | 0.06 | 2.1 | 0.24 | 6.24 | 22.28 | 0 |
| 8A | 5.89 | 24.13 | 0.31 | 5.59 | 0.24 | 9.06 | 36.01 | 0.52 |
| 9A | 19.9 | 50.32 | 0.41 | 19.48 | 0.4 | 7.65 | 48.56 | 3.51 |
| 10A | 19.43 | 49.05 | 0.46 | 18.97 | 0.4 | 7.03 | 44.11 | 3.54 |
| 11A | 1.8 | 7.89 | 0.22 | 1.58 | 0.23 | 4.46 | 3.68 | 3.61 |
| 12A | 4.43 | 10.73 | 0.46 | 3.97 | 0.41 | 8.14 | 8.67 | 0.88 |
| 13A | 1.39 | 9.17 | 0.16 | 1.22 | 0.15 | 5.91 | 14.09 | 0.09 |
| iMAD | ||||||||
| 1B | 6.22 | 21.2 | 0.28 | 5.93 | 0.29 | 1.47 | 13.72 | 6.68 |
| 2B | 6.1 | 16.71 | 0.52 | 5.57 | 0.36 | 2.83 | 16.87 | 8.84 |
| 3B | 5.95 | 12.8 | 0.87 | 5.08 | 0.47 | 7.63 | 15.02 | 0 |
| PPNAD | ||||||||
| 1C | 1.48 | 7.8 | 0.15 | 1.33 | 0.19 | 5.36 | 8.35 | 4.11 |
| 2C | 7.5 | 27.76 | 0.6 | 6.9 | 0.27 | 7.32 | 11.68 | 8.6 |
| 3C | 11.65 | 49.52 | 0.47 | 11.18 | 0.24 | 10.35 | 15.71 | 4.51 |
| 4C | 11.2 | 33.43 | 0.83 | 10.37 | 0.33 | 11.81 | 23.9 | 19.57 |
| 5C | 12.34 | 45.59 | 0.49 | 11.85 | 0.27 | 2.75 | 9.18 | 9.17 |
| 6C | 9.17 | 33.98 | 0.19 | 8.98 | 0.27 | 2.56 | 0.89 | 1.87 |
| 7C | 1.53 | 37.13 | 0 | 1.65 | 0.04 | 6.67 | 9.41 | 3 |
| 8C | 11.05 | 88.65 | 0.09 | 10.97 | 0.12 | 2.26 | 0 | 2.06 |
| CPA | ||||||||
| 1D | 15.89 | 46.32 | 0.28 | 15.62 | 0.34 | 3.02 | 37.43 | 1.72 |
| 2D | 8.59 | 20.76 | 0.25 | 8.34 | 0.41 | 9.74 | 24.71 | 3.76 |
| 3D | 7.14 | 18.48 | 0.42 | 6.73 | 0.39 | 6.12 | 40.22 | 0.94 |
| APA | ||||||||
| 1E | 11.36 | 9.29 | 0 | 2.79 | 0.29 | 17.23 | 27.89 | 5.14 |
| 2E | 24.26 | 37.21 | 0.03 | 12.65 | 0.34 | 7.46 | 12.24 | 0 |
| 3E | 6.54 | 39.23 | 0 | 10.27 | 0.26 | 6.9 | 33.46 | 4.35 |
| 4E | 8.12 | 23.98 | 0.23 | 11.14 | 0.47 | 12.33 | 13.01 | 5.39 |
| 5E | 2.7 | 58.4 | 0.39 | 33.04 | 0.57 | 14.73 | 0 | 5.21 |
| Normal | ||||||||
| 1F | 12.68 | 23.88 | 0.35 | 11.01 | 0.48 | 2.6 | 15.09 | 5.79 |
| 2F | 10.18 | 43.48 | 0.09 | 24.17 | 0.56 | 2.74 | 8.57 | 8.12 |
| 3F | 11.38 | 22.79 | 0.12 | 6.42 | 0.29 | 2.72 | 9.83 | 3.46 |
| 4F | 33.43 | 23.67 | 0.25 | 7.87 | 0.34 | 2.16 | 17.02 | 6.12 |
BAHs, bilateral adrenal hyperplasias; PRKAR1A, PKA regulatory subunit 1A; MMAD, massive macronodular adrenocortical disease; PPNAD, primary pigmented adrenocortical disease; iMAD, isolated micronodular adrenocortical disease; CPA, cortisol-producing adenoma; APA, aldosterone-producing adenoma. Units, cAMP levels, arbitrary units/mg; cAMP-binding activity, c.p.m./μg of protein; PDE activity, arbitrary units/mg.
Figure 1.

(A–D) PKA activity ratio, cAMP levels, cAMP-binding, and PDE activity in BAHs without PRKAR1A mutations, PPNAD (due to PRKAR1A mutations), CPAs, APAs, and normal adrenal tissue (*statistically significant compared with normal samples); (E and F) staining for PDE11A, a tissue affected by PPNAD caused by a PRKAR1A-inactivating mutation (5× and 40× respectively); the nodular (Nod) cells stain strongly for PDE11A; (G) the opposite is true in a sporadic CPA: the nodular tissue (Nod) stains for PDE11A less strongly than the surrounding normal cortical (C) tissue.
All adrenal hyperplasias (6.73±5.4c.p.m./μg) had statistically significant lower basal PKA levels than normal adrenal (16.92±11.05c.p.m./μg, P=0.0059) and lower basal PKA levels than APAs (10.59±8.25c.p.m./μg, P>0.1). Upon stimulation with cAMP, there were no significant differences (P>0.05); APAs: 24.62±12.2 c.p.m./μg, normal adrenal gland: 39.7±1.4c.p.m./μg, CPA: 28.5+15.4c.p.m./μg, iMAD: 16.9+4.2c.p.m./μg, MMAD/AIMAH: 23.3+16.3c.p.m./μg; PPNAD had the highest stimulation of PKA activity in response to cAMP, as we have published elsewhere 8: 40.4+23.2c.p.m./μg. Overall, adrenal hyperplasias without PRKAR1A mutations (0.25+0.12c.p.m./μg) as well as PPNAD (0.21+0.09c.p.m./μg) had significantly lower PKA activity ratio in comparison with normal adrenal (0.42+0.14c.p.m./μg; P=0.033 and P=0.016 respectively). PKA activity ratio was higher in CPAs (0.38+0.04c.p.m./μg) than in PPNAD (P=0.02). Within BAHs, PPNAD had lower PKA activity ratio than iMAD (0.37+0.08; P=0.033).
cAMP levels and cAMP binding
All lesions had higher cAMP levels than normal adrenal tissue, including APAs (Fig. 1B; Table 2). When subgrouped, CPAs (6.3±3.4 arbitrary units/mg) were not different from BAHs without PRKAR1A mutations (6.6±2.3 arbitrary units/mg) or PPNAD caused by PRKAR1A mutations (6.1±3.6 arbitrary units/mg).
In addition, all lesions associated with AICS (with the exception of PPNAD) had higher cAMP-binding activity (Fig. 1C; Table 2) than normal adrenal tissue (12.6±4.07c.p.m./μg). CPAs had the highest cAMP-binding activity (34.1±8.3c.p.m./μg; P=0.006). PPNAD, a disease that is caused by inactivating mutations of the most important cAMP receptor, PRKAR1A, had the lowest cAMP binding activity (9.78±7.8c.p.m./μg); both CPAs (34.1±8.307c.p.m./μg; P=0.001) and BAHs without PRKAR1A mutations (24.17±13.2; P=0.01) had statistically significant higher cAMP-binding activity than PPNAD.
PDE activity
CPAs had the lowest PDE activity (Fig. 1D, Table 2) compared with normal adrenal (2.1±1.4 vs 5.8±2 arbitrary units/mg, P=0.037) and APAs (3.64±3.1 vs 2.1±1.4 arbitrary units/mg, P>0.1). Adrenal hyperplasias as a group (3.79±3.29 arbitrary units/mg) had lower PDE activity from normal tissue (P>0.1); MMAD/AIMAH was closer to CPAs and tended to be different from normal adrenal (P=0.1).
These data suggested that lesions with PRKAR1A mutations have higher PDE activity, whereas sporadic CPAs have lower PDE activity. We then used an antibody for a highly expressed PDE in the adrenal, PDE11A, to stain paraffin-fixed slides from five PPNAD specimens and three sporadic CPAs; consistent with the above biochemical data, all PPNADs showed high expression of PDE11A (Fig. 1E) and especially within the cortisol-producing nodules (Fig. 1F). In contrast, all three sporadic CPAs (that did not have any mutations in any of the tested genes) had lower PDE11A expression within the tumor tissue compared with surrounding normal adrenal cortex (Fig. 1G).
CREB and P-CREB western blotting and IHC
We assessed CREB and P-CREB in all tissues by western blotting; there were no statistically significant different ratios of P-CREB versus CREB between the groups, including CPAs and APAs (data not shown). Since this was inconsistent with the above data, we wanted to explore this further. We then specifically analyzed three tumors with known protein-truncating PDE11A mutations that we have published elsewhere (12), which clearly had decreased PDE activity and increased cAMP levels (12, 14, 19), and compared their data with the lesions studied here (Fig. 2). The P-CREB/CREB ratio was significantly increased in iMAD with PDE11A mutations compared with all other lesions tested and the normal adrenal samples, as shown in Fig. 2.
Figure 2.

P-CREB to CREB ratio (measured by optical density of western blots) using tissue lysates from normal adrenals, iMAD associated with PDE11A-inactivating mutations, PPNAD due to PRKAR1A mutations, and MMAD/AIMAH.
Immunostaining for CREB and P-CREB of CPA (3), iMAD without PDE11A mutations (3), iMAD with PDE11A mutations (4), and PPNAD with PRKAR1A mutations (5) showed that, indeed, only tumors with PDE11A mutations (described in 12) have a significantly higher CREB and P-CREB expression (Fig. 3).
Figure 3.
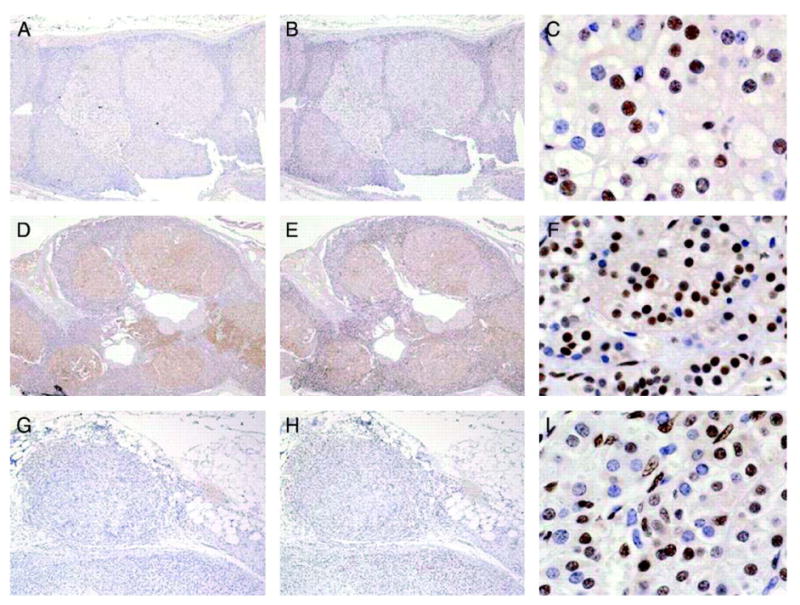
Immunostaining for CREB (A, D, and G) and P-CREB (B, C, E, F, H and I) of adrenocortical tissue from a patient with AICS and multiple CPAs who did not have any germline or somatic mutations in any of the genes tested (A–C), a patient with a germline PDE11A protein-truncating mutation (D–F), and a patient with PPNAD due to a PRKAR1A-inactivating mutation.
Discussion
In the present study, we investigated cAMP levels, cAMP-binding activity, PKA, and PDE activities in lesions associated with AICS (adrenal hyperplasias and adenomas) that had no mutations in the GNAS, PRKAR1A, PDE11A, and PDE8B genes and compared these data with those derived from samples with PPNAD and PRKAR1A mutations, and a limited number of normal adrenal glands and APAs. The data show that these benign tumors have abnormalities in the cAMP pathway; for example all ADTs tested had higher cAMP levels than normal adrenal tissue (Table 2). Likewise, cAMP-binding activity was higher than normal adrenal tissue in all non-PRKAR1A-related lesions, independently of the histology, CPAs, and BAHs without PRKAR1A mutations (MMAD/AIMAH and iMAD). PPNAD that is due to inactivating PRKAR1A mutations 8, 9, 10 was expected to have low cAMP-binding activity and it had so.
These tumors were not tested for aberrant expression of GPCR, but none of these lesions bore any of the known mutations of the cAMP pathway. Although aberrant GPCR expression could explain the higher cAMP levels 3, 6 in at least MMAD/AIMAH, cAMP-binding activity should not be affected by this phenomenon 20, 21, 22. Alterations of cAMP endocellular receptors, as well as changes in the kinetics of cAMP responsiveness, are probably responsible for this observation.
Furthermore, the PKA activity ratio was decreased in BAHs compared with normal tissue. This finding was expected for samples bearing PRKAR1A mutations but not for samples from other BAHs, such as iMAD and MMAD/AIMAH that did not contain PRKAR1A or 17q22–24 defects 16. PKA activity ratio indicates the amount of PKA that is in its active form 8, 20, 21, 22. These data suggest that functional abnormalities of the PKA enzyme may be present in BAHs even when PRKAR1A and its chromosomal locus are not involved.
PDE activity was measured for the first time in CPAs; our data indicated a marked decrease in PDE activity in CPAs and a (non-significant) decrease in BAHs without PRKAR1A mutations in comparison with normal adrenal. These were lesions that did not have mutations in the known PDE genes that are mutated in ADTs (PDE11A and PDE8B), indicating that, perhaps, additional PDEs or genes that regulate the expression of the latter are involved in the pathogenesis of benign adrenocortical lesions. In fact, one of the reasons that we had so few CPAs in this study was that we had very few that did not have even benign polymorphisms of the PDE11A gene. PDE11A gene variants are very frequent in ADTs 14, 17, 18, but the present data suggest that additional PDEs may be involved.
If abnormalities of the cAMP-signaling pathway are present in adrenal lesions associated with AICS, then CREB and P-CREB should be altered in these lesions; our data showed that this was not the case by western blotting in tissue lysates; only adrenal lesions with germline PDE11A mutations had an increased P-CREB/CREB ratio (Fig. 2). Even samples with known PRKAR1A mutations (PPNAD), which have a known up-regulation of the cAMP-signaling/PKA system 8, 9, 10, 11, had P-CREB/CREB ratio comparable with normal adrenal tissue.
We suspect that the reason for this discrepancy is that in CPAs with low PDE activity the postulated defect is somatic, present only within the tumor tissue and not in the surrounding tissue. In a tissue lysate, it is impossible to separate the tumor tissue from surrounding cells, but within the nodule by IHC one does see an increase in nuclear staining of P-CREB (Fig. 3A–C). Patients with PDE11A germline defects have abnormal PDE activity in all cells that is clearly leading to up-regulation of the CREB and P-CREB as shown in Fig. 3(D–F) in all cells, both adenomatous and of the surrounding cortex. On the other hand, patients with germline PRKAR1A defects have up-regulation of the PKA system (and consequently of the CREB and P-CREB molecules) only after loss of the normal allele; this occurs only within the nodules and not in the surrounding cortex (Fig. 3G–I).
We conclude that almost all types of benign lesions of the adrenal gland that were tested in this study exhibited functional abnormalities of the cAMP-signaling pathway. These data indicate that more genetic defects of regulatory molecules of this pathway exist and remain to be identified; they also suggest that pharmacological modulation of the cAMP-signaling pathway could potentially be explored in the treatment of CS caused by benign adrenocortical lesions.
Acknowledgments
The present work was supported by the United States National Institutes of Health, Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) intramural project Z01-HD-000642-04 to Dr C A Stratakis. Dr Bimpaki was, in part, supported by the Faculty of Medicine, University of Crete, Heraklion, Crete, Greece, Postgraduate & Master Program on the Molecular Basis of Human Disease.
References
- 1.Stratakis CA. Cushing syndrome caused by adrenocortical tumors and hyperplasias (corticotropin-independent Cushing syndrome) Endocrine Development. 2008;13:117–132. doi: 10.1159/000134829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stratakis CA, Boikos SA. Genetics of adrenal tumors associated with Cushing's syndrome: a new classification for bilateral adrenocortical hyperplasias. Nature Clinical Practice. Endocrinology and Metabolism. 2007;3:748–757. doi: 10.1038/ncpendmet0648. [DOI] [PubMed] [Google Scholar]
- 3.Bourdeau I, Lampron A, Costa MH, Tadjine M, Lacroix A. Adrenocorticotropic hormone-independent Cushing's syndrome. Current Opinion in Endocrinology, Diabetes and Obesity. 2007;14:219–225. doi: 10.1097/MED.0b013e32814db842. [DOI] [PubMed] [Google Scholar]
- 4.Boston BA, Mandel S, LaFranchi S, Bliziotes M. Activating mutation in the stimulatory guanine nucleotide-binding protein in an infant with Cushing's syndrome and nodular adrenal hyperplasia. Journal of Clinical Endocrinology and Metabolism. 1994;79:890–893. doi: 10.1210/jcem.79.3.8077378. [DOI] [PubMed] [Google Scholar]
- 5.Fragoso MC, Domenice S, Latronico AC, Martin RM, Pereira MA, Zerbini MC, Lucon AM, Mendonca BB. Cushing's syndrome secondary to adrenocorticotropin-independent macronodular adrenocortical hyperplasia due to activating mutations of GNAS1 gene. Journal of Clinical Endocrinology and Metabolism. 2003;88:2147–2151. doi: 10.1210/jc.2002-021362. [DOI] [PubMed] [Google Scholar]
- 6.Mircescu H, Jilwan J, N'Diaye N, Bourdeau I, Tremblay J, Hamet P, Lacroix A. Are ectopic or abnormal membrane hormone receptors frequently present in adrenal Cushing's syndrome? Journal of Clinical Endocrinology and Metabolism. 2000;85:3531–3536. doi: 10.1210/jcem.85.10.6865. [DOI] [PubMed] [Google Scholar]
- 7.Bertherat J, Contesse V, Louiset E, Barrande G, Duparc C, Groussin L, Emy P, Bertagna X, Kuhn JM, Vaudry H, Lefebvre H. In vivo and in vitro screening for illegitimate receptors in adrenocorticotropin-independent macronodular adrenal hyperplasia causing Cushing's syndrome: identification of two cases of gonadotropin/gastric inhibitory polypeptide-dependent hypercortisolism. Journal of Clinical Endocrinology and Metabolism. 2005;90:1302–1310. doi: 10.1210/jc.2004-1256. [DOI] [PubMed] [Google Scholar]
- 8.Kirschner LS, Carney JA, Pack SD, Taymans SE, Giatzakis C, Cho YS, Cho-Chung YS, Stratakis CA. Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nature Genetics. 2000;26:89–92. doi: 10.1038/79238. [DOI] [PubMed] [Google Scholar]
- 9.Groussin L, Jullian E, Perlemoine K, Louvel A, Leheup B, Luton JP, Bertagna X, Bertherat J. Mutations of the PRKAR1A gene in Cushing's syndrome due to sporadic primary pigmented nodular adrenocortical disease. Journal of Clinical Endocrinology and Metabolism. 2002;87:4324–4329. doi: 10.1210/jc.2002-020592. [DOI] [PubMed] [Google Scholar]
- 10.Groussin L, Kirschner LS, Vincent-Dejean C, Perlemoine K, Jullian E, Delemer B, Zacharieva S, Pignatelli D, Carney JA, Luton JP, Bertagna X, Stratakis CA, Bertherat J. Molecular analysis of the cyclic AMP-dependent protein kinase A (PKA) regulatory subunit 1A (PRKAR1A) gene in patients with Carney complex and primary pigmented nodular adrenocortical disease (PPNAD) reveals novel mutations and clues for pathophysiology: augmented PKA signaling is associated with adrenal tumorigenesis in PPNAD. American Journal of Human Genetics. 2002;71:1433–1442. doi: 10.1086/344579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Groussin L, Horvath A, Jullian E, Boikos S, Rene-Corail F, Lefebvre H, Cephise-Velayoudom FL, Vantyghem MC, Chanson P, Conte-Devolx B, Lucas M, Gentil A, Malchoff CD, Tissier F, Carney JA, Bertagna X, Stratakis CA, Bertherat J. A PRKAR1A mutation associated with primary pigmented nodular adrenocortical disease in 12 kindreds. Journal of Clinical Endocrinology and Metabolism. 2006;91:1943–1949. doi: 10.1210/jc.2005-2708. [DOI] [PubMed] [Google Scholar]
- 12.Horvath A, Boikos S, Giatzakis C, Robinson-White A, Groussin L, Griffin KJ, Stein E, Levine E, Delimpasi G, Hsiao HP, Keil M, Heyerdahl S, Matyakhina L, Libè R, Fratticci A, Kirschner LS, Cramer K, Gaillard RC, Bertagna X, Carney JA, Bertherat J, Bossis I, Stratakis CA. A genome-wide scan identifies mutations in the gene encoding phosphodiesterase 11A4 (PDE11A) in individuals with adrenocortical hyperplasia. Nature Genetics. 2006;38:794–800. doi: 10.1038/ng1809. [DOI] [PubMed] [Google Scholar]
- 13.Horvath A, Mericq V, Stratakis CA. Mutation in PDE8B, a cAMP-specific phosphodiesterase in adrenal hyperplasia. New England Journal of Medicine. 2008;358:750–752. doi: 10.1056/NEJMc0706182. [DOI] [PubMed] [Google Scholar]
- 14.Horvath A, Giatzakis C, Robinson-White A, Boikos S, Levine E, Griffin K, Stein E, Kamvissi V, Soni P, Bossis I, de Herder W, Carney JA, Bertherat J, Gregersen PK, Remmers EF, Stratakis CA. Adrenal hyperplasia and adenomas are associated with inhibition of phosphodiesterase 11A in carriers of PDE11A sequence variants that are frequent in the population. Cancer Research. 2006;66:11571–11575. doi: 10.1158/0008-5472.CAN-06-2914. [DOI] [PubMed] [Google Scholar]
- 15.Bertherat J, Groussin L, Sandrini F, Matyakhina L, Bei T, Stergiopoulos S, Papageorgiou T, Bourdeau I, Kirschner LS, Vincent-Dejean C, Perlemoine K, Gicquel C, Bertagna X, Stratakis CA. Molecular and functional analysis of PRKAR1A and its locus (17q22–24) in sporadic adrenocortical tumors: 17q losses, somatic mutations, and protein kinase A expression and activity. Cancer Research. 2003;63:5308–5319. [PubMed] [Google Scholar]
- 16.Bourdeau I, Matyakhina L, Stergiopoulos SG, Sandrini F, Boikos S, Stratakis CA. 17q22–24 chromosomal losses and alterations of protein kinase a subunit expression and activity in adrenocorticotropin-independent macronodular adrenal hyperplasia. Journal of Clinical Endocrinology and Metabolism. 2006;91:3626–3632. doi: 10.1210/jc.2005-2608. [DOI] [PubMed] [Google Scholar]
- 17.Libe R, Fratticci A, Coste J, Tissier F, Horvath A, Groussin L, Rene-Corail F, Bertagna X, Raffin-Sanson ML, Stratakis CA, Bertherat J. Phosphodiesterase 11A4 (PDE11A4) and genetic predisposition to adrenocortical tumors. Clinical Cancer Research. 2008;14:4016–4024. doi: 10.1158/1078-0432.CCR-08-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boikos S, Horvath A, Heyerdahl S, Stein E, Robinson-White A, Bossis I, Bertherat J, Stratakis CA. Phosphodiesterase 11A expression in the adrenal cortex, primary pigmented nodular adrenocortical disease and other corticotropin-independent lesions. Hormone and Metabolic Research. 2008;40:347–353. doi: 10.1055/s-2008-1076694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Horvath A, Giatzakis C, Tsang K, Greene E, Osorio P, Boikos S, Libè R, Patronas Y, Robinson-White A, Remmers E, Bertherat J, Nesterova M, Stratakis CA. A cAMP-specific phosphodiesterase (PDE8B) that is mutated in adrenal hyperplasia is expressed widely in human and mouse tissues: a novel PDE8B isoform in human adrenal cortex. European Journal of Human Genetics. 2008;16:1245–1253. doi: 10.1038/ejhg.2008.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gilman AG. A protein binding assay for adenosine 3′:5′-cyclic monophosphate. PNAS. 1970;67:305–312. doi: 10.1073/pnas.67.1.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nesterova MV, Sashchenko LP, Vasiliev VY, Severin ES. A cyclic adenosine 3′,5′-monophosphate-dependent histone kinase from pig brain, Purification and some properties of the enzyme. Biochimica et Biophysica Acta. 1975;377:271–281. doi: 10.1016/0005-2744(75)90309-5. [DOI] [PubMed] [Google Scholar]
- 22.Nesterova M, Bossis I, Wen F, Horvath A, Matyakhina L, Stratakis CA. An immortalized human cell line bearing a PRKAR1A-inactivating mutation: effects of overexpression of the wild-type allele and other protein kinase A subunits. Journal of Clinical Endocrinology and Metabolism. 2008;93:565–571. doi: 10.1210/jc.2007-1902. [DOI] [PMC free article] [PubMed] [Google Scholar]