

Abstract
Chlorine (Cl2) is a reactive oxidant gas used extensively in industrial processes. Exposure of both humans and animals to high concentrations of Cl2 results in acute lung injury, which may resolve spontaneously or progress to acute respiratory failure. Injury to airway and alveolar epithelium may result from chemical reactions of Cl2, from HOCl (the hydrolysis product of Cl2), and/or from the various reaction products, such as chloramines, that are formed from the reactions of these chlorinating species with biological molecules. Subsequent reactions may initiate self-propagating reactions and induce the production of inflammatory mediators compounding injury to pulmonary surfactant, ion channels, and components of lung epithelial and airway cells. Low-molecular-weight antioxidants, such as ascorbate, glutathione, and urate, present in the lung epithelial lining fluid and tissue, remove Cl2 and HOCl and thus decrease injury to critical target biological targets. However, levels of lung antioxidants of animals exposed to Cl2 in concentrations likely to be encountered in the vicinity of industrial accidents decrease rapidly and irreversibly. Our measurements show that prophylactic administration of a mixture containing ascorbate and desferal N-acetyl-cysteine, a precursor of reduced glutathione, prevents Cl2-induced injury to the alveolar epithelium of rats exposed to Cl2. The clinical challenge is to deliver sufficient quantities of antioxidants noninvasively, after Cl2 exposure, to decrease morbidity and mortality.
Keywords: ascorbate, N-acetyl-cysteine, chlorine, alveolar epithelium, hypochlorous acid
CL2 AND ITS REACTIVE INTERMEDIATES: CHEMISTRY
Chlorine (Cl2) is a dense, acrid, pungent, greenish-yellow gas that is easily recognized by both color and odor at very low concentrations. It is noncombustible and moderately soluble at room temperature and atmospheric pressure. It is the ninth largest produced chemical by volume in the United States, transported mainly by rail to manufacturing plants for pulp bleaching, waste sanitation, and pharmaceutical manufacturing. It is also added in swimming pools to limit the number of pathogens (1). However, accidental or deliberate release of Cl2 into the atmosphere during industrial accidents has been associated with significant morbidity and mortality (1, 2). During the last few years, Cl2 cylinders have been bundled with traditional explosives, raising significant concerns regarding the possible reemergence of this agent as a chemical weapon against both combatants and civilians (3). The density of Cl2 is greater than that of air; therefore, it has a tendency to settle in low-lying areas, which may affect exposures on a microenvironmental scale.
Cl2 is a highly reactive oxidant gas and has higher solubility in water than nitrogen dioxide and ozone, two well-known environmental pollutants (4). When inhaled, Cl2 dissolves in the epithelial lung lining fluid (ELF), a thin layer of fluid covering the apical surfaces of airway and distal lung epithelial cells, and then reacts with biological molecules, such as low-molecular-weight antioxidants (5). As the concentrations of antioxidants are decreased, Cl2 undergoes hydrolysis to generate hypochlorous (HOCl) and hydrochloric (HCl) acids according to the following reactions:
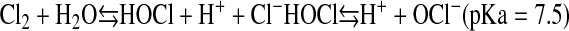 |
Bicarbonate in the ELF buffers and neutralizes HCl, limiting its toxicity (6). Consequently, Cl2 toxicity has been attributed to HOCl and its conjugate base (7). We have recently shown that Cl2 reacts with low-molecular-weight antioxidants in the ELF with rates exceeding its hydrolysis (5). Both Cl2 and HOCl/OCl− may contribute to the initiation and propagation of lung injury.
REACTIONS OF CL2 AND ITS REACTIVE INTERMEDIATES WITH BIOLOGICAL TARGETS
The main targets of Cl2, HOCl, and OCl− are functional groups of proteins and amino acids such as cysteine (8, 9), methionine, side-chain amino groups in amino acids, terminal amino groups, yielding the corresponding chloramines (10), and aromatic amino acids (such as tyrosine), yielding 3-chlorotyrosine (11–13). Chloramines are toxic and both contribute to and compound the injurious effects of Cl2/HOCl/OCl− to biological tissues (14–17). Accidental inhalation of chloramines formed by mixing cleaning products containing sodium hypochlorite with ammonia may cause respiratory injury that may require emergency treatment (18). HOCl also damages DNA and poly-ADP-ribose, a DNA repair enzyme (19, 20).
There is also evidence to suggest that chloramines and HOCl may activate inflammatory cascades. For example, HOCl activates the mitogen-activated protein kinase pathway (21), while the cell-permeable glycine and taurine chloramines regulate NF-κB activity via oxidation of its inhibitor IκBa (22). Activated inflammatory cells, attracted into the alveolar space by chemokines and cytokines, release myeloperoxidase and large amounts of HOCl (0.1–1 mM) in the vicinity of airway and alveolar epithelial cells (23). Also, exposure of mice to Cl2 gas up-regulates the inducible form of nitric oxide synthase (iNOS) in alveolar epithelial cells and macrophages (24). Thus, exposure to Cl2 gas initiates a series of biochemical reactions that could be detrimental to the host, even after the end of the exposure period.
Inhalation of Cl2 at concentrations less than 50 ppm causes reversible bronchospasm, increased mucous production and airway resistance, and decreased respiratory rate in humans and animals (25–30). There is little alveolar injury, since most Cl2 penetrates no deeper than the upper airways (31). Bessac and coworkers (32, 33) showed that low concentrations of aerosolized HOCl, as well as other oxidants, activate transient receptor potential ankyrin 1 (TRPA1) ion channels, located in a subset of airway sensory neurons, through covalent modification of the channel protein. Trpa1−/− mice exhibited reduced respiratory depression in response to low concentrations of aerosolized HOCl compared with wild-type mice (32, 34). These findings suggest that activation of TRPA1 channels by low concentrations of Cl2, HOCl, and other oxidants contributes to the neurological and airway responses mentioned above. In the short term, decreased respiratory rate can limit the amount of inhaled Cl2 and thus lower the extent of lung injury. The role of TRPA1 channels in the initiation and progression of lung injury in mice exposed to higher concentrations of Cl2 (> 100 ppm) is currently under investigation.
At higher concentrations (those encountered during industrial accidents or terrorist attacks), inhaled Cl2 molecules penetrate into distal lung regions (31) and may damage critical components of alveolar epithelial cells and the pulmonary surfactant system, resulting in severe hypoxemia, pulmonary edema, and even death from respiratory failure (28, 35–40). In additional studies, we exposed surfactant protein (SP)-A to various concentrations of HOCl. This resulted in oxidized and chlorinated amino acids in its carbohydrate recognition domain and decreased its ability to bind mannose residues, an important step in the binding and killing of pathogens (41). SP-D, like SP-A, is a lectin and plays diverse and important roles in innate immunity and pulmonary homeostasis. Exposure of SP-D to HOCl decreased it ability to aggregate pathogens, and resulted in the generation of abnormal disulfide cross-linked oligomers (42). These findings indicate that animals exposed to Cl2 may be at risk of bacterial infections due to compromised innate immunity.
Lung epithelial cells actively transport Na+ ions from the alveolar to the interstitial spaces: Na+ ions enter the apical membranes of epithelial cells mainly through amiloride-sensitive Na+ (ENaC) channels and are transported across the basolateral membranes by the Na+,K+-ATPAse (43). Active Na+ transport limits the degree of alveolar edema following damage to the alveolar epithelium; patients with acute lung injury that maintain the ability to concentrate alveolar proteins (as a result of active Na+ reabsorption) have better prognosis than those who cannot (44, 45). Exposure of C57BL/6 and BALB/c mice to Cl2 (50–500 ppm for 30 min) decreased Na+-dependent alveolar fluid clearance (AFC) from 28 ± 0.7% to 15 ± 4% (% of instilled volume/30 min, n = 3–23, P < 0.05), at 1 hour after exposure. This decrease was due to specific injury to ENaC, the rate-limiting pathways in the vectorial transport of Na+ ions across epithelial cells (17). Na+,K+-ATPAse was most likely damaged as well. Major lung airway and alveolar epithelial targets of Cl2 are shown in Figure 1.
Figure 1.

Schematic representation of lung epithelial targets and reactive intermediates formed in the lung epithelial fluids during inhalation of Cl2. Diagram on top shows sites of penetration of Cl2 (upper airways for inhaled Cl2 < 50 ppm; distal lung regions for Cl2 > 50 ppm). The depths of the airway (in blue color) and alveolar (yellow color) epithelial lining fluids are not in scale (thickness of ALF and epithelial lining fluid [ELF] = 5–10 and 0.1–0.24 μm, respectively; reviewed in Reference 61). Some potential cellular targets such as cilia, surfactant monolayer, tubular myelin, surfactant protein A and D, ion channels, Na+,K+-ATPase, airway and alveolar cells, alveolar macrophages, and NF-κB are shown in the diagram. GSH = reduced glutathione; ASC = ascorbate; X = secondary reactive intermediates (such as chloramines) formed by the interaction of HOCl and Cl2 with proteins and lipids. A sensory nerve that contains TRPA1 channels (red dots) is also shown. Cl2 molecules (prior to being hydrolyzed to HOCl) may attack components of the surfactant monolayer at the surface of the alveolar epithelial lining fluid, tubular myelin, surfactant proteins, and proteins on the surface of alveolar type II cells because of the closer proximity of these targets to the air–liquid interface (5).
Recent findings suggest that inhaled Cl2 inhibits eNOS-dependent vessel dilation via decreased eNOS expression (Rakesh P. Patel, University of Alabama; personal communication). It has been reported that exposure of mice to ozone causes vascular dysfunction, oxidative stress, mitochondrial damage, and atherogenesis (46). The deleterious effects of inhaled oxidant gases may extend to extrapulmonary sites, resulting in long-term systemic abnormities.
LUNG ANTIOXIDANT DEFENSES
During normal metabolism, steady-state levels of reactive oxygen and nitrogen species are kept at very low levels by the existence of various antioxidant enzymes (such as superoxide dismutase, glutathione peroxidase, glutathione-S-transferase, catalase, and thioredoxin) as well as low-molecular-weight antioxidants (such as glutathione, urate, ascorbate, Vitamin E, etc.) (47–50). Significant levels of enzymatic antioxidants (such as the extracellular form of superoxide dismutase), as well as low-molecular-weight scavengers (reduced glutathione, ascorbate, and urate) also exist in the ELF and help decrease steady-state concentrations of extracellular and intracellular oxidants to low levels (49, 51). A variety of transition metal ion binding molecules, such as ceruloplasmin, transferrin, ferritin, and bilirubin, prevent the formation of hydroxyl radicals and secondary self-propagating reactions by maintaining very low levels of free iron. Normal ELF contains plasma proteins (mainly albumin), the concentration of which increase significantly after inhalation of Cl2 (35). Albumin has significant antioxidant capacity (52), and administration of albumin to patients with acute lung injury decreased carbonyl content in their plasma, an index of oxidative stress (53). Concentrations of major antioxidants in the human and rodent bronchoalveolar lavage fluid (BAL) are shown in Table 1.
TABLE 1.
CONCENTRATIONS OF LOW-MOLECULAR-WEIGHT ANTIOXIDANTS IN THE BRONCHOALVEOLAR LAVAGE OF RATS, RABBITS, AND HUMANS
| Rats (35*) | Rabbits (62) | Humans (63) | Humans (62) | Humans (64) | |
|---|---|---|---|---|---|
| Asc (μM) | 17 ± 1.5 | 2.5 ± 0.1 | 0.5 ± 0.05 | 0.5 ± 0.05 | 0.64 (0.4–0.9) |
| Urate (μM) | 0.7 ± 0.2 | 0.4 ± 0.04 | 0.7 ± 0.04 | 0.7 ± 0.04 | 0.74 (0.6–0.8) |
| GSH (mM) | 0.5 ± 0.05 | 1.1 ± 0.2 | 1.6 ± 0.2 | 1.5 ± 0.2 | 0.7 (0.45–0.9) |
| (GSH/GSSG) | 10.5 ± 2 | 2.2 | 49 | 50 | 68 |
| Protein (mg/100 ml) | 16 ± 1.5 | – | 7 | 7 ± 1 | 8 |
Definition of abbreviations: Asc = ascorbate, GSH = reduced glutathione; GSH/GSSG = ratio of reduced to oxidized glutathione.
Values are means ± 1 SEM or means with ranges shown in parentheses.
Numbers in parentheses in column headings indicate reference numbers.
There is significant interest in identifying FDA-approved agents that decrease acute lung injury. Vitamin C or L-ascorbic acid is the natural L-enantiomer of ascorbic acid. Ascorbic acid is a facile reductant and an important hydrophilic antioxidant. At physiological pH most of the ascorbic acid is present as ascorbate, acting as an antioxidant by donating one or two electrons to oxidants or free radicals. Ascorbate reacts directly with hydroxyl radicals and reduces Vitamin E radicals (generated during reduction of lipid hydroperoxides or peroxyl radicals [54]). It also reduces HOCl, yielding Cl− and H2O. Reduced glutathione (GSH) is the most abundant of the intracellular free thiols, is present in high concentrations in the normal ELF (51), and reduces lipid peroxides and hydrogen peroxide via a reaction catalyzed by glutathione peroxidase. In the process GSH is oxidized to its disulfide (GSSG) (55). A decrease of GSH/GSSG ratio is considered an index of generation of oxidative stress and has been shown to be a risk factor for the development of acute respiratory distress syndrome in alcoholics (56). N-acetyl-L-cysteine (NAC) is a precursor for GSH synthesis, interacts with electrophiles via its cysteine content, and reduces cystine to cysteine, an important mechanism for intracellular lung GSH elevation in vivo (57). Administration of NAC mitigates oxidative stress in a number of organs (reviewed in References 57 and 58). Ascorbate is the predominant low-molecular-weight antioxidant in rats and rabbits (which synthesize ascorbate), while GSH plays an important role in the human lung (see Table 1). A detailed account of the various enzymatic and nonenzymatic antioxidant systems in the lung can be found in a number of recent reviews (57).
EXPOSURE TO CL2 DEPLETES LUNG ASCORBATE LEVELS AND DAMAGES THE ALVEOLAR EPITHELIUM
To test the hypothesis that lung antioxidants play an important role in the responses of animals to Cl2, we pretreated rats with a mixture of antioxidants and exposed them to 184 or 400 ppm Cl2 for 30 minutes in an environmental chamber in a whole body mode (59). They were then returned to room air and the extent of lung injury was assessed by measuring the partial pressure of arterial oxygen and the concentration of protein in BAL fluid. One hour after Cl2 exposure, ascorbate levels and GSH/GSSG ratios in BAL and tissue compartments were decreased. In rats exposed to 400 ppm Cl2, ascorbate levels in BAL and lung tissues, as well as GSH/GSSG ratios in lung tissues, remained low at 24 hours after exposure, which may compromise the ability of these animals to survive a subsequent exposure to Cl2 (35).
Based on these observations and extensive literature regarding antioxidant treatment of animals with various forms of acute lung injury (48), we tested the hypothesis that enhancing lung antioxidant defenses in rats may decrease the extent of Cl2-induced lung injury. Rats were injected twice with 0.3 ml saline containing ascorbate (80 mg/kg body weight), NAC (Acetadote; 150 mg/kg), and deferoxamine (deferoxamine mesylate; 15 mg/kg). The first injection was administered intramuscularly 18 hours before Cl2 exposure, the second intravenously 1 hour before Cl2 exposure. Deferoxamine limits the production of hydroxyl radicals by scavenging iron. Both deferoxamine and NAC help maintain ascorbate in its reduced state. As shown in Figure 2 and reported previously (35), rats developed significant levels of hypoxemia and hypercapnia and increased protein levels in their BAL fluid (albumin, IgG, and IgM) 1 hour after exposure to 184 ppm Cl2 for 30 minutes. Levels of ascorbate in BAL fluid also decreased significantly. Prophylactic administration of antioxidants as described above restored PaO2 and ascorbate to normal levels and decreased BAL fluid protein concentration by about 35% (Figure 2). These studies provide proof of concept that prophylactic administration of antioxidants (commonly administered in patients for a variety of conditions) blunts Cl2-induced injury to the alveolar epithelium.
Figure 2.

Systemic administration of antioxidants decreases lung injury. Rats were injected with a mixture of antioxidants (ascorbate, NAC, and deferoxamine) or an equivalent amount of vehicle (saline), 18 hours and 1 hour before being exposed to 184 ppm Cl2 for 30 minutes, returned to room air, and killed 1 hour later. Values are means ± 1 SEM. In each case, open bars represent values from rats exposed to air, solid bars from rats exposed to Cl2 and pretreated with saline, and hatched bars from rats pretreated with antioxidants and then exposed to Cl2. PaO2 was measured in arterial samples from the carotid artery drawn during anesthesia; protein and ascorbate concentrations were measured in BAL samples. *P < 0.05 compared with the corresponding saline values. Adapted by permission from Reference 35.
ANTIOXIDANTS, ADMINISTERED AFTER CL2 EXPOSURE, REPLENISH DEPLETED STORES OF RAT LUNGS
Prophylactic administration of antioxidants for the prevention of Cl2-induced lung injury may benefit first responders and other personnel likely to be exposed to Cl2 in the immediate future. There is also a need to develop effective treatments for people that have been exposed to Cl2. We are assessing the efficacy of low-molecular-weight scavengers in decreasing lung injury when administered to animals at various intervals after Cl2 exposure. We exposed rats to 300 ppm Cl2 for 30 minutes, and then returned them to room air. At the end of exposure respiration was labored, with flaring of the nostrils during inspiration. Within 5 to 10 minutes upon return to room air, rats received an intravenous (tail vein) injection of 200 μl of sterile saline containing 20 mg ascorbic acid. One hour later, the rats were killed, a blood sample was drawn from the left ventricle, and their lungs were lavaged with 8 ml of normal saline as previously described (35). Concentrations of ascorbate in the BAL fluid and lung tissues were measured by high-pressure liquid chromatography as previously described (35), and total amounts of ascorbate in the BAL were calculated. In addition, we calculated the volume of the ELF by measuring the concentrations of urea in both the plasma and recovered BAL fluid as previously described (60). From these data we calculated the ascorbate concentration in the ELF by the following formula:
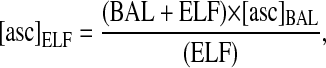 |
where BAL is the volume of saline used for lavage, [asc]BAL is the concentration of ascorbate in the BAL, and ELF is the volume of the epithelial lining fluid calculated from the following formula as previously described (60):
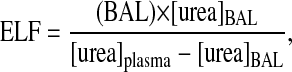 |
where [urea]BAL and [urea]plasma are the concentrations of urea in the BAL and plasma, respectively.
As shown in Figure 3, intravenous injection of ascorbate in rats after Cl2 exposure returned ELF ascorbate to control levels. Currently, we are evaluating whether replenishing decreased ascorbate levels in the alveolar spaces and lung tissues of Cl2-exposed rats and mice, by post-exposure administration of ascorbate, decreases the extent of alveolar and airway injury up to 7 days after exposure.
Figure 3.

Augmentation of lung ascorbate levels by post-exposure, intravenous administration of ascorbate. Rats were exposed to 300 ppm Cl2 for 30 minutes and then returned to room air. Within 10 minutes after exposure, they received a single injection of 20 mg ascorbic acid in 0.2 ml of normal saline. They were killed 1 hour later, their lungs were lavaged, and a blood sample was drawn from the left ventricle. Urea concentrations were measured in both the bronchoalveolar lavage (BAL) fluid and the plasma and ELF volume was calculated from the dilution of urea as previously described (60). The amounts of ascorbate in the BAL fluid and lung tissues were then measured by HPLC as previously described (35). Values are means ± 1 SEM for n = 6 rats in each group. *Significantly different from the corresponding saline group under the same conditions; #significantly different from the corresponding variable in the air group.
CONCLUSIONS
Inhalation of Cl2 concentrations likely to be encountered in the vicinity of major industrial accidents results in severe lung injury, which may cause death. Ascorbate, an important lung antioxidant in rodents, forms the first line of defense and limits injury to vital components of the alveolar epithelium such as surfactant, epithelial tight junctions, ion channels, cilia, and so on. However, lung ascorbate levels are depleted, which decreases the ability of the pulmonary system to defend from subsequent oxidant insult. Prophylactic administration of ascorbate prevents the decrease of lung ascorbate and blunts lung injury by exposure to Cl2 at concentrations likely to be encountered in industrial accidents. It remains to be seen whether post-exposure administration of antioxidants will prevent the progression of lung injury in animal models. The clinical challenge is to find noninvasive routes of antioxidant administration, and optimization of delivery methods to mass casualties within a reasonable time frame after an exposure, provided that preliminary work in animal models suggest benefit for the validation of human studies.
Acknowledgments
The authors thank Jo Anne Balanay for technical assistance; Teri Potter for editorial assistance; and Drs. Rakesh P. Patel, Sven-Eric Jordt, Jack Lancaster, Jr., Asta Jurkuvenaite, and Sotirios Zarogiannis for many helpful discussions.
The research is supported by the CounterACT Program, National Institutes Of Health Office of the Director, and the National Institute of Environmental Health Sciences, Grant Number 5P01ES011617.
Supported by PHS grants 2R01HL031197, 5 U01ES015676, and 5 U54ES017218.
Conflict of Interest Statement: A.K.Y. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. A.B. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. S.F.D. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. M.L. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. G.L.S. received more than $100,001 from the National Institutes of Health (NIH) in sponsored grants, serving as a co-investigator on various grants; $1,001–$5,000 from the NIOSH in sponsored grants, serving as a co-investigator; and more than $100,001 from Philip Morris in sponsored grants, serving as a co-investigator from 2000–2003. G.L.S. attests that the funding for this paper was not derived from funds from Philip Morris. E.M.P. received more than $100,001 from the NIH and $10,001–$50,000 from Philip Morris in sponsored grants. E.M.P. assumed the role of PI of the existing PM grant for the departing UAB faulty member so that funds to support research in South America could continue. E.M.P. attests that no research conducted in the laboratory and no authorships associated with manuscript were derived from funding from Philip Morris. S.M. served as a consultant for Sepracor, Inc., receiving a $2,000 honorarium for participating in their annual scientific meeting; $10,001–$50,000 from Talecris in sponsored grants to identify the interactions of alpha antitrypsin with ion channels (grant expired last year); $50,001–$100,000 in sponsored grants from Inspire Pharmaceuticals to identify the role of P2Y inhibitors on RSV injury to the respiratory system (grant expired 2 years ago); and more than $100,001 from Sepracor, Inc. in sponsored grants to test the efficacy of BROVANA in chlorine-induced injury (grant expired in June 2009). S.M. has a pending patent from the UAB Research Foundation for methods for using pyrimidine synthesis inhibitors to increase airway epithelial cell fluid uptake, and received $5,001–$10,000 from Elsevier for editing a book on free radical effects on membranes. S.M. has a pending patent from Purdue University for DPPC formulations and methods for using DPPC as a surfactant.
References
- 1.Evans RB. Chlorine: state of the art. Lung 2005;183:151–167. [DOI] [PubMed] [Google Scholar]
- 2.Sexton JD, Pronchik DJ. Chlorine inhalation: the big picture. J Toxicol Clin Toxicol 1998;36:87–93. [DOI] [PubMed] [Google Scholar]
- 3.Cave D, Fadam A. Iraq insurgents employ chlorine in bomb attacks. New York Times, February 22, 2007.
- 4.Sander R. Compilation of Henry's law constants for inorganic and organic species of potential importance in environmental chemistry. [Accessed May 2010.] Available from: http://www.mpch-mainz.mpg.de/∼sander/res/henry.html.
- 5.Squadrito G, Postlethwait EM, Matalon S. Elucidating mechanisms of chlorine toxicity: reaction kinetics, thermodynamics, and physiological implications. Am J Physiol Lung Cell Mol Physiol (In press) [DOI] [PMC free article] [PubMed]
- 6.Nielson DW. Electrolyte composition of pulmonary alveolar subphase in anesthetized rabbits. J Appl Physiol 1986;60:972–979. [DOI] [PubMed] [Google Scholar]
- 7.Barrow CS, Alarie Y, Warrick JC, Stock MF. Comparison of the sensory irritation response in mice to chlorine and hydrogen chloride. Arch Environ Health 1977;32:68–76. [DOI] [PubMed] [Google Scholar]
- 8.den Hartog GJ, Haenen GR, Vegt E, van der Vijgh WJ, Bast A. Efficacy of HOCl scavenging by sulfur-containing compounds: antioxidant activity of glutathione disulfide? Biol Chem 2002;383:709–713. [DOI] [PubMed] [Google Scholar]
- 9.Hawkins CL, Pattison DI, Davies MJ. Hypochlorite-induced oxidation of amino acids, peptides and proteins. Amino Acids 2003;25:259–274. [DOI] [PubMed] [Google Scholar]
- 10.Weiss SJ, Klein R, Slivka A, Wei M. Chlorination of taurine by human neutrophils. evidence for hypochlorous acid generation. J Clin Invest 1982;70:598–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crow JP. Measurement and significance of free and protein-bound 3-nitrotyrosine, 3-chlorotyrosine, and free 3-nitro-4-hydroxyphenylacetic acid in biologic samples: a high-performance liquid chromatography method using electrochemical detection. Methods Enzymol 1999;301:151–160. [DOI] [PubMed] [Google Scholar]
- 12.Hazen SL, Hsu FF, Mueller DM, Crowley JR, Heinecke JW. Human neutrophils employ chlorine gas as an oxidant during phagocytosis. J Clin Invest 1996;98:1283–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hazen SL, Heinecke JW. 3-Chlorotyrosine, a specific marker of myeloperoxidase-catalyzed oxidation, is markedly elevated in low density lipoprotein isolated from human atherosclerotic intima. J Clin Invest 1997;99:2075–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Foote CS, Goyne TE, Lehrer RI. Assessment of chlorination by human neutrophils. Nature 1983;301:715–716. [DOI] [PubMed] [Google Scholar]
- 15.Pattison DI, Hawkins CL, Davies MJ. Hypochlorous acid-mediated protein oxidation: how important are chloramine transfer reactions and protein tertiary structure? Biochemistry 2007;46:9853–9864. [DOI] [PubMed] [Google Scholar]
- 16.Robaszkiewicz A, Bartosz G, Soszynski M. N-chloroamino acids cause oxidative protein modifications in the erythrocyte membrane. Mech Ageing Dev 2008;129:572–579. [DOI] [PubMed] [Google Scholar]
- 17.Song W, Wei S, Shou Y, Lazrak A, Liu G, Londino JD, Squadrito GL, Matalon S. Inhibition of lung fluid clearance and epithelial Na+ channels by chlorine, hypochlorous acid and chloramines. J Biol Chem 2010;285:9716–9728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pascuzzi TA, Storrow AB. Mass casualties from acute inhalation of chloramine gas. Mil Med 1998;163:102–104. [PubMed] [Google Scholar]
- 19.Whiteman M, Jenner A, Halliwell B. Hypochlorous acid-induced base modifications in isolated calf thymus DNA. Chem Res Toxicol 1997;10:1240–1246. [DOI] [PubMed] [Google Scholar]
- 20.Van Rensburg CE, Van Staden AM, Anderson R. Inactivation of poly (ADP-ribose) polymerase by hypochlorous acid. Free Radic Biol Med 1991;11:285–291. [DOI] [PubMed] [Google Scholar]
- 21.Midwinter RG, Vissers MC, Winterbourn CC. Hypochlorous acid stimulation of the mitogen-activated protein kinase pathway enhances cell survival. Arch Biochem Biophys 2001;394:13–20. [DOI] [PubMed] [Google Scholar]
- 22.Midwinter RG, Cheah FC, Moskovitz J, Vissers MC, Winterbourn CC. IkappaB is a sensitive target for oxidation by cell-permeable chloramines: inhibition of NF-kappaB activity by glycine chloramine through methionine oxidation. Biochem J 2006;396:71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weiss SJ. Tissue destruction by neutrophils. N Engl J Med 1989;320:365–376. [DOI] [PubMed] [Google Scholar]
- 24.Martin JG, Campbell HR, Iijima H, Gautrin D, Malo JL, Eidelman DH, Hamid Q, Maghni K. Chlorine-induced injury to the airways in mice. Am J Respir Crit Care Med 2003;168:568–574. [DOI] [PubMed] [Google Scholar]
- 25.D'Alessandro A, Kuschner W, Wong H, Boushey HA, Blanc PD. Exaggerated responses to chlorine inhalation among persons with nonspecific airway hyperreactivity. Chest 1996;109:331–337. [DOI] [PubMed] [Google Scholar]
- 26.Demnati R, Fraser R, Martin JG, Plaa G, Malo JL. Effects of dexamethasone on functional and pathological changes in rat bronchi caused by high acute exposure to chlorine. Toxicol Sci 1998;45:242–246. [DOI] [PubMed] [Google Scholar]
- 27.Demnati R, Fraser R, Ghezzo H, Martin JG, Plaa G, Malo JL. Time-course of functional and pathological changes after a single high acute inhalation of chlorine in rats. Eur Respir J 1998;11:922–928. [DOI] [PubMed] [Google Scholar]
- 28.Morris JB, Wilkie WS, Shusterman DJ. Acute respiratory responses of the mouse to chlorine. Toxicol Sci 2005;83:380–387. [DOI] [PubMed] [Google Scholar]
- 29.Bonetto G, Corradi M, Carraro S, Zanconato S, Alinovi R, Folesani G, Da DL, Mutti A, Baraldi E. Longitudinal monitoring of lung injury in children after acute chlorine exposure in a swimming pool. Am J Respir Crit Care Med 2006;174:545–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kennedy SM, Enarson DA, Janssen RG, Chan-Yeung M. Lung health consequences of reported accidental chlorine gas exposures among pulpmill workers. Am Rev Respir Dis 1991;143:74–79. [DOI] [PubMed] [Google Scholar]
- 31.Nodelman V, Ultman JS. Longitudinal distribution of chlorine absorption in human airways: comparison of nasal and oral quiet breathing. J Appl Physiol 1999;86:1984–1993. [DOI] [PubMed] [Google Scholar]
- 32.Bessac BF, Sivula M, von Hehn CA, Escalera J, Cohn L, Jordt SE. TRPA1 is a major oxidant sensor in murine airway sensory neurons. J Clin Invest 2008;118:1899–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bessac BF, Jordt SE. Sensory irritation by toxic gases: mechanisms, health effects and countermeasures. Proc Am Thorac Soc 2010;7:269–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bessac BF, Jordt SE. BreathtakingTRP channels: TRPA1 and TRPV1 in airway chemosensation and reflex control. Physiology (Bethesda) 2008;23:360–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leustik M, Doran S, Bracher A, Williams S, Squadrito GL, Schoeb TR, Postlethwait E, Matalon S. Mitigation of chlorine-induced lung injury by low-molecular-weight antioxidants. Am J Physiol Lung Cell Mol Physiol 2008;295:L733–L743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Batchinsky AI, Martini DK, Jordan BS, Dick EJ, Fudge J, Baird CA, Hardin DE, Cancio LC. Acute respiratory distress syndrome secondary to inhalation of chlorine gas in sheep. J Trauma 2006;60:944–956. [DOI] [PubMed] [Google Scholar]
- 37.Wang J, Oldner A, Winskog C, Edston E, Walther SM. Effects of endothelin receptor antagonism on acute lung injury induced by chlorine gas. Crit Care Med 2006;34:1731–1737. [DOI] [PubMed] [Google Scholar]
- 38.Wang J, Winskog C, Edston E, Walther SM. Inhaled and intravenous corticosteroids both attenuate chlorine gas-induced lung injury in pigs. Acta Anaesthesiol Scand 2005;49:183–190. [DOI] [PubMed] [Google Scholar]
- 39.Wang J, Zhang L, Walther SM. Administration of aerosolized terbutaline and budesonide reduces chlorine gas-induced acute lung injury. J Trauma 2004;56:850–862. [DOI] [PubMed] [Google Scholar]
- 40.Bell DG. Management of acute respiratory distress syndrome (ARDS) following chlorine exposure [abstract]. Am J Respir Crit Care Med 2008;176:A314. [Google Scholar]
- 41.Davis IC, Zhu S, Sampson JB, Crow JP, Matalon S. Inhibition of human surfactant protein A function by oxidation intermediates of nitrite. Free Radic Biol Med 2002;33:1703–1713. [DOI] [PubMed] [Google Scholar]
- 42.Crouch EC, Hirche TO, Shao B, Boxio R, Wartelle J, Benabid R, McDonald B, Heinecke JW, Matalon S, Belaaouaj A. Myeloperoxidase-dependent inactivation of surfactant protein D in vitro and in vivo. J Biol Chem 2010;285:16757–16770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matalon S, O'Brodovich H. Sodium channels in alveolar epithelial cells: molecular characterization, biophysical properties, and physiological significance. Annu Rev Physiol 1999;61:627–661. [DOI] [PubMed] [Google Scholar]
- 44.Ware LB, Matthay MA. Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med 2001;163:1376–1383. [DOI] [PubMed] [Google Scholar]
- 45.Matthay MA, Wiener-Kronish JP. Intact epithelial barrier function is critical for the resolution of alveolar edema in humans. Am Rev Respir Dis 1990;142:1250–1257. [DOI] [PubMed] [Google Scholar]
- 46.Chuang GC, Yang Z, Westbrook DG, Pompilius M, Ballinger CA, White CR, Krzywanski DM, Postlethwait EM, Ballinger SW. Pulmonary ozone exposure induces vascular dysfunction, mitochondrial damage, and atherogenesis. Am J Physiol Lung Cell Mol Physiol 2009;297:L209–L216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Freeman BA, Crapo JD. Biology of disease: free radicals and tissue injury. Lab Invest 1982;47:412–426. [PubMed] [Google Scholar]
- 48.Lang JD, McArdle PJ, O'Reilly PJ, Matalon S. Oxidant-antioxidant balance in acute lung injury. Chest 2002;122:314S–320S. [DOI] [PubMed] [Google Scholar]
- 49.Bowler RP, Crapo JD. Oxidative stress in airways: is there a role for extracellular superoxide dismutase? Am J Respir Crit Care Med 2002;166:S38–S43. [DOI] [PubMed] [Google Scholar]
- 50.Rahman I, MacNee W. Oxidant/antioxidant imbalance in smokers and chronic obstructive pulmonary disease. Thorax 1996;51:348–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cantin AM, North SL, Hubbard RC, Crystal RG. Normal alveolar epithelial lining fluid contains high levels of glutathione. J Appl Physiol 1987;63:152–157. [DOI] [PubMed] [Google Scholar]
- 52.Roche M, Rondeau P, Singh NR, Tarnus E, Bourdon E. The antioxidant properties of serum albumin. FEBS Lett 2008;582:1783–1787. [DOI] [PubMed] [Google Scholar]
- 53.Quinlan GJ, Mumby S, Martin GS, Bernard GR, Gutteridge JM, Evans TW. Albumin influences total plasma antioxidant capacity favorably in patients with acute lung injury. Crit Care Med 2004;32:755–759. [DOI] [PubMed] [Google Scholar]
- 54.Ballinger CA, Cueto R, Squadrito G, Coffin JF, Velsor LW, Pryor WA, Postlethwait EM. Antioxidant-mediated augmentation of ozone-induced membrane oxidation. Free Radic Biol Med 2005;38:515–526. [DOI] [PubMed] [Google Scholar]
- 55.Antonicelli F, Brown D, Parmentier M, Drost EM, Hirani N, Rahman I, Donaldson K, MacNee W. Regulation of LPS-mediated inflammation in vivo and in vitro by the thiol antioxidant nacystelyn. Am J Physiol Lung Cell Mol Physiol 2004;286:L1319–L1327. [DOI] [PubMed] [Google Scholar]
- 56.Moss M, Steinberg KP, Guidot DM, Duhon GF, Treece P, Wolken R, Hudson LD, Parsons PE. The effect of chronic alcohol abuse on the incidence of ARDS and the severity of the multiple organ dysfunction syndrome in adults with septic shock: an interim and multivariate analysis. Chest 1999;116:97S–98S. [PubMed] [Google Scholar]
- 57.Rahman I. Antioxidant therapeutic advances in COPD. Ther Adv Respir Dis 2008;2:351–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Atkuri KR, Mantovani JJ, Herzenberg LA, Herzenberg LA. N-acetylcysteine–a safe antidote for cysteine/glutathione deficiency. Curr Opin Pharmacol 2007;7:355–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cheng YC, Bowen L, Rando RJ, Squadrito GL, Matalon S, Postlethwait EM. Exposing animals to oxidant gases: nose only vs. whole body. Proc Am Thorac Soc (In press). [DOI] [PMC free article] [PubMed]
- 60.Rennard SI, Basset G, Lecossier D, O'Donnell KM, Pinkston P, Martin PG, Crystal RG. Estimation of volume of epithelial lining fluid recovered by lavage using urea as marker of dilution. J Appl Physiol 1986;60:532–538. [DOI] [PubMed] [Google Scholar]
- 61.Widdicombe J. Airway and alveolar permeability and surface liquid thickness: theory. J Appl Physiol 1997;82:3–12. [DOI] [PubMed] [Google Scholar]
- 62.Markart P, Luboeinski T, Korfei M, Schmidt R, Wygrecka M, Mahavadi P, Mayer K, Wilhelm J, Seeger W, Guenther A, et al. Alveolar oxidative stress is associated with elevated levels of nonenzymatic low-molecular-weight antioxidants in patients with different forms of chronic fibrosing interstitial lung diseases. Antioxid Redox Signal 2009;11:227–240. [DOI] [PubMed] [Google Scholar]
- 63.Schmidt R, Luboeinski T, Markart P, Ruppert C, Daum C, Grimminger F, Seeger W, Gunther A. Alveolar antioxidant status in patients with acute respiratory distress syndrome. Eur Respir J 2004;24:994–999. [DOI] [PubMed] [Google Scholar]
- 64.Behndig AF, Blomberg A, Helleday R, Duggan ST, Kelly FJ, Mudway IS. Antioxidant responses to acute ozone challenge in the healthy human airway. Inhal Toxicol 2009;21:933–942. [DOI] [PubMed] [Google Scholar]