

Abstract
Exposure to chlorine gas (Cl2) primarily causes injury to the lung and is characterized by inflammation and oxidative stress mediated by reactive chlorine species. Reducing lung injury and improving respiratory function are the principal therapeutic goals in treating individuals exposed to Cl2 gas. Less is known on the potential for Cl2 gas exposure to cause injury to extrapulmonary tissues and specifically to mediate endothelial dysfunction. This concept is forwarded in this article on the basis that (1) many irritant gases whose reactivity is limited to the lung have now been shown to have effects that promote endothelial dysfunction in the systemic vasculature, and as such lead to the acute and chronic cardiovascular disease events (e.g., myocardial infarctions and atherosclerosis); and (2) that endogenously produced reactive chlorine species are now considered to be central in the development of cardiovascular diseases. This article discusses these two areas with the view of providing a framework in which potential extrapulmonary toxic effects of Cl2 gas exposure may be considered.
Keywords: nitric oxide, endothelial dysfunction, chlorination, inflammation
Exposure to chlorine (Cl2) gas remains an ongoing health concern, both via its possible use in chemical warfare and via accidental exposure during industrial manufacturing and transport. Indeed, approximately 15million tons of Cl2 are produced annually in the United States for a variety of industrial purposes (e.g., water purification, pharmaceutical and disinfectant development) and is transported predominantly by rail to all cities. Therefore there is potential to expose large numbers of civilians to Cl2 gas, and this is underscored by incidents related to large-scale Cl2-induced toxicity after accidental release (1–4). Cl2 gas–mediated toxicity is complex, consisting of an initial injury to the lungs that continues even after cessation of Cl2 exposure, ultimately leading to pulmonary dysfunction, hypoxemia and compromised oxygen delivery, vital organ perfusion, and function. Understanding the mechanisms by which Cl2 gas exposure causes lung injury are central to the development of therapeutics that can be administered after Cl2 exposure in both civilian and military casualty scenarios. In this article we focus on the potential for Cl2 gas exposure to promote injury to extrapulmonary tissues and suggest that this relatively underappreciated aspect of Cl2 toxicity requires consideration especially in the context of development of post-exposure therapeutics.
INHALED TOXICANTS, EXTRAPULMONARY INJURY, AND ENDOTHELIAL DYSFUNCTION: AN EMERGING PARADIGM
An emerging theme in pulmonary and vascular toxicology is the concept that an insult compartmentalized in the lung can result in extrapulmonary vascular injury. This is exemplified by environmental exposure to inhaled irritants and the subsequent increased susceptibility to cardiovascular disease (e.g., atherosclerosis) (5–8). Key examples in the latter case include the association of cigarette smoke, particulate matter, or ozone exposure with the acceleration of atherosclerosis, a chronic inflammatory disease of the vessel wall that underlies many cardiovascular diseases and that contributes substantially to morbidity and mortality worldwide. Clinical and epidemiologic studies together with recent experimental studies have definitively linked exposure to inhaled irritants with cardiovascular disease (5–12). Like most inhaled toxicants, the direct reactivity between the inhaled species and biological molecules is restricted to the lung compartment, suggesting that the effects on systemic vascular function that lead to enhanced atherogenesis are mediated by formation of secondary and diffusible species. This is further indicated by the fact that in this setting vascular dysfunction is a chronic process (years) compared with the relatively fast (seconds to minutes) reactivity between inhaled species and lung biomolecules. The precise mediators and mechanisms linking inhalation of toxicants with extrapulmonary vascular dysfunction are still under investigation and depend on the nature of the inhaled irritant(s) involved. However, one common mechanism by which diverse environmental/inhaled stressors may predispose the systemic vasculature to inflammatory disease is to induce dysfunction in the endothelial nitric oxide (NO) signaling pathway leading to endothelial dysfunction, a clinical term that is fast becoming synonymous with inflammatory vascular disease (13, 14).
Nitric oxide produced by the endothelial isoform of nitric oxide synthase from L-arginine has diverse physiologic roles in the vasculature, including regulating approximately 25% of basal blood flow (NO is a vasodilator) in humans, regulating cellular respiration, maintaining an anti-inflammatory, antithrombotic, antioxidant, and anti–smooth muscle proliferation state (15, 16). NO is central therefore in vascular homeostasis mechanisms. Aberrant NO signaling is a common feature in endothelial dysfunction and may contribute to the development of inflammatory diseases. In general this occurs by either decreased NO synthesis and/or redirection of NO from “physiologic” to “pathologic” (or proinflammatory) signaling processes. An in-depth review of the mechanisms that lead to this scenario is beyond the purview of this article, with discussion below focused on potential mechanisms of endothelial dysfunction associated with increased exposure to reactive chlorine species.
REACTIVE CHLORINE SPECIES AND VASCULAR INJURY
Several lines of evidence implicate a role for reactive chlorine species in the development of vascular diseases, with most attention focused on myeloperoxidase (MPO)-derived hypochlorous acid (HOCl) (17–20) and subsequent generation of reactive chlorine species (e.g., reactive chloramines) (21). Myeloperoxidase is primarily expressed in neutrophils, and during the latter's activation it catalyzes the formation of HOCl (Equation 1), a component of the phagocytic arsenal that is responsible for killing invading pathogens.
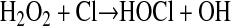 |
(1) |
HOCl (pKa = 7.5) and its derivative salts (e.g., NaOCl, the reactive component of bleach), react with multiple biological molecules, including lipids, amino acids, and nucleic acids, at relatively rapid rates, forming a variety of chlorination and oxidation products (22–30). In addition, HOCl can promote nitration of biomolecules (e.g., forming 3-nitrotyrosine) by reacting with nitrite to form the nitrating species nitrylchloride (31, 32). Depending on the tissue/cellular environment and dose of HOCl, these reactivities can result in a spectrum of modifications with diverse functional effects that encompass killing invading pathogens to promoting cytotoxicity of host tissues and perturbation of signaling pathways that regulate inflammation and hence vascular disease. A specific role for HOCl in the development of vascular disease is evidenced by (1) detection of chlorinated lipids, sterols, and proteins in atherosclerotic lesions (33–36); (2) reagent HOCl promoting vascular endothelial dysfunction (37–42) (discussed in more detail below); (3) decreased inflammation in MPO-deficient mice (43), increased atherosclerosis in MPO-transgenic mice (44, 45), and lack of coronary heart disease in humans who are MPO deficient (46); and (4) significant associations with degree of MPO activity and severity of myocardial infarctions (18). In summary, HOCl is considered one of the key reactive species that can promote oxidation, chlorination, and nitration of biomolecules, thereby leading to the development of inflammatory disease in the cardiovascular system.
MECHANISMS OF HOCL-INDUCED ATHEROSCLEROSIS AND ENDOTHELIAL DYSFUNCTION
In discussing the mechanisms by which HOCl may promote vascular injury, we will focus on atherosclerosis and coronary artery disease for which most evidence exists. Many studies have identified sites on specific proteins that are modified by HOCl that are associated a pro-atherogenic function (reviewed in Reference 18). This is exemplified by low- and high-density lipoproteins (LDL and HDL, respectively), on which markers indicating modification by HOCl have been detected in human atheroma. Importantly, the specific modifications have been shown to convert both LDL and HDL into pro-atherogenic particles (47–50). Furthermore, increased chlorotyrosine levels measured on HDL in outpatient cardiology subjects is significantly associated with an elevated risk for cardiovascular disease (18). Ongoing studies are identifying a diverse array of proteins that are modified by HOCl and that lead to altered function and ultimately a proinflammatory state. With regard to endothelial dysfunction specifically, several studies have documented that exposure of endothelial cells to physiologically relevant doses of HOCl promotes dysfunction in eNOS-derived NO signaling (37–42). Our group has shown that HOCl chlorinates L-Arginine, with the resultant mono- and di-chlorinated products being competitive inhibitors with native L-arginine for eNOS-dependent NO production (41, 42). Other groups have demonstrated that HOCl, either directly or via stimulation of reactive oxygen species from NADPH oxidase, compromises eNOS structural stability and hence inhibits NO formation (38, 39). Similarly, HOCl-dependent oxidation of lipids can result in aldehydic products that in turn may inhibit eNOS (51). Finally, HOCl-dependent protein modification forms epitopes that are recognized by the receptor for advanced glycation end products (RAGE) (52). Activation of these receptors on the endothelium is proinflammatory in part via inhibition of eNOS-derived NO signaling (53). In other words, via multiple distinct mechanisms HOCl promotes dysfunction in the eNOS-derived NO-signaling pathway.
CHLORINE GAS–INDUCED TOXICITY: DOES EXTRAPULMONARY VASCULAR INJURY OCCUR, AND IF SO, HOW?
Much of the lung injury caused by Cl2 gas exposure is thought to occur via its hydrolysis to form HOCl (discussed in other articles in this issue) via mechanisms similar to those described above. The potential for Cl2 gas to induce extrapulmonary vascular injury and specifically endothelial dysfunction has not been investigated. As indicated above, however, the paradigm for inhaled irritants mediating extrapulmonary injury is established, and a role for reactive chlorine species in vascular disease clearly indicated. It is interesting to speculate, therefore, that Cl2 gas exposure will also fall into the category of inhaled reactive gases that can have detrimental effects on the systemic vasculature, and preliminary studies in our laboratory using a rat model exposed to Cl2 gas indeed support this concept (unpublished observations). Moreover, case reports of Cl2 inhalation toxicity also support this potential with incidences of hypertension and liver toxicity being cited (2, 54). Clearly, a direct cause and effect of Cl2 exposure and extrapulmonary tissue injury from case studies is difficult to conclude. We cite these simply to highlight the need for consideration of extrapulmonary effects of Cl2 gas exposure and note that these, similar to lung injury, may also manifest both on acute and chronic time scales.
The question then arises: how could Cl2 gas exposure result in extrapulmonary injury? This question also applies to the myriad of inhaled toxicants (see above) that also cause dysfunction in systemic tissues. Common mechanisms may include stress responses associated with a lung irritation/toxicity and stimulation of inflammation. Due the reactive nature of inhaled irritants, the latter is likely to be mediated by activation of alveolar macrophages and epithelial cells in the lung compartment, resulting in the up-regulation and secretion of proinflammatory cytokines into the circulation. Interestingly, some of these (e.g., TNF-α) can in turn promote endothelial dysfunction by inhibiting eNOS-dependent signaling (55). Whereas HOCl formation is unique to Cl2 gas exposure compared with non–Cl2 gas inhaled irritants, due to its high reactivity this reactive species is unlikely to be able to migrate away from the lung to directly cause injury to distal tissues. However, HOCl-derived products that would be unique to Cl2 gas exposure that could exit the lung and mediate effects in other tissues can be proposed. These include a variety of chlorinated products which can have relatively higher stabilities than HOCl. One example pertinent to endothelial dysfunction is chlorination of L-arginine (41), for example, as discussed above.
PERSPECTIVES
A focus of this issue on Cl2 gas toxicity underscores the potential for Cl2 gas to cause injury in civilian and military settings and highlights the need for more mechanistic information on how Cl2 gas exposure promotes toxicity. This information is vital for the development and use of effective therapeutics. The lung is the primary target, damage to which manifests in both acute and chronic symptoms. The goal of this article was to highlight the potential for Cl2 gas toxicity to promote injury to extrapulmonary tissues also. This concept (illustrated in Figure 1) is based on observations that (1) Cl2-derived species are critical in development of inflammatory vascular disease, and (2) chemically diverse inhaled irritants are now established risk factors for the development of cardiovascular disease with causative mechanisms beginning to be identified. Cl2 gas exposure can fit into both categories. Clearly, the hypothesis that Cl2 gas promotes extrapulmonary dysfunction requires vigorous testing, and ongoing efforts are beginning to address this. We feel that emerging data do suggest a role for Cl2 gas in promoting endothelial dysfunction. The prevalence of endothelial dysfunction, coupled with its key role in the development of a cardiovascular diseases, mandates a better understanding of the potential for Cl2 gas to mediate endothelial dysfunction which in turn will lead to more efficacious and targeted preventive and treatment therapeutics.
Figure 1.

Hypothesis: inhaled chlorine gas induction of extrapulmonary vascular dysfunction. Chlorine gas inhalation causes progressive lung damage resulting in acute lung injury (ALI), adult respiratory distress syndrome (ARDS), and reactive airway syndrome (RAS). Proposed mechanisms of injury include increased exposure to hypochlorous acid (HOCl) and secondary formation of proinflammatory cytokines. In addition, we hypothesize that inhaled chlorine gas also causes extrapulmonary vascular dysfunction and specifically endothelial dysfunction secondary to formation of chlorinated products and release of proinflammatory cytokines into the circulation.
The research is supported by the CounterACT Program, National Institutes Of Health Office of the Director, and the National Institute of Environmental Health Sciences, Grant Number U54ES017218.
Conflict of Interest Statement: A.S. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. J.H. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. C.R.W. received grant support from the National Institutes of Health (NIH) (more than $100,001), and his spouse/life partner received grant support from the NIH ($50,001–$100,000). R.P.P. received grant support from Ikaria (formerly iNO Therapeutics) and the NIH (more than $100,001).
References
- 1.Evans RB. Chlorine: state of the art. Lung 2005;183:151–167. [DOI] [PubMed] [Google Scholar]
- 2.Van Sickle D, Wenck MA, Belflower A, Drociuk D, Ferdinands J, Holguin F, Svendsen E, Bretous L, Jankelevich S, Gibson JJ, et al. Acute health effects after exposure to chlorine gas released after a train derailment. Am J Emerg Med 2009;27:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wenck MA, Van Sickle D, Drociuk D, Belflower A, Youngblood C, Whisnant MD, Taylor R, Rudnick V, Gibson JJ. Rapid assessment of exposure to chlorine released from a train derailment and resulting health impact. Public Health Rep 2007;122:784–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Almagro Nievas D, Acuna Castillo R, Hernandez Jerez A, Robles Montes A. [Investigation of an outbreak of acute respiratory illness due to exposure to chlorine gas in a public swimming pool.] Gac Sanit 2008;22:287–290. [DOI] [PubMed] [Google Scholar]
- 5.Ying Z, Kampfrath T, Thurston G, Farrar B, Lippmann M, Wang A, Sun Q, Chen LC, Rajagopalan S. Ambient particulates alter vascular function through induction of reactive oxygen and nitrogen species. Toxicol Sci 2009;111:80–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chuang GC, Yang Z, Westbrook DG, Pompilius M, Ballinger CA, White CR, Krzywanski DM, Postlethwait EM, Ballinger SW. Pulmonary ozone exposure induces vascular dysfunction, mitochondrial damage, and atherogenesis. Am J Physiol Lung Cell Mol Physiol 2009;297:L209–L216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang Z, Harrison CM, Chuang GC, Ballinger SW. The role of tobacco smoke induced mitochondrial damage in vascular dysfunction and atherosclerosis. Mutat Res 2007;621:61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lippmann M, Frampton M, Schwartz J, Dockery D, Schlesinger R, Koutrakis P, Froines J, Nel A, Finkelstein J, Godleski J, et al. The U.S. Environmental protection agency particulate matter health effects research centers program: a midcourse report of status, progress, and plans. Environ Health Perspect 2003;111:1074–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ruidavets JB, Cournot M, Cassadou S, Giroux M, Meybeck M, Ferrieres J. Ozone air pollution is associated with acute myocardial infarction. Circulation 2005;111:563–569. [DOI] [PubMed] [Google Scholar]
- 10.Knight-Lozano CA, Young CG, Burow DL, Hu ZY, Uyeminami D, Pinkerton KE, Ischiropoulos H, Ballinger SW. Cigarette smoke exposure and hypercholesterolemia increase mitochondrial damage in cardiovascular tissues. Circulation 2002;105:849–854. [DOI] [PubMed] [Google Scholar]
- 11.Borja-Aburto VH, Castillejos M, Gold DR, Bierzwinski S, Loomis D. Mortality and ambient fine particles in southwest Mexico City, 1993–1995. Environ Health Perspect 1998;106:849–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Penn A, Snyder CA. 1,3 butadiene, a vapor phase component of environmental tobacco smoke, accelerates arteriosclerotic plaque development. Circulation 1996;93:552–557. [DOI] [PubMed] [Google Scholar]
- 13.Munzel T, Sinning C, Post F, Warnholtz A, Schulz E. Pathophysiology, diagnosis and prognostic implications of endothelial dysfunction. Ann Med 2008;40:180–196. [DOI] [PubMed] [Google Scholar]
- 14.Widlansky ME, Gokce N, Keaney JF Jr, Vita JA. The clinical implications of endothelial dysfunction. J Am Coll Cardiol 2003;42:1149–1160. [DOI] [PubMed] [Google Scholar]
- 15.Moncada S. Nitric oxide: discovery and impact on clinical medicine. J R Soc Med 1999;92:164–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Napoli C, Ignarro LJ. Nitric oxide and pathogenic mechanisms involved in the development of vascular diseases. Arch Pharm Res 2009;32:1103–1108. [DOI] [PubMed] [Google Scholar]
- 17.Lau D, Baldus S. Myeloperoxidase and its contributory role in inflammatory vascular disease. Pharmacol Ther 2006;111:16–26. [DOI] [PubMed] [Google Scholar]
- 18.Nicholls SJ, Hazen SL. Myeloperoxidase, modified lipoproteins, and atherogenesis. J Lipid Res 2009;50:S346–S351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pennathur S, Heinecke JW. Oxidative stress and endothelial dysfunction in vascular disease. Curr Diab Rep 2007;7:257–264. [DOI] [PubMed] [Google Scholar]
- 20.Sirpal S. Myeloperoxidase-mediated lipoprotein carbamylation as a mechanistic pathway for atherosclerotic vascular disease. Clin Sci (Lond) 2009;116:681–695. [DOI] [PubMed] [Google Scholar]
- 21.Hazell LJ, Davies MJ, Stocker R. Secondary radicals derived from chloramines of apolipoprotein b-100 contribute to HOCl-induced lipid peroxidation of low-density lipoproteins. Biochem J 1999;339:489–495. [PMC free article] [PubMed] [Google Scholar]
- 22.Malle E, Marsche G, Arnhold J, Davies MJ. Modification of low-density lipoprotein by myeloperoxidase-derived oxidants and reagent hypochlorous acid. Biochim Biophys Acta 2006;1761:392–415. [DOI] [PubMed] [Google Scholar]
- 23.Pitt AR, Spickett CM. Mass spectrometric analysis of hocl- and free-radical-induced damage to lipids and proteins. Biochem Soc Trans 2008;36:1077–1082. [DOI] [PubMed] [Google Scholar]
- 24.Dever GJ, Benson R, Wainwright CL, Kennedy S, Spickett CM. Phospholipid chlorohydrin induces leukocyte adhesion to apoe−/− mouse arteries via upregulation of p-selectin. Free Radic Biol Med 2008;44:452–463. [DOI] [PubMed] [Google Scholar]
- 25.Hawkins CL, Pattison DI, Davies MJ. Hypochlorite-induced oxidation of amino acids, peptides and proteins. Amino Acids 2003;25:259–274. [DOI] [PubMed] [Google Scholar]
- 26.Hazen SL, Gaut JP, Hsu FF, Crowley JR, d'Avignon A, Heinecke JW. P-hydroxyphenylacetaldehyde, the major product of L-tyrosine oxidation by the myeloperoxidase-H2O2-chloride system of phagocytes, covalently modifies epsilon-amino groups of protein lysine residues. J Biol Chem 1997;272:16990–16998. [DOI] [PubMed] [Google Scholar]
- 27.Hazen SL, Hsu FF, Duffin K, Heinecke JW. Molecular chlorine generated by the myeloperoxidase-hydrogen peroxide-chloride system of phagocytes converts low density lipoprotein cholesterol into a family of chlorinated sterols. J Biol Chem 1996;271:23080–23088. [DOI] [PubMed] [Google Scholar]
- 28.Jerlich A, Pitt AR, Schaur RJ, Spickett CM. Pathways of phospholipid oxidation by hocl in human LDL detected by LC-MS. Free Radic Biol Med 2000;28:673–682. [DOI] [PubMed] [Google Scholar]
- 29.Pattison DI, Hawkins CL, Davies MJ. Hypochlorous acid-mediated oxidation of lipid components and antioxidants present in low-density lipoproteins: absolute rate constants, product analysis, and computational modeling. Chem Res Toxicol 2003;16:439–449. [DOI] [PubMed] [Google Scholar]
- 30.Shen Z, Wu W, Hazen SL. Activated leukocytes oxidatively damage DNA, RNA, and the nucleotide pool through halide-dependent formation of hydroxyl radical. Biochemistry 2000;39:5474–5482. [DOI] [PubMed] [Google Scholar]
- 31.Boersma BJ, Patel RP, Kirk M, Jackson PL, Muccio D, Darley-Usmar VM, Barnes S. Chlorination and nitration of soy isoflavones. Arch Biochem Biophys 1999;368:265–275. [DOI] [PubMed] [Google Scholar]
- 32.Eiserich JP, Cross CE, Jones AD, Halliwell B, van der Vliet A. Formation of nitrating and chlorinating species by reaction of nitrite with hypochlorous acid: a novel mechanism for nitric oxide-mediated protein modification. J Biol Chem 1996;271:19199–19208. [DOI] [PubMed] [Google Scholar]
- 33.Daugherty A, Dunn JL, Rateri DL, Heinecke JW. Myeloperoxidase, a catalyst for lipoprotein oxidation, is expressed in human atherosclerotic lesions. J Clin Invest 1994;94:437–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hazen SL, Heinecke JW. 3-chlorotyrosine, a specific marker of myeloperoxidase-catalyzed oxidation, is markedly elevated in low density lipoprotein isolated from human atherosclerotic intima. J Clin Invest 1997;99:2075–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Malle E, Waeg G, Schreiber R, Grone EF, Sattler W, Grone HJ. Immunohistochemical evidence for the myeloperoxidase/H2O2/halide system in human atherosclerotic lesions: colocalization of myeloperoxidase and hypochlorite-modified proteins. Eur J Biochem 2000;267:4495–4503. [DOI] [PubMed] [Google Scholar]
- 36.Woods AA, Linton SM, Davies MJ. Detection of HOCl-mediated protein oxidation products in the extracellular matrix of human atherosclerotic plaques. Biochem J 2003;370:729–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jaimes EA, Sweeney C, Raij L. Effects of the reactive oxygen species hydrogen peroxide and hypochlorite on endothelial nitric oxide production. Hypertension 2001;38:877–883. [PubMed] [Google Scholar]
- 38.Stocker R, Huang A, Jeranian E, Hou JY, Wu TT, Thomas SR, Keaney JF Jr. Hypochlorous acid impairs endothelium-derived nitric oxide bioactivity through a superoxide-dependent mechanism. Arterioscler Thromb Vasc Biol 2004;24:2028–2033. [DOI] [PubMed] [Google Scholar]
- 39.Xu J, Xie Z, Reece R, Pimental D, Zou MH. Uncoupling of endothelial nitric oxidase synthase by hypochlorous acid: role of nad(p)h oxidase-derived superoxide and peroxynitrite. Arterioscler Thromb Vasc Biol 2006;26:2688–2695. [DOI] [PubMed] [Google Scholar]
- 40.Yang J, Cheng Y, Ji R, Zhang C. Novel model of inflammatory neointima formation reveals a potential role of myeloperoxidase in neointimal hyperplasia. Am J Physiol Heart Circ Physiol 2006;291:H3087–H3093. [DOI] [PubMed] [Google Scholar]
- 41.Zhang C, Patel R, Eiserich JP, Zhou F, Kelpke S, Ma W, Parks DA, Darley-Usmar V, White CR. Endothelial dysfunction is induced by proinflammatory oxidant hypochlorous acid. Am J Physiol Heart Circ Physiol 2001;281:H1469–H1475. [DOI] [PubMed] [Google Scholar]
- 42.Zhang C, Reiter C, Eiserich JP, Boersma B, Parks DA, Beckman JS, Barnes S, Kirk M, Baldus S, Darley-Usmar VM, et al. L-arginine chlorination products inhibit endothelial nitric oxide production. J Biol Chem 2001;276:27159–27165. [DOI] [PubMed] [Google Scholar]
- 43.Zhang R, Brennan ML, Shen Z, MacPherson JC, Schmitt D, Molenda CE, Hazen SL. Myeloperoxidase functions as a major enzymatic catalyst for initiation of lipid peroxidation at sites of inflammation. J Biol Chem 2002;277:46116–46122. [DOI] [PubMed] [Google Scholar]
- 44.Castellani LW, Chang JJ, Wang X, Lusis AJ, Reynolds WF. Transgenic mice express human MPO -463g/a alleles at atherosclerotic lesions, developing hyperlipidemia and obesity in -463g males. J Lipid Res 2006;47:1366–1377. [DOI] [PubMed] [Google Scholar]
- 45.McMillen TS, Heinecke JW, LeBoeuf RC. Expression of human myeloperoxidase by macrophages promotes atherosclerosis in mice. Circulation 2005;111:2798–2804. [DOI] [PubMed] [Google Scholar]
- 46.Kutter D, Devaquet P, Vanderstocken G, Paulus JM, Marchal V, Gothot A. Consequences of total and subtotal myeloperoxidase deficiency: risk or benefit? Acta Haematol 2000;104:10–15. [DOI] [PubMed] [Google Scholar]
- 47.Bergt C, Pennathur S, Fu X, Byun J, O'Brien K, McDonald TO, Singh P, Anantharamaiah GM, Chait A, Brunzell J, et al. The myeloperoxidase product hypochlorous acid oxidizes HDL in the human artery wall and impairs abca1-dependent cholesterol transport. Proc Natl Acad Sci USA 2004;101:13032–13037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McCall MR, Carr AC, Forte TM, Frei B. LDL modified by hypochlorous acid is a potent inhibitor of lecithin-cholesterol acyltransferase activity. Arterioscler Thromb Vasc Biol 2001;21:1040–1045. [DOI] [PubMed] [Google Scholar]
- 49.Zheng L, Nukuna B, Brennan ML, Sun M, Goormastic M, Settle M, Schmitt D, Fu X, Thomson L, Fox PL, et al. Apolipoprotein a-i is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. J Clin Invest 2004;114:529–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zheng L, Settle M, Brubaker G, Schmitt D, Hazen SL, Smith JD, Kinter M. Localization of nitration and chlorination sites on apolipoprotein a-i catalyzed by myeloperoxidase in human atheroma and associated oxidative impairment in abca1-dependent cholesterol efflux from macrophages. J Biol Chem 2005;280:38–47. [DOI] [PubMed] [Google Scholar]
- 51.Marsche G, Heller R, Fauler G, Kovacevic A, Nuszkowski A, Graier W, Sattler W, Malle E. 2-chlorohexadecanal derived from hypochlorite-modified high-density lipoprotein-associated plasmalogen is a natural inhibitor of endothelial nitric oxide biosynthesis. Arterioscler Thromb Vasc Biol 2004;24:2302–2306. [DOI] [PubMed] [Google Scholar]
- 52.Marsche G, Semlitsch M, Hammer A, Frank S, Weigle B, Demling N, Schmidt K, Windischhofer W, Waeg G, Sattler W, et al. Hypochlorite-modified albumin colocalizes with rage in the artery wall and promotes MCP-1 expression via the RAGE-ERK1/2 map-kinase pathway. FASEB J 2007;21:1145–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Linden E, Cai W, He JC, Xue C, Li Z, Winston J, Vlassara H, Uribarri J. Endothelial dysfunction in patients with chronic kidney disease results from advanced glycation end products (AGE)-mediated inhibition of endothelial nitric oxide synthase through rage activation. Clin J Am Soc Nephrol 2008;3:691–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Leube G, Kreiter H. [Acute chlorine poisoning: case reports of 90 patients with acute poisoning.] Med Klin 1971;66:354–357. [PubMed] [Google Scholar]
- 55.Gao X, Xu X, Belmadani S, Park Y, Tang Z, Feldman AM, Chilian WM, Zhang C. TNF-alpha contributes to endothelial dysfunction by upregulating arginase in ischemia/reperfusion injury. Arterioscler Thromb Vasc Biol 2007;27:1269–1275. [DOI] [PubMed] [Google Scholar]