

Abstract
Protein kinase B (PKB, Akt) is a Ser/Thr kinase involved in the regulation of cell survival, proliferation, and metabolism and is activated by dual phosphorylation on Thr308 in the activation loop and Ser473 in the hydrophobic motif. It plays a contributory role to platelet function, although little is known about its regulation. In this study, we investigated the role of the mammalian target of rapamycin complex (mTORC)-2 in Akt regulation using the recently identified small molecule ATP competitive mTOR inhibitors PP242 and Torin1. Both PP242 and Torin1 blocked thrombin and insulin-like growth factor 1-mediated Akt Ser473 phosphorylation with an IC50 between 1 and 5 nm, whereas the mTORC1 inhibitor rapamycin had no effect. Interestingly, PP242 and Torin1 had no effect on Akt Thr308 phosphorylation, Akt1 activity, and phosphorylation of the Akt substrate glycogen synthase kinase 3β, indicating that Ser473 phosphorylation is not necessary for Thr308 phosphorylation and maximal Akt1 activity. In contrast, Akt2 activity was significantly reduced, concurrent with inhibition of PRAS40 phosphorylation, in the presence of PP242 and Torin1. Other signaling pathways, including phospholipase C/PKC and the MAPK pathway, were unaffected by PP242 and Torin1. Together, these results demonstrate that mTORC2 is the kinase that phosphorylates Akt Ser473 in human platelets but that this phosphorylation is dispensable for Thr308 phosphorylation and Akt1 activity.
Keywords: Adipocyte, Akt PKB, Glycogen Synthase Kinase 3, Insulin, Insulin-like Growth Factor (IGF), mTOR Complex (mTORC), PI 3-Kinase, Platelet, PRAS40, p70S6 Kinase
Introduction
When a blood vessel becomes damaged, platelets adhere to exposed collagen and are rapidly activated, promoting degranulation and exposure of fibrinogen binding sites on integrin αIIbβ3. Subsequent binding of fibrinogen couples the platelets together, resulting in platelet aggregation and thrombus formation. Platelet activation also results in the exposure of phosphatidylserine on the cell membrane, resulting in formation of the prothrombinase complex and subsequent thrombin formation. Thrombin proteolytically cleaves fibrinogen, resulting in a fibrin network that consolidates the thrombus and activates protease-activated receptor (PAR)3 1 and PAR4 on the platelet surface, resulting in further platelet activation and recruitment to the site of injury.
PAR receptors are G protein-coupled receptors that activate phospholipase C, resulting in the hydrolysis of phosphoinositol (PI) 4,5-bisphosphate into diacylglycerol and inositol 1,4,5-trisphosphate (IP3). IP3 subsequently mobilizes Ca2+ from intracellular stores and together with diacylglycerol activates protein kinase C (PKC) isoforms, leading to platelet activation. In addition, platelet activation by a variety of agonists results in the activation of phosphoinositide 3-kinase (PI3), leading to an increase in PI (3,4)P2 and PI (3,4,5)P3 concentrations in the cell. PIP3 accumulation at the membrane recruits the pleckstrin homology domain containing proteins Akt and phosphoinositide-dependent kinase 1. This results in phosphorylation of Akt on Thr308 by phosphoinositide-dependent kinase 1. Subsequent phosphorylation of Ser473 results in maximal Akt activity (1). Platelets express Akt1 and Akt2, with Akt1 the dominant isoform in human platelets and Akt2 in mouse platelets (2, 3). Akt is activated by a variety of platelet agonists (thrombin, collagen, ADP, and thromboxane A2) and priming agents (IGF-1, thrombopoietin and VEGF) (4). Priming agents are unable to induce aggregation by themselves but potentiate the effects of platelet agonists. Evidence that Akt supports platelet function comes from Akt1- and Akt2-deficient mouse models (2, 5). Akt1-deficient mice have increased tail bleeding times and reduced platelet function in response to thrombin and collagen (5), whereas Akt2-deficient mice are resistant to in vivo thrombosis and demonstrate reduced platelet responsiveness (2). So far, little is known about Akt regulation in human platelets, and the kinase that phosphorylates Akt Ser473 has not been identified. The identity of the kinase that phosphorylates Ser473 in other cell types has also been the subject of considerable debate. Proposed candidates include MAPKAP-K2, ILK, PKC, and modified phosphoinositide-dependent kinase 1 (1, 6–8). However, recent evidence suggests that two members of the PI3K-related superfamily, mTORC2 (mammalian target of rapamycin complex 2) and DNA-activated protein kinase are largely responsible for the phosphorylation of Akt Ser473 in most cellular contexts (9). mTORC2 is a multimeric kinase composed of mTOR, Rictor, Protor, mLST8, and mSin1. This complex, in contrast to mTORC1, is insensitive to inhibition by rapamycin, although extended treatment may inhibit new mTORC2 formation (10).
Knockdown or genetic ablation of mTOR, Rictor, or mLST8 blocked insulin-stimulated Ser473 phosphorylation whereas deficiency of the mTORC1 component Raptor had no effect (11, 12). Additionally, purified mTORC2 could directly phosphorylate recombinant Akt Ser473 but not Thr308. Indeed, insulin-stimulated Akt Ser473 phosphorylation in classical insulin-responsive cell types such as 3T3-L1 adipocytes is mediated by mTORC2 (13). Recently, several groups have independently developed potent and selective inhibitors of mTOR, which prevent insulin- and serum-stimulated Ser473 phosphorylation but also reduce Thr308 phosphorylation in a Ser473-dependent manner (14–17). This was a surprising result because previous work demonstrated that Thr308 is still phosphorylated in cells expressing the Akt S473A mutant and in cells in which mTORC2 activity is ablated by deletion of mTORC2 components (1, 11, 12, 18). This raised the question of whether Thr308 phosphorylation is dependent on Ser473 phosphorylation in cells in which mTORC2 is acutely inhibited.
In this study we wished to investigate the role of mTORC2 in Akt regulation in human platelets. We have used newly developed inhibitors of mTOR, PP242 and Torin1, and the mTORC1-selective inhibitor rapamycin to address the role of mTORC2 in Akt regulation and compared it with Akt regulation in primary adipocytes, the latter known to be dependent on mTORC2. Our findings demonstrate that (i) mTORC2 phosphorylates Ser473 in both platelets and adipocytes; (ii) Thr308 phosphorylation is independent of Ser473 phosphorylation; and (iii) that, in contrast to primary adipocytes, Ser473 phosphorylation is not required for Akt1 activity in human platelets.
EXPERIMENTAL PROCEDURES
Materials
Male Wistar rats (220–250 g) were fed ad libitum with a stock diet (CRM; Bioshore, Manea, Cambridgeshire, UK). pThr389 p70S6K, pSer473 Akt, pThr308 Akt, pSer9 GSK3β, pThr246 PRAS40, PKC phospho-motif (used for analysis of pleckstrin phosphorylation), pThr202/Tyr204 ERK, pThr180/Tyr182 p38, and Akt2 (L79BZ) antibodies were from Cell Signaling Technologies (New England Biolabs, Hitchin, UK). Akt1(B-1) and p70S6K (H-9) antibodies were from Santa Cruz (Insight Biotechnology, Wembley, UK). The Akt1 rabbit mAb (AW24) was from Upstate. The Akt2 antibody was raised against amino acids 453–470 of murine Akt2 in rabbits and kindly provided by Dick Denton and Kelly Moule (School of Biochemistry, University of Bristol). Microcystin-LR was from Axxora (Nottingham, UK). Akti1/2 was from Merck Chemicals. Rapamycin was from Tocris (Avonmouth, UK). Torin1 was a kind gift from David Sabatini (Whitehead Institute for Biomedical Research, Cambridge, MA) and Nathanael Gray (Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA). PP242 was from Sigma. PAR1-activating peptide (SFLLRN-NH2) was from Bachem (Weil am Rhein, Germany). [γ-32P]ATP was from PerkinElmer Life Sciences. Enhanced chemiluminescent detection reagents were from GE Healthcare. Peroxidase-conjugated secondary antibodies were from Jackson Immunoresearch (Stratech, Newmarket, UK). NuPAGE SDS-PAGE sample buffer was from Invitrogen. RPRAATF was synthesized by Graham Bloomberg (University of Bristol). All other reagents were from Sigma unless otherwise indicated.
Isolation and Incubation of Platelets
Human platelets were isolated from whole blood as previously described (19) and resuspended at 4 × 108/ml in modified HEPES-Tyrode buffer (145 mm NaCl, 3 mm KCl, 0.5 mm Na2HPO4, 1 mm MgSO4, 10 mm HEPES, pH 7.2, 0.1% (w/v) d-glucose, 0.02 unit/ml apyrase, and 10 μm indomethacin). Platelets were treated with vehicle (0.2% DMSO) or compound for 15 min, stimulated as indicated, and lysed directly in 4× NuPAGE sample buffer (whole cell lysate). Alternatively, platelets were extracted with an equal volume of ice-cold (i) radioimmune precipitation assay buffer (50 mm HEPES, pH 7.4, 400 mm NaCl, 2 mm EDTA, 2% (v/v) IGEPAL CA-630, 1% (w/v) sodium deoxycholate, 0.2% (w/v) SDS, 40 mm sodium β-glycerol phosphate, 20 mm sodium pyrophosphate, 2 mm benzamidine, 2 μm microcystin-LR, 10 mm sodium orthovanadate, and 2 μg/ml each pepstatin, antipain, and leupeptin) for immunoprecipitation of p70S6K and Akt or (ii) Triton X-100 extraction buffer (50 mm HEPES, pH 7.4, 2% (v/v) Triton X-100, 2 mm EDTA, 40 mm sodium β-glycerol phosphate, 20 mm sodium pyrophosphate, 2 mm benzamidine, 20% (v/v) glycerol, 10 mm sodium orthovanadate, 2 μm microcystin-LR, and 2 μg/ml each pepstatin, antipain, and leupeptin) for Akt activity assays.
Preparation and Incubation of Epididymal Fat Cells
Adipocytes were isolated from the epididymal fat pads of Wistar rats as previously described (20). Adipocytes were treated with vehicle (0.2% DMSO) or compound for 15 min at 37 °C and stimulated as indicated prior to extraction in an equal volume of ice-cold Triton X-100 extraction buffer followed by snap freezing in liquid nitrogen.
Immunoprecipitation of p70S6K and Akt from Platelets
p70S6K, Akt1, or Akt2 was immunoprecipitated from radioimmune precipitation assay lysates by incubation with 2 μg of anti-p70S6K (H-9), anti-Akt1 (B1), or anti-Akt2 (antiserum 1.1) and 10 μl of protein G-Sepharose for 2 h at 4 °C. Immune complexes were washed with extraction buffer prior to elution with 2× NuPAGE sample buffer.
Western Blotting
Immunoprecipitates (IPs) and whole cell lysates were analyzed by SDS-PAGE/Western blotting using 8% bis-tris gels as previously described (21). Membranes containing Akt1 (B1) and Akt2 (antiserum 1.1) IPs were incubated with antibodies raised in rabbits and mice, respectively.
Akt Activity Assay
Akt1 and Akt2 were immunoprecipitated from adipocyte and platelet lysates by tumbling with 2 μg of anti-Akt1 (B1) and 10 μl of protein G-Sepharose or with 2 μg of anti-Akt2 (antiserum 1.1) and 10 μl of protein A-Sepharose, respectively, for 2 h at 4 °C. Immune complexes were washed twice with 1 × Triton X-100 extraction buffer + 500 mm NaCl, twice with wash buffer (20 mm HEPES, pH 7.4, 0.1 mm EGTA, 0.03% Brij-35, 1 mm sodium orthovanadate, 1 mm DTT), and twice with assay buffer (20 mm HEPES, pH 7.4, 10 mm MgCl2, 20 mm sodium β-glycerol phosphate, 1 mm EGTA, and 1 mm DTT). Akt activity was assayed in the presence of 50 μm ATP (2 μCi of [γ-32P]ATP), 0.5 mg/ml RPRAATF peptide (22), and 2.5 μm PKA inhibitor (IP-20). Reactions were incubated for 30 min at 30 °C with continuous shaking and terminated by spotting onto P81 phosphocellulose paper. Filters were washed with 0.75% orthophosphoric acid before measuring the incorporation of [γ-32P]ATP by scintillation counting. Results are expressed as percentage of the signal obtained with thrombin, IGF-1, or insulin alone.
Aggregometry
Washed platelets (2 × 108/ml) were incubated with vehicle (0.2% DMSO) or the indicated compounds for 15 min, and aggregation was stimulated with 0.7 μm SFLLRN-NH2 in a Chronolog 490–4D aggregometer as previously described (23).
Statistics
All data are presented as the mean ± S.E. of at least three independent observations. Data presented with statistical analysis were tested using a one-way ANOVA with Dunnett's post test using GraphPad Prism version 4.02.
RESULTS
mTOR Inhibitors Selectively Inhibit Thrombin-stimulated Akt Serine 473 Phosphorylation in Human Platelets
To evaluate the role of mTOR in Akt phosphorylation, we used two recently identified small molecule mTOR inhibitors, PP242 and Torin1. These inhibitors are highly potent and selective inhibitors of the mTORC1 and mTORC2 complexes with little effect on the related PI3K family (15–17). As expected, both PP242 and Torin1 dose-dependently inhibited thrombin-induced phosphorylation of the mTORC1 substrate p70S6K, with maximal inhibition reached at 5 nm (Fig. 1, A and B). Incubation with PP242 and Torin1 also resulted in loss of Akt Ser473 phosphorylation, demonstrating that Akt Ser473 is phosphorylated downstream of mTOR. Slightly higher doses were required for inhibition of Akt Ser473 phosphorylation compared with p70S6K phosphorylation (Fig. 1, A and B). Interestingly, we found no effect of the mTOR inhibitors on Akt Thr308 phosphorylation (Fig. 1, A and B). Thrombin-stimulated phosphorylation of the Akt substrate GSK3β was also left intact, suggesting that sufficient Akt activity remains for substrate phosphorylation. Furthermore, PP242 and Torin1 had no effect on phosphorylation of pleckstrin, a substrate for the closely related AGC kinase PKC, and the MAPK family members ERK and p38.
FIGURE 1.

mTOR inhibitors selectively inhibit phosphorylation of Akt Ser473 in human platelets. Washed platelets were incubated with vehicle (0.2% DMSO) or the indicated concentrations of PP242 (A and C) and Torin1 (B and D) for 15 min prior to stimulation with 0.1 unit/ml thrombin for 15 min (A and B) or 100 nm IGF-1 for 3 min (C and D). The phosphorylation of the indicated proteins was analyzed by SDS-PAGE/Western blotting of immunoprecipitates (p70S6K) or whole cell lysates (Akt, GSK3, pleckstrin, ERK, p38 MAPK). Membranes were stripped and reprobed for α-tubulin as a loading control. Results are representative of three independent experiments.
mTOR Inhibitors Have No Effect on Insulin/IGF-1-stimulated Akt Threonine 308 Phosphorylation
Three recent independent studies showed that mTOR inhibitors not only affect Ser473 Akt phosphorylation, but also inhibit Thr308 Akt phosphorylation (14–16). Because these studies used serum, insulin, and/or IGF-1, the discrepancy with our study may be explained by the different stimuli used. Fig. 1, C and D, demonstrate that IGF-1-stimulated Akt Ser473 phosphorylation in platelets is equally sensitive to the mTOR inhibitors PP242 and Torin1 as for thrombin. Similarly, IGF-1-stimulated Akt Thr308 phosphorylation and phosphorylation of the downstream substrate GSK3β were unaffected by mTOR inhibitors (Fig. 1, C and D). To investigate the possibility that the discrepancy with previously published reports (14–16) is due to the use of cell lines versus primary cells in the present study, we next studied Akt phosphorylation in rat primary adipocytes, which are highly insulin-responsive cells. Higher concentrations of inhibitors were used to compensate for the high fat content of adipocytes (Fig. 2). PP242 and Torin1 reduced both insulin-stimulated p70S6K and Akt Ser473 phosphorylation, whereas they did not attenuate Thr308 phosphorylation (Fig. 2). Together, these results show that Akt Thr308 phosphorylation in response to insulin/IGF-1 is not affected by mTOR inhibitors in both platelets and adipocytes.
FIGURE 2.
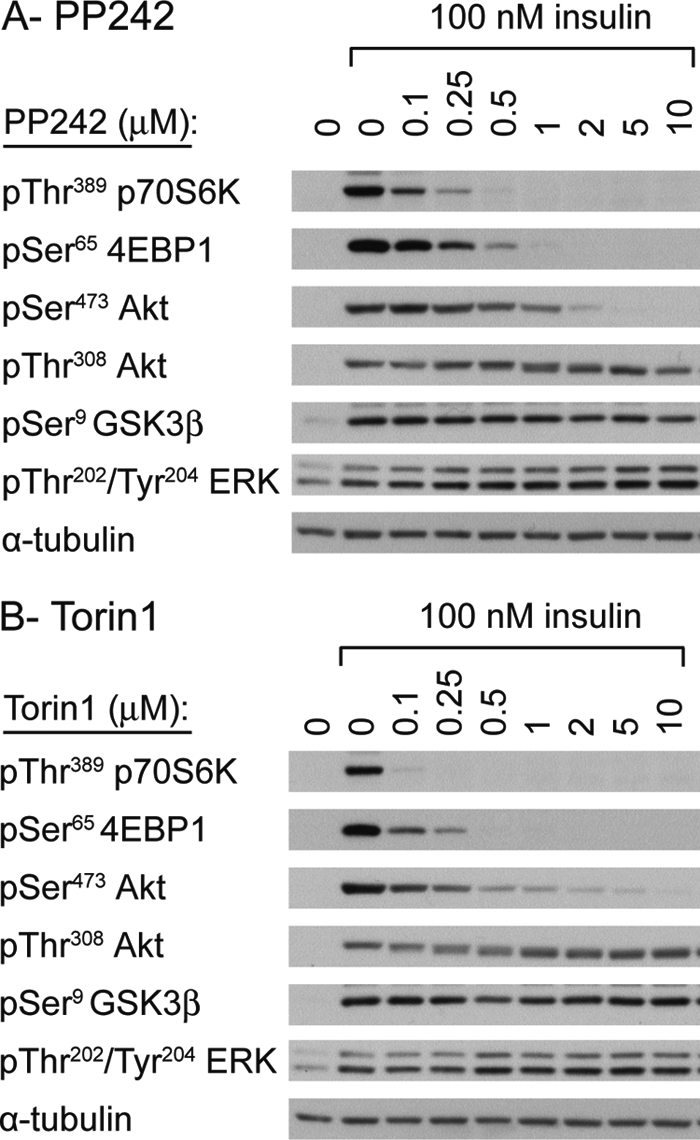
mTOR inhibitors selectively inhibit phosphorylation of Akt Ser473 in primary adipocytes. Washed adipocytes were incubated with vehicle (0.2% DMSO) or the indicated concentrations of PP242 (A) and Torin1 (B) for 15 min prior to stimulation with 100 nm insulin for 15 min. The phosphorylation of the indicated proteins was analyzed by SDS-PAGE/Western blotting of whole cell lysates. Membranes were stripped and reprobed for α-tubulin as a loading control. Results are representative of three independent experiments.
mTORC2-mediated Akt Serine 473 Phosphorylation Is Dispensable for Phosphorylation of GSK3β, but Not PRAS40
To confirm that Akt Ser473 phosphorylation is mediated by mTORC2, and not mTORC1, we studied Akt phosphorylation in the presence of the mTORC1 inhibitor rapamycin and the mTORC1/2 inhibitors PP242 and Torin1. As expected, phosphorylation of the mTORC1 substrates p70S6K and 4E-BP1 is blocked by all compounds in both platelets and primary adipocytes (Fig. 3, A and C, top). Surprisingly, IGF-1 was unable to induce p70S6K phosphorylation in platelets (Fig. 3B, data not shown). Rapamycin had no effect on Akt Ser473 phosphorylation, whereas PP242 and Torin1 prevented phosphorylation in all cell types (Fig. 3, A–C). These results confirm that mTORC2, and not mTORC1, is the kinase upstream of Akt Ser473 phosphorylation in human platelets and primary adipocytes.
FIGURE 3.

mTORC2-mediated Akt Ser473 phosphorylation is dispensable for phosphorylation of GSK3β, but not PRAS40. Washed platelets were incubated with vehicle (0.2% DMSO) or the indicated compounds for 15 min prior to stimulation with 0.1 unit/ml thrombin for 15 min (A) or 100 nm IGF-1 for 3 min (B). Alternatively, primary adipocytes were incubated with vehicle (0.2% DMSO) or with the indicated compounds for 15 min prior to stimulation with 100 nm insulin for 15 min (C). The phosphorylation of the indicated proteins was analyzed by SDS-PAGE/Western blotting of immunoprecipitates (p70S6K, A) or whole cell lysates (Akt, GSK3, PRAS40, pleckstrin, ERK, p38 MAPK). Membranes were stripped and reprobed for α-tubulin as a loading control. Results are representative of three independent experiments. The bar graphs represent quantification of Thr246 phosphorylation of PRAS40 (ratio phosphorylated/total) expressed as a percentage of the signal obtained with thrombin (A), IGF-1 (B) and insulin (C) alone (means ± S.E. (error bars), n = 3).
Phosphorylation of the endogenous Akt substrate, GSK3β (Fig. 3, A–C), was largely unaffected by PP242 and Torin1. GSK3β is presently the only verified Akt substrate in human platelets (24). Here, we identify an additional Akt substrate in platelets, PRAS40. Phosphorylation of Thr246 PRAS40 is completely blocked in the presence of the Akt inhibitor Akti1/2 (Fig. 3, A–C, bar graphs). Interestingly, in contrast to GSK3β, PRAS40 phosphorylation is significantly reduced by PP242 and Torin1 in both platelets and adipocytes (Fig. 3, A–C, bar graphs). The reduction in PRAS40 phosphorylation was mTORC2-dependent as the mTORC1 inhibitor rapamycin had no significant inhibitory effect (Fig. 3, A–C).
mTORC2-mediated Akt Serine 473 Phosphorylation Is Not Required for Akt1 Activity in Human Platelets
To evaluate to what extent the mTOR inhibitors affect phosphorylation and kinase activity of the individual Akt isoforms, Akt1 and Akt2 were immunoprecipitated from thrombin and IGF-1-stimulated platelet lysate and insulin-stimulated adipocyte lysates. The relative expression levels of Akt1 and Akt2 were determined by comparing the Akt1/Akt2 kinase activity ratios in platelets versus primary adipocytes. In vitro kinase assays showed that the Akt1/Akt2 activity ratios were 1.40 ± 0.15 (thrombin-stimulated platelets), 2.22 ± 0.28 (IGF-1-stimulated platelets), and 0.09 ± 0.03 (insulin-stimulated rat adipocytes), confirming earlier studies that Akt2 is the major isoform in primary adipocytes (2, 3). As expected, PP242 and Torin1 blocked Akt1 Ser473 phosphorylation, with no effect on Akt1 Thr308 phosphorylation in platelets and adipocytes (Fig. 4Ai, Ci, and Ei). Interestingly, thrombin and IGF-1-stimulated Akt1 activity was unaffected by PP242 and Torin1, but blocked by Akti1/2 (Fig. 4, Aii and Cii), demonstrating that Ser473 phosphorylation is not required for Akt1 activity in human platelets. In contrast, mTOR inhibitors significantly reduced Akt1 activity in primary adipocytes (Fig. 4Eii). Similar to Akt1, Akt2 Ser473 phosphorylation was blocked by PP242 and Torin1, whereas Akt2 Thr308 phosphorylation was still present (Fig. 4, Bi, Di, and Fi). Note that Ser473 phosphorylation of Akt2, but not Akt1, results in a band shift (see arrows). Akt2 activity was significantly reduced by PP242 and Torin1 in both platelets and adipocytes (Fig. 4, Bii, Dii, and Fii) and showed strong correlation with PRAS40 phosphorylation (compare Fig. 4 with the bar graphs in Fig. 3). Together, these results demonstrate that mTORC2 is the kinase that phosphorylates Akt Ser473 in both platelets and primary adipocytes and contributes to Akt2 activity. However, Akt Ser473 phosphorylation is dispensable for Akt1 activity in platelets, but not adipocytes.
FIGURE 4.

mTORC2-mediated Akt Ser473 phosphorylation is not required for Akt1 activity in human platelets. Washed platelets were incubated with vehicle (0.2% DMSO) or the indicated compounds for 15 min prior to stimulation with 0.1 unit/ml thrombin for 15 min (A and B) or 100 nm IGF-1 for 3 min (C and D). Alternatively, primary adipocytes were incubated with vehicle (0.2% DMSO) or with the indicated compounds for 15 min prior to stimulation with 100 nm insulin for 15 min (E and F). Akt1 (A, C, and E) and Akt2 (B, D, and F) were immunoprecipitated (IP) from detergent lysates and phosphorylation of Thr308 and Ser473 analyzed by SDS-PAGE/Western blotting (i) and in vitro activity analyzed using the peptide substrate, RPRAATF (ii). Activity results are expressed as a percentage of the signal obtained with thrombin (A and B), IGF-1 (C and D), and insulin (E and F) alone (n = 3, mean ± S.E. (error bars); **, p < 0.001).
mTOR Inhibitors Have No Effect on PAR1-stimulated Platelet Aggregation
To evaluate the contribution of mTOR to platelet aggregation, we incubated platelets with increasing concentrations of PP242, Torin1, and rapamycin and measured aggregation in response to EC50 stimulation of the thrombin receptor PAR1 with the peptide agonist, SFLLRN (Fig. 5). PP242 had no significant effect on platelet aggregation up to a concentration of 100 nm (Fig. 5A), under which conditions both p70S6K and Akt Ser473 phosphorylation were blocked (see Fig. 1). Torin1 had no effect at 1–10 nm, although a small potentiatory effect was seen at higher concentrations (Fig. 5B). The discrepancy between the effects of PP242 and Torin1 and lack of correlation with p70S6K and Akt Ser473 phosphorylation suggest that this effect is unrelated to mTOR inhibition. Finally, inhibition of mTORC1 with rapamycin had no significant effect on platelet aggregation. Together, these results suggest that mTOR does not contribute directly to platelet aggregation.
FIGURE 5.
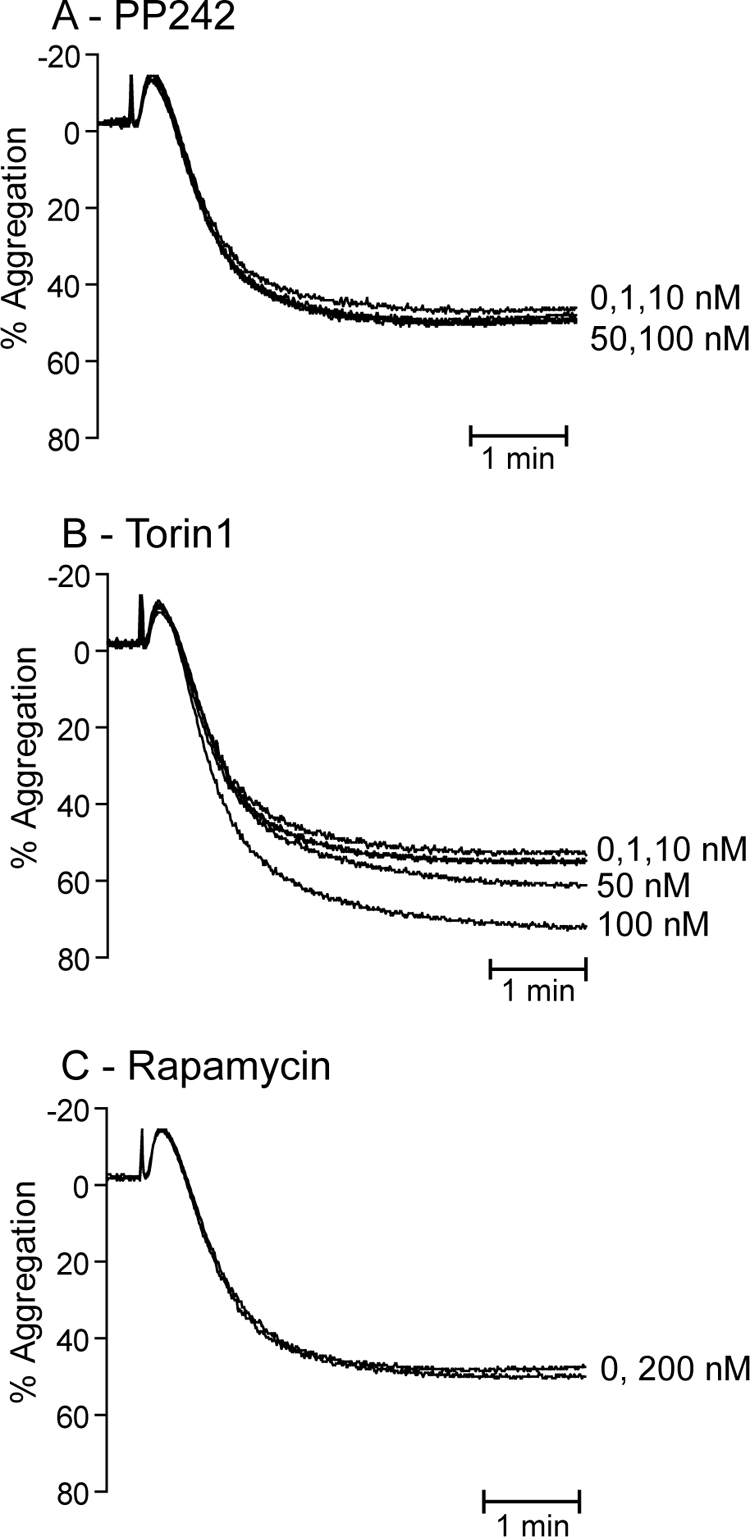
mTOR activity does not contribute to platelet aggregation. Washed platelets (2 × 108/ml) were incubated with the indicated concentrations of PP242 (A), Torin1 (B), or 200 nm rapamycin (C) for 15 min prior to stimulation of PAR1 with 0.7 μm SFLLRN. The resulting aggregation was recorded for 5 min and is expressed as percent aggregation. Results are representative of three independent experiments.
DISCUSSION
Akt, a major effector of PI3K, is involved in the regulation of multiple essential biological processes, with its dysregulation being linked to a wide range of disease processes (25). Mouse knock-out studies demonstrated that Akt plays a supporting role in platelet function by increasing fibrinogen binding to integrin αIIbβ3, secretion of α and dense granules, aggregation, and thrombus formation (2, 5). The role of Akt in human platelets is less defined, mainly due to the lack of specific inhibitors, but it is clear that platelet primers that activate the PI3K/Akt pathway potentiate platelet function (26–29). Because little is known about the regulation of Akt in human platelets, this study aimed to identify the role of mTORC2 through the use of recently developed potent and specific mTOR inhibitors (15–17).
We determined that PP242 and Torin1 are able to inhibit both thrombin and IGF-1-induced phosphorylation of Akt Ser473 in human platelets whereas they had no effect on activation of the closely related AGC kinase PKC, and the MAPK members ERK and p38. We obtained similar results with Ku-0063794 developed by Astra-Zeneca (14), which became commercially available during the course of this study (results not shown). Furthermore, acute treatment of platelets with rapamycin had no effect on the phosphorylation of Akt, demonstrating that mTORC2 is the Akt Ser473 kinase in platelets. We demonstrate that loss of Ser473 phosphorylation in platelets: (i) is not accompanied by a concurrent loss of Akt Thr308 phosphorylation, (ii) results in a partial loss of PRAS40 Thr246 phosphorylation but has no effect on GSK3β Ser9 phosphorylation and (iii) resulted in a selective reduction in Akt2 but not Akt1 activity.
Our finding that both PP242 and Torin1 failed to induce a concurrent loss of Akt Thr308 phosphorylation was in contrast to the original studies using these inhibitors. The latter studies suggested that Akt Ser(P)473 can facilitate Akt Thr(P)308 (15, 16) and are in agreement with previous reports (14, 30, 31). The lack of effect of the mTOR inhibitors on Akt Thr308 phosphorylation in platelets cannot be explained by differences in cellular context (thus cell-specific) or differences in receptor tyrosine kinase versus G protein-coupled receptor signaling as we found identical results in platelets and primary adipocytes and for SFLLRN (activating the G protein-coupled receptor PAR-1) and IGF-1 (activating a receptor tyrosine kinase). These results are in agreement with a recent study using pyrazolopyrimidine ATP-competitive mTOR inhibitors, which also found no effect on Thr(P)308 at concentrations that blocked Ser(P)473 (32). Indeed, the original studies on Akt phosphorylation in Brian Hemmings' laboratory employing alanine mutants of Akt Thr308 and Ser473 demonstrated that IGF-1-stimulated phosphorylation of these sites can occur independently (1). Furthermore, our findings in adipocytes are also in agreement with more recent conditional animal knock-out studies, where the mTORC2 component Rictor was specifically ablated in adipose tissue (33).
One possibility is that the use of primary cells in our study (platelets and adipocytes) versus cell lines in the original studies may explain the differences observed. Although the finding that PP242 reduced insulin-stimulated phosphorylation of both Ser473 and Thr308 in vivo in perigenital fat and liver in mice would argue against this (15), it cannot be ruled out that the latter results may be due to adverse effects of PP242 at higher doses (14). Together, our results and previously published data clearly demonstrate that Thr308 phosphorylation can occur independently of Ser473 in a wide range of cellular contexts.
We found that Ser9 phosphorylation of the Akt substrate GSK3β in both human platelets and adipocytes was unaffected by inhibition of mTOR. This is in agreement with previous studies where GSK3β phosphorylation was at most only moderately affected in the absence of mTORC2 activity (14, 33). Potentially this lack of inhibition of GSK3β phosphorylation could be attributed to its promiscuity as a kinase substrate (34–36). In this study, we identified PRAS40 as a novel Akt substrate in platelets which can therefore be used as an alternative Akt reporter to GSK3β. Phosphorylation of PRAS40 at Thr246 was significantly reduced in platelets and adipocytes in the presence of the mTOR inhibitors. To correlate endogenous substrate phosphorylation with Akt kinase activity, we performed in vitro Akt1 and Akt2 activity assays using the Akt-selective substrate peptide RPRAATF (22). Interestingly, we found no effect of mTOR inhibitors on Akt1 activity, whereas Akt2 activity was strongly reduced. In contrast, both Akt1 and Akt2 activity were inhibited in primary adipocytes.
The data on GSK3β and PRAS40 phosphorylation can potentially be explained by Akt isoforms phosphorylating different substrates. The finding that Akt1 activity is not affected in platelets, but PRAS40 phosphorylation is reduced, suggests that PRAS40 is predominantly an Akt2 substrate in vivo. Indeed, we found a strong correlation between PRAS40 phosphorylation and Akt2, but not Akt1, activity (compare Fig. 3 bar graphs with Fig. 4). Similarly, GSK3β may be predominantly an Akt1 substrate because its phosphorylation is left intact by mTOR inhibitors. However, in primary adipocytes, both Akt1 and Akt2 activity and PRAS40 phosphorylation were strongly reduced by mTORC2 inhibitors, whereas GSK3β phosphorylation was unaffected. It is therefore unlikely that GSK3β phosphorylation is regulated by one particular Akt isoform. Indeed, recent studies suggest that GSK3β phosphorylation is controlled by all three Akt isoforms (37). Interestingly, although Akt3 has not been previously detected in mouse and human platelets (2, 3), a recent abstract by the Du group suggests that Akt3 may be the major isoform responsible for GSK3β phosphorylation in mouse platelets (38).
Alternatively, substrates may require different levels of Akt activity for maximal phosphorylation and downstream effects. Previous work by the groups of Alessi and Sutherland, for example, showed that maximal insulin repression of PEPCK and G6Pase genes in the liver requires <10% Akt activity (39). Furthermore, GSK3 can be phosphorylated by many different kinases (34–36), creating the possibility that other kinases phosphorylate GSK3 under conditions where Akt activity is restricted. PRAS40 phosphorylation, however, mirrored Akt activity closely (39). PRAS40 can also be phosphorylated by mTORC1 on Ser183, Ser212, and Ser221 (40), which may indirectly influence the phosphorylation status of the Akt phosphorylation site Thr246. However, the mTORC1 inhibitor rapamycin did not affect Thr246 PRAS40 phosphorylation, making this is an unlikely scenario.
Previous work where mTORC2 components have been genetically ablated has shown that mTORC2 may regulate the substrate specificity of Akt (11, 12). Ablation of any of the mTORC2 components mSin1, mLST8, or Rictor reduced Foxo1/3, but not GSK3 and TSC2 phosphorylation (11, 12). Similarly, in our study GSK3β phosphorylation was unaffected, but PRAS40 phosphorylation was reduced in the absence of mTORC2 activity. Thus, GSK3β may only require Akt Thr308 phosphorylation, whereas PRAS40 requires both Thr308 and Ser473 phosphorylation for optimal substrate phosphorylation. Interestingly, we found that Akt2 Ser473 phosphorylation, but not Akt1 phosphorylation, results in an Akt band shift.
This band shift was complete in adipocytes, demonstrating dual phosphorylation of Akt2. In contrast, the band shift was only partial in platelets with both single and dual phosphorylated Akt2 present upon stimulation (Fig. 4).
The novel finding of our study is that Akt1 activity is not affected by the absence of Ser473 phosphorylation in human platelets. This is interesting in the context that previous studies showed a significant inhibitory effect on Akt1 activity in the absence of mTORC2 activity, and indeed Ser473 is believed to increase Akt1 activity by 4–5-fold (41). These results furthermore suggest that Akt1 and Akt2 activity can be differentially regulated by mTORC2 in platelets. One explanation for this may be that Akt1 and Akt2 bind to different intracellular proteins that regulate their substrate specificity or activity. Indeed, many Akt-interacting proteins have been identified, some of which are isoform-selective (42, 43). The finding that Akt1 activity in primary adipocytes, in contrast to platelets, is strongly reduced by mTOR inhibitors, suggests that it is a cell-specific observation. This may be explained by differences in Akt interactome between these cell types.
mTORC1 is involved in platelet fibrin-dependent clot retraction, but contributes little to platelet aggregation (44). In our study we also found no significant effect of the mTORC1 inhibitor rapamycin and the mTOR inhibitors PP242 and Torin1 on platelet aggregation. The latter may be explained by the lack of effect mTOR inhibitors on Akt1 activity. Because aggregation studies represent a complex measurement of platelet function in which the role of mTOR may not be apparent, further studies are presently under way to investigate the role of mTOR in responses such as fibrinogen binding, secretion, and thrombosis.
In conclusion, we have demonstrated that although mTORC2 is the Ser473 kinase in platelets, there was no impact on Akt1 activity or phosphorylation of the downstream substrate, GSK3β, whereas Akt2 activity and PRAS40 phosphorylation were significantly reduced. This was at odds with the regulation of Akt in primary adipocytes, a classical model of Akt signaling, where the loss of Ser473 phosphorylation led to a significant reduction in both Akt1 and Akt2 activity. Therefore, our data clearly demonstrate that Ser473 phosphorylation is not absolutely required for Akt1 activity, but has a cell/tissue-specific function.
Acknowledgments
We thank David Sabatini (Whitehead Institute of Biomedical Research) and Nathanael Gray (Dana-Farber Cancer Institute) for the Torin1, Dick Denton and Kelly Moule (School of Biochemistry, University of Bristol) for the anti-Akt2 antiserum, Igor Khaliulin and Phil Pasdois (School of Biochemistry, University of Bristol) for help with tissue collection, and the blood donors within the Medical Sciences Building for their generous donations.
This work was supported by British Heart Foundation Grants PG/08/113/25515, FS/05/047/18875, and PG/08/056/25325 (to I. H.).
- PAR
- protease-activated receptor
- bis-tris
- bis(2-hydroxyethyl)aminotris(hydroxymethyl)methane
- DMSO
- dimethyl sulfoxide
- GSK3β
- glycogen synthase kinase 3β
- IGF
- insulin-like growth factor
- mTORC
- mammalian target of rapamycin complex.
REFERENCES
- 1. Alessi D. R., Andjelkovic M., Caudwell B., Cron P., Morrice N., Cohen P., Hemmings B. A. (1996) EMBO J. 15, 6541–6551 [PMC free article] [PubMed] [Google Scholar]
- 2. Woulfe D., Jiang H., Morgans A., Monks R., Birnbaum M., Brass L. F. (2004) J. Clin. Invest. 113, 441–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kroner C., Eybrechts K., Akkerman J. W. (2000) J. Biol. Chem. 275, 27790–27798 [DOI] [PubMed] [Google Scholar]
- 4. Gresele P., Falcinelli E., Momi S. (2008) Trends Pharmacol. Sci. 29, 352–360 [DOI] [PubMed] [Google Scholar]
- 5. Chen J., De S., Damron D. S., Chen W. S., Hay N., Byzova T. V. (2004) Blood 104, 1703–1710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Balendran A., Casamayor A., Deak M., Paterson A., Gaffney P., Currie R., Downes C. P., Alessi D. R. (1999) Curr. Biol. 9, 393–404 [DOI] [PubMed] [Google Scholar]
- 7. Rane M. J., Coxon P. Y., Powell D. W., Webster R., Klein J. B., Pierce W., Ping P., McLeish K. R. (2001) J. Biol. Chem. 276, 3517–3523 [DOI] [PubMed] [Google Scholar]
- 8. Delcommenne M., Tan C., Gray V., Rue L., Woodgett J., Dedhar S. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 11211–11216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bozulic L., Hemmings B. A. (2009) Curr. Opin. Cell Biol. 21, 256–261 [DOI] [PubMed] [Google Scholar]
- 10. Sarbassov D. D., Ali S. M., Sengupta S., Sheen J. H., Hsu P. P., Bagley A. F., Markhard A. L., Sabatini D. M. (2006) Mol. Cell 22, 159–168 [DOI] [PubMed] [Google Scholar]
- 11. Jacinto E., Facchinetti V., Liu D., Soto N., Wei S., Jung S. Y., Huang Q., Qin J., Su B. (2006) Cell 127, 125–137 [DOI] [PubMed] [Google Scholar]
- 12. Guertin D. A., Stevens D. M., Thoreen C. C., Burds A. A., Kalaany N. Y., Moffat J., Brown M., Fitzgerald K. J., Sabatini D. M. (2006) Dev. Cell 11, 859–871 [DOI] [PubMed] [Google Scholar]
- 13. Hresko R. C., Mueckler M. (2005) J. Biol. Chem. 280, 40406–40416 [DOI] [PubMed] [Google Scholar]
- 14. García-Martínez J. M., Moran J., Clarke R. G., Gray A., Cosulich S. C., Chresta C. M., Alessi D. R. (2009) Biochem. J. 421, 29–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Feldman M. E., Apsel B., Uotila A., Loewith R., Knight Z. A., Ruggero D., Shokat K. M. (2009) PLoS Biol. 7, e38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Thoreen C. C., Kang S. A., Chang J. W., Liu Q., Zhang J., Gao Y., Reichling L. J., Sim T., Sabatini D. M., Gray N. S. (2009) J. Biol. Chem. 284, 8023–8032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu Q., Chang J. W., Wang J., Kang S. A., Thoreen C. C., Markhard A., Hur W., Zhang J., Sim T., Sabatini D. M., Gray N. S. (2010) J. Med. Chem. 53, 7146–7155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shiota C., Woo J. T., Lindner J., Shelton K. D., Magnuson M. A. (2006) Dev. Cell 11, 583–589 [DOI] [PubMed] [Google Scholar]
- 19. Hunter R. W., Hers I. (2009) J. Thromb. Haemost. 7, 2123–2130 [DOI] [PubMed] [Google Scholar]
- 20. Hers I., Tavaré J. M. (2005) Biochem. J. 388, 713–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hunter R. W., Mackintosh C., Hers I. (2009) J. Biol. Chem. 284, 12339–12348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Alessi D. R., Caudwell F. B., Andjelkovic M., Hemmings B. A., Cohen P. (1996) FEBS Lett. 399, 333–338 [DOI] [PubMed] [Google Scholar]
- 23. Hunter R. W., Harper M. T., Hers I. (2008) J. Thromb. Haemost. 6, 1923–1932 [DOI] [PubMed] [Google Scholar]
- 24. Barry F. A., Graham G. J., Fry M. J., Gibbins J. M. (2003) FEBS Lett. 553, 173–178 [DOI] [PubMed] [Google Scholar]
- 25. Dummler B., Hemmings B. A. (2007) Biochem. Soc. Trans. 35, 231–235 [DOI] [PubMed] [Google Scholar]
- 26. Hers I. (2007) Blood 110, 4243–4252 [DOI] [PubMed] [Google Scholar]
- 27. Campus F., Lova P., Bertoni A., Sinigaglia F., Balduini C., Torti M. (2005) J. Biol. Chem. 280, 24386–24395 [DOI] [PubMed] [Google Scholar]
- 28. Pasquet J. M., Gross B. S., Gratacap M. P., Quek L., Pasquet S., Payrastre B., van Willigen G., Mountford J. C., Watson S. P. (2000) Blood 95, 3429–3434 [PubMed] [Google Scholar]
- 29. Angelillo-Scherrer A., Burnier L., Flores N., Savi P., DeMol M., Schaeffer P., Herbert J. M., Lemke G., Goff S. P., Matsushima G. K., Earp H. S., Vesin C., Hoylaerts M. F., Plaisance S., Collen D., Conway E. M., Wehrle-Haller B., Carmeliet P. (2005) J. Clin. Invest. 115, 237–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sarbassov D. D., Guertin D. A., Ali S. M., Sabatini D. M. (2005) Science 307, 1098–1101 [DOI] [PubMed] [Google Scholar]
- 31. Scheid M. P., Marignani P. A., Woodgett J. R. (2002) Mol. Cell. Biol. 22, 6247–6260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yu K., Toral-Barza L., Shi C., Zhang W. G., Lucas J., Shor B., Kim J., Verheijen J., Curran K., Malwitz D. J., Cole D. C., Ellingboe J., Ayral-Kaloustian S., Mansour T. S., Gibbons J. J., Abraham R. T., Nowak P., Zask A. (2009) Cancer Res. 69, 6232–6240 [DOI] [PubMed] [Google Scholar]
- 33. Kumar A., Lawrence J. C., Jr., Jung D. Y., Ko H. J., Keller S. R., Kim J. K., Magnuson M. A., Harris T. E. (2010) Diabetes 59, 1397–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang H. H., Lipovsky A. I., Dibble C. C., Sahin M., Manning B. D. (2006) Mol. Cell 24, 185–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Frame S., Cohen P. (2001) Biochem. J. 359, 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hayashi H., Sudo T. (2009) Thromb. Haemost. 102, 327–335 [DOI] [PubMed] [Google Scholar]
- 37. Brognard J., Sierecki E., Gao T., Newton A. C. (2007) Mol. Cell 25, 917–931 [DOI] [PubMed] [Google Scholar]
- 38. O'Brien K., Stojanovic-Terpo A., Hay N., Du X. (2010) Circulation 122, A19993 [Google Scholar]
- 39. Logie L., Ruiz-Alcaraz A. J., Keane M., Woods Y. L., Bain J., Marquez R., Alessi D. R., Sutherland C. (2007) Diabetes 56, 2218–2227 [DOI] [PubMed] [Google Scholar]
- 40. Wang L., Harris T. E., Lawrence J. C., Jr. (2008) J. Biol. Chem. 283, 15619–15627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Andjelkoviæ M., Alessi D. R., Meier R., Fernandez A., Lamb N. J., Frech M., Cron P., Cohen P., Lucocq J. M., Hemmings B. A. (1997) J. Biol. Chem. 272, 31515–31524 [DOI] [PubMed] [Google Scholar]
- 42. Du K., Tsichlis P. N. (2005) Oncogene 24, 7401–7409 [DOI] [PubMed] [Google Scholar]
- 43. Fayard E., Xue G., Parcellier A., Bozulic L., Hemmings B. A. (2010) Curr. Top. Microbiol. Immunol. 346, 31–56 [DOI] [PubMed] [Google Scholar]
- 44. Weyrich A. S., Denis M. M., Schwertz H., Tolley N. D., Foulks J., Spencer E., Kraiss L. W., Albertine K. H., McIntyre T. M., Zimmerman G. A. (2007) Blood 109, 1975–1983 [DOI] [PMC free article] [PubMed] [Google Scholar]