

Abstract
The 5-hydroxytryptamine 2A receptor (5-HT2AR) undergoes constitutive and agonist-dependent internalization. Despite many advances in our understanding of G protein-coupled receptor trafficking, the exact mechanism of endocytic sorting of G protein-coupled receptors remains obscure. Recently, we have reported a novel finding documenting a global role for the ubiquitin ligase c-Cbl in regulating vesicular sorting of epidermal growth factor receptor (Baldys, A., Göoz, M., Morinelli, T. A., Lee, M. H., Raymond, J. R., Jr., Luttrell, L. M., and Raymond, J. R., Sr. (2009) Biochemistry 48, 1462–1473). Thus, we tested the hypothesis that c-Cbl might play a role in 5-HT2AR recycling. In this study, we demonstrated an association of 5-HT2AR with c-Cbl. Furthermore, down-regulation of c-Cbl by RNA interference blocked efficient recycling of 5-HT2AR to the plasma membrane. Immunofluorescence microscopy revealed that 5-HT2A receptors were trapped in early endosome antigen 1- and Rab11-positive sorting endosomes in cells overexpressing c-Cbl mutants lacking carboxyl termini. This inhibitory effect was associated with a relative decrease in association of c-Cbl truncation proteins with the 5-HT2AR, compared with that observed for the full-length c-Cbl fusion protein. Consistent with the delayed recycling, 5-HT2AR resensitization was greatly attenuated in the presence of c-Cbl mutants lacking carboxyl termini, as detected by changes in the cytosolic calcium. Taken together, these studies have led to the discovery that the C-terminal region of c-Cbl plays a crucial role in the temporal and spatial control of 5-HT2AR recycling.
Keywords: Receptor Desensitization, Receptor Recycling, Receptor Regulation, Trafficking, Ubiquitin Ligase, Serotonin
Introduction
G protein-coupled receptors (GPCR)2 belong to the largest superfamily of plasma membrane proteins, accounting for >1% of the human genome (2). GPCRs transduce extracellular signals into intracellular effectors and control a number of physiological responses, including vision, cognition, metabolism, cardiac function, and neurotransmission (3). Abnormal and/or dysregulated GPCR signaling has been associated with many human diseases, including various types of cancer.
GPCRs are activated at the plasma membrane by agonist binding. Following a change in the conformation of the receptors, GPCRs couple to heterotrimeric G proteins and initiate a variety of downstream signaling pathways (4). Receptor responsiveness is subsequently attenuated, a concept defined as receptor desensitization, by receptor phosphorylation, arrestin binding, and clathrin-mediated endocytosis (5). Interestingly, in addition to signal transduction cascades initiated at the plasma membrane, some receptors may continue to signal from endocytic compartments in a G protein-independent manner. It is now well established that GPCR trafficking is a critical factor that determines spatial and temporal control of GPCR signaling.
Once internalized, GPCRs are sorted in the early endosomes to either recycling pathways leading to receptor resensitization, or sorted for down-regulation in lysosomes, which permanently terminates receptor signaling. Lysosomal sorting of a number of GPCRs involves a covalent modification of receptors with ubiquitin (6). Although some ubiquitin ligases have been identified, future work is required to further characterize and define the specific details of sorting of ubiquitinated GPCRs. In that regard, it has been reported that ubiquitination and down-regulation of protease-activated receptor 2 is mediated by c-Cbl (7), the ubiquitin ligase that is known to regulate sorting of receptor tyrosine kinases to lysosomes (8). Receptor recycling to the plasma membrane, on the other hand, was initially thought to occur via bulk membrane flow. Recent studies, however, revealed that GPCR recycling was a highly specific process. It appeared that specific amino acids residues of GPCRs and interactions with certain sorting proteins were required for efficient trafficking of internalized receptors to the plasma membrane (9).
Previously, others have identified some GPCR interacting proteins involved in determining the fate of GPCRs. However, many aspects of GPCR intracellular trafficking remain mysterious. We have recently characterized a novel role for c-Cbl (ubiquitin ligase) in the active sorting of the epidermal growth factor receptor (EGFR) to recycling endosomes. Although, c-Cbl was implicated in mediating lysosomal degradation of some GPCRs (7), possible roles for c-Cbl in GPCR recycling have not yet been addressed. We report here that c-Cbl interacts the 5-HT2A receptor and that the carboxyl-terminal domain of c-Cbl regulates efficient 5-HT2AR recycling to the plasma membrane and receptor resensitization. Taken together, these results implicate c-Cbl in a novel sorting pathway of 5-HT2AR intracellular trafficking.
EXPERIMENTAL PROCEDURES
Materials
Human embryonic kidney (HEK293) cells were purchased from American Type Culture Collection (Manassas, VA). Serotonin, cyclohexamide, and probenecid were purchased from Sigma. Rabbit polyclonal anti-Rab11, mouse monoclonal anti-v5, rabbit polyclonal anti-GFP antibodies, and Alexa Fluor-conjugated secondary antibodies were from Invitrogen. Rabbit polyclonal anti-early endosome antigen 1 (EEA1) (clone H-300) antibody was from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse monoclonal anti-c-Cbl antibody was from BD Biosciences. Rabbit polyclonal anti-lysosome-associated membrane protein (LAMP) and rabbit polyclonal mannose 6-phosphate receptor (M6PR) antibodies were from Abcam (Cambridge, MA). Mouse monoclonal anti-ubiquitin (clone FK2) antibody was from Biomol (Plymouth Meeting, PA). Mouse monoclonal anti-β-actin (clone AC-15) antibody was from Sigma. Peroxidase-labeled secondary antibodies were from Jackson ImmunoResearch Laboratories (West Grove, PA). SDS-PAGE molecular weight markers were from Bio-Rad.
Cell Culture, RNA Interference Experiments, and DNA Transfection
HEK293 cells were grown in Eagle's minimum essential medium supplemented with 10% (v/v) heat-inactivated fetal bovine serum (Invitrogen) at 37 °C in a humidified atmosphere of 95% air and 5% CO2. Cells were grown to ∼75% confluence and placed in serum-deprived binding media (minimum essential medium supplemented with 0.1% bovine serum albumin) for 24 h prior to treatments. In all experiments 100 μg/ml of cycloheximide, a protein synthesis inhibitor, was used 3 h prior to and during treatments. For synchronized ligand pulse experiments, cells at 4 °C were incubated with an agonist for 60 min in serum-deprived binding medium containing 20 mm HEPES, pH 7.4. Then, the cells were rinsed with ice-cold PBS to remove unbound ligand, following which the bound ligand stimulated 5-HT2AR upon exposure to pre-warmed ligand-free medium at 37 °C.
A mixture of four SMARTselection-designed siRNAs targeting one gene (Thermo Fisher Scientific, Lafayette, CO) were transfected using Oligofectamine (Invitrogen) reagent according to the manufacturer's instructions. A pool of 4 siGenome non-targeting siRNAs, designated as scrambled (SCR) siRNA, was used as control. 72 h following transfection, cell lysates were assayed for silencing effectiveness by Western blotting and immunofluorescence staining.
The expression constructs pcDNA3GFP-Cbl-WT, pcDNA3GFP-Cbl-(1–436), and pcDNA3GFP-Cbl-N have been described previously and kindly provided by Dr. Hamid Band (10, 11). 5-HT2AR was cloned in-frame with the v5 epitope into the pEF6/V5-His TOPO vector using a pEF6/V5-His TOPO TA Expression Kit (Invitrogen). The 5-HT2AR-YFP receptor construct was generated by cloning the cDNA sequence of human 5-HT2AR into XhoI and BamHI sites of EYFP-N1 vector (Clontech, BD Biosciences, Franklin Lakes, NJ). HEK293 cells were transiently transfected with the above constructs using Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen). Initial comparison revealed no difference between mock- and non-transfected cells. Thus, non-transfected cells were subsequently used as controls.
Western Blotting
After treatments, cells were rinsed briefly with PBS and extracted with RIPA buffer (150 mm NaCl, 1 mm EDTA, 0.25% sodium deoxycholate, 1% Nonidet P-40, 0.1% SDS in PBS) containing protease inhibitors. Cells were subsequently sonicated, and protein concentrations were determined by the BCA assay (Pierce). Equal amounts of proteins were separated by SDS-PAGE on 4–12% polyacrylamide gels (Invitrogen), transferred to PVDF membranes, and blocked with 5% milk in PBS for 1 h at room temperature. Following several washes with PBS containing 0.1% Tween, the membranes were incubated with the appropriate dilutions of primary and peroxidase-conjugated secondary antibodies (as directed by the manufacturer) in blocking solution. Immunoblotted proteins were detected using ECL reagents (GE Healthcare).
Immunofluorescence Staining and Confocal Microscopy
Cells were grown on 35-mm lysine-coated, glass-bottom culture dishes (MatTek Corporation, Ashland, MA). After treatments, cells were fixed with freshly prepared 3.7% paraformaldehyde in PBS for 15 min at room temperature. Subsequently, cells were permeabilized with 0.1% Triton X-100 (Sigma) in PBS for 5 min, following which nonspecific binding sites were blocked with 3% normal serum (Santa Cruz Biotechnologies) in PBS for 1 h. Incubations with the appropriate dilutions of primary and Alexa Fluor-conjugated secondary antibodies (as directed by the manufacturer) were performed in blocking solution. Confocal microscopy was performed using a Zeiss LSM 510 META laser-scanning microscope (Carl Zeiss, Inc., Thornwood, NY) equipped with a ×60 objective, using the following laser wavelengths: excitation 488 nm, emission 505–530 nm; excitation 543 nm, emission 560–615 nm; excitation 633 nm, emission 630–700 nm. Quantifications of the colocalization coefficients, derived from measured pixel overlaps between 5-HT2AR and EEA1, Rab11, M6PR, and LAMP, were performed using Zeiss LSM 510 colocalization analysis software. The mean values were averaged from at least 16 independent single cell images.
Immunoprecipitation
After the indicated treatments, HEK293 cells grown in 100-mm dishes were scraped into ice-cold PBS and centrifuged at 10,000 × g. Pellets were lysed in 1 ml of RIPA lysis buffer supplemented with protease and phosphatase inhibitors, as described above. Equal amounts of proteins were precleared by incubation for 60 min at 4 °C with normal mouse or rabbit IgG (Santa Cruz Biotechnologies, Santa Cruz, CA). After a brief centrifugation, the supernatants were removed and incubated overnight at 4 °C with either anti-v5 or anti-GFP antibodies. Immunoprecipitates were captured with 50 μl of either protein A/G-agarose (Santa Cruz Biotechnology), or TrueBlot Ig IP beads (eBioscience, San Diego, CA) at 4 °C for 1 h. The samples were then centrifuged and washed 4 times with 1 ml of RIPA buffer. Proteins were eluted from the beads using Laemmli sample buffer. Samples were subsequently analyzed by SDS-PAGE and Western blotting. The membranes were incubated with the appropriate dilutions of primary and TrueBlot peroxidase-conjugated secondary antibodies (eBioscience, San Diego, CA) (as directed by the manufacturer) in blocking solution.
Cell Surface Reversible Biotinylation
Attenuated receptor recycling was measured biochemically by the gain of intracellular receptors, which had been labeled with disulfide-linked (cleavable) biotin, following biotin stripping. Briefly, HEK293 cells were surface biotinylated with 0.5 mg/ml of cell-impermeable sulfo-NHS-S-S biotin (Pierce) in PBS for 30 min at 4 °C. Excess biotin was subsequently quenched by washing with ice-cold 15 mm glycine. Cells were washed with PBS and incubated in serum-free medium at 37 °C for 1 h prior to treatments. Cells were then incubated on ice with 1 μm 5-HT for 60 min, washed free of unbound ligand, and either collected following stripping of cell surface biotin (biotin removed), or collected without biotin stripping (cell surface receptors), or collected following exposure to prewarmed ligand-free medium at 37 °C for 180 min and cell surface biotin stripping (intracellular receptors). Stripping of cell surface-biotinylated receptors was performed at 4 °C by washing the cells three times for 5 min with ice-cold GSH cleavage buffer (50 mm GSH, 75 mm NaCl, 1 mm EDTA, 1% BSA, 0.75% 10 n NaOH). Cell lysates were subsequently subjected to avidin precipitation and immunoblotting.
Intracellular Calcium Measurements
Cells were grown in lysine-coated 96-well clear-bottom black plates (Greiner Bio-One, Monroe, NC). After treatments, cells were loaded with Calcium 3 dye (Molecular Devices, Sunnyvale, CA) for 60 min at 37 °C in Hanks' balanced salt solution containing 2.5 mm probenecid. The changes in intracellular calcium were detected with fluorometric imaging plate reader (Molecular Devices, Sunnyvale, CA).
Statistical Analyses
Statistical significance was determined using analysis of variance with Dunnett's or Bonferroni post-test to correct for multiple comparisons (GraphPad Prism, version 4). p values of <0.05 were considered statistically significant.
RESULTS
Effects of Serotonin (5-HT) on 5-HT2AR Trafficking
First, we evaluated the ability of serotonin to induce 5-HT2AR endocytosis and intracellular trafficking in HEK293 cells. To minimize the effects of simultaneous 5-HT2AR processing, i.e. receptor endocytosis, sorting, recycling, and/or degradation, we performed a synchronized ligand pulse experiment, as described previously (12), with some modifications successfully characterized by us (1). This protocol allowed us to track a single cohort of liganded receptors. Briefly, the cells transiently transfected with v5-tagged 5-HT2AR were allowed to bind the ligand (1 μm 5-HT) at 4 °C for 60 min, following which the unbound ligand was removed and the cells were transferred to 37 °C to initiate synchronous 5-HT2AR processing (Fig. 1A). We observed that at 0 min 5-HT2AR was mostly localized to the plasma membrane (Fig. 1A, 0 min). Some 5-HT2A receptors also were detected intracellularly, which is indicative of constitutive receptor activity (13). Ten min after the synchronous ligand pulse, there was considerable movement of the 5-HT-stimulated 5-HT2AR into intracellular compartments (Fig. 1A, 10 min), with nearly complete internalization after 30 min (Fig. 1A, 30 min). Three hours following rewarming of the cells, the majority of 5-HT2AR re-appeared on the plasma membrane, with no detectable receptor degradation (Fig. 1A, 180 min). There was minimal internalization of 5-HT2AR in the absence of ligand (not shown), so the internalization of the receptors was the result of stimulation by 5-HT.
FIGURE 1.

Effects of serotonin on 5-HT2AR trafficking. A, serum-deprived HEK293 cells, transiently transfected with 5-HT2AR-v5 for 24 h, were incubated on ice with 1 μm 5-HT for 60 min, washed free of unbound ligand, and exposed to prewarmed ligand-free medium at 37 °C for 0, 10, 30, and 180 min. Cells then were fixed, stained with anti-v5 antibody (visualized with Alexa Fluor 568-conjugated secondary antibody (red)), and analyzed by confocal microscopy. The bar is 5 μm. B, serum-deprived HEK293 cells, which either were NT or transiently transfected with 5-HT2AR-v5 for 24 h, and treated at 37 °C with 1 μm 5-HT for 0 or 180 min. After being washed with ice-cold PBS, cell lysates were subjected to immunoprecipitation (IP) with an antibody to 5-HT2AR-v5, followed by immunoblotting (IB) with an antibody to 5-HT2AR-v5. Data shown in panels A are representative of three independent experiments. Results in panel B are mean ± S.E. of three separate experiments.
To confirm our microscopy observations, we sought to determine the degree of 5-HT2AR down-regulation by means of immunoprecipitation with a monoclonal antibody directed against v5-tagged 5-HT2AR (Fig. 1B). This analysis revealed that treatment with 1 μm 5-HT for 180 min resulted in little or no change in the immunoreactivity of 5-HT2A receptors.
We later focused our efforts on determining the intracellular localization of 5-HT2A receptors following a 30-min synchronized pulse of 5-HT. For these studies, we utilized various markers of subcellular organelles, i.e. EEA1 for early endosomes, Rab11 for the perinuclear slow recycling compartment, M6PR for late endosomes, and LAMP for lysosomes (Fig. 2A). These data show partial and high colocalization patterns of 5-HT2AR with EEA1 and Rab11, respectively, and no apparent colocalization with either M6PR or LAMP. Our microscopy observations were confirmed by evaluation of the colocalization coefficients, which measured pixel overlap between the 5-HT2A receptors and EEA1, Rab11, M6PR, or LAMP, respectively (Fig. 2B). Taken together, these results demonstrate that 5-HT-activated 5-HT2A receptors undergo endocytosis and are recycled via EEA1- and Rab11-positive intracellular compartments back to the plasma membrane.
FIGURE 2.

Effects of serotonin on 5-HT2AR intracellular localization. A, serum-deprived HEK293 cells, transiently transfected with 5-HT2AR-v5 for 24 h, were incubated on ice with 1 μm 5-HT for 60 min, washed free of unbound ligand, and exposed to prewarmed ligand-free medium at 37 °C for 30 min. Cells then were fixed, stained with anti-v5 antibody (visualized with Alexa Fluor 568-conjugated secondary antibody (red)), and anti-EEA1, -Rab11, -M6PR, or -LAMP antibodies (visualized with Alexa Fluor 488-conjugated secondary antibody (green)), and analyzed by confocal microscopy. Data shown are representative of three independent experiments. Yellow indicates colocalization. White arrows pinpoint partial EEA1/5-HT2AR colocalization. The bar is 5 μm. B, the colocalizations between the 5-HT2AR-v5 and EEA1, Rab11, M6PR, or LAMP observed in panels A were quantified using Zeiss LSM 510 META colocalization analysis software. The mean colocalization coefficients, averaged from at least 16 independent single-cell images, represent pixel overlap between the 5-HT2AR-v5 and the respective markers. The coefficients varied from 0 to 1, with 0 corresponding to non-overlapping images and 1 corresponding to 100% colocalization. ## indicate a p of <0.001 versus M6PR and LAMP; ** indicate a p of <0.001 versus EEA1.
Interaction of 5-HT2AR with c-Cbl
Previously, we documented a global role for c-Cbl in the regulation of vesicular sorting of EGFR (1). Thus, in this work we considered the hypothesis that c-Cbl might affect recycling of 5-HT2A receptors. To study this possibility, we first determined whether 5-HT2A receptors were associated with c-Cbl. Fig. 3 shows results from co-immunoprecipitation experiments, in which non-transfected HEK293 (NT) cells or cells transiently transfected with 5-HT2AR were subjected to immunoprecipitation, followed by Western blotting of the c-Cbl. We did not observe any c-Cbl immunoreactivity in the immunoprecipitates from NT cells. In contrast, the endogenous c-Cbl was robustly co-immunoprecipitated with transiently expressed 5-HT2A receptors (Fig. 3, second lane, top panel), indicating an interaction between the two proteins. Our work does not distinguish whether this is a direct interaction, or whether it occurs through an intermediate biding partner.
FIGURE 3.
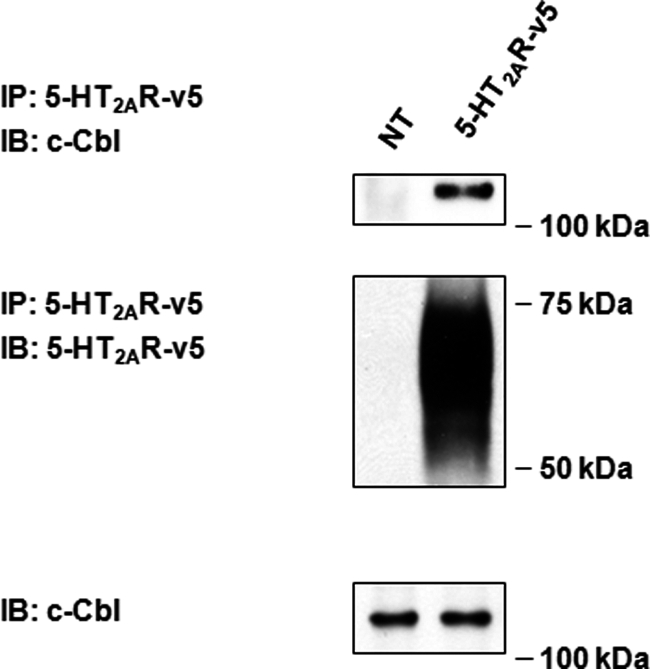
Interaction of 5-HT2AR with c-Cbl. Serum-deprived HEK293 cells either were NT or transiently transfected with 5-HT2AR-v5 for 24 h. After being washed with ice-cold PBS, cell lysates were subjected to immunoprecipitation (IP) with an antibody to 5-HT2AR-v5, followed by immunoblotting (IB) with an antibody to c-Cbl. Blots were then stripped and reprobed for the total amount of 5-HT2AR-v5. Cell lysates used for immunoprecipitation were also probed for the total amount of c-Cbl to normalize for protein expression. Insets are representative of three independent experiments.
Role of c-Cbl in Agonist-induced 5-HT2AR Trafficking
We next sought to determine whether the association of 5-HT2A receptors with c-Cbl had any functional consequences on recycling. To test the possibility that c-Cbl might influence the recycling of 5-HT2AR, we used a siRNA knockdown approach to selectively deplete endogenous c-Cbl (Fig. 4). As shown in Fig. 4A, a pool of four different siRNA duplexes against c-Cbl considerably suppressed the levels of c-Cbl protein, as measured by Western blotting analysis. We then assessed the ability of 5-HT2AR to recycle under control and c-Cbl knockdown conditions in HEK293 cells transiently expressing 5-HT2AR. Fig. 4B demonstrates that 3 h after a synchronized pulse of 5-HT, 5-HT2AR recycling was not affected by scrambled siRNA (SCR siRNA, top panels), as the majority of receptors were found on the plasma membrane. In striking contrast, c-Cbl-knockdown significantly impaired the ability of 5-HT2AR to recycle as long as 3 h after a pulse of 5-HT. This was evidenced by accumulation of 5-HT2A receptors in the intracellular compartments (c-Cbl siRNA, bottom panels). These results imply that c-Cbl is required for efficient trafficking of the 5-HT2A receptors. To our knowledge this is the first evidence showing that c-Cbl is involved in GPCR recycling.
FIGURE 4.

Role of c-Cbl in agonist-induced 5-HT2AR trafficking. Serum-deprived HEK293 cells, transiently transfected with control SCR or c-Cbl siRNA for 72 h (A), or transiently transfected with SCR or c-Cbl siRNA for 72 h and 5-HT2AR-v5 for 24 h (B), were immunoblotted with anti-c-Cbl or -β-actin antibodies (A) or 180 min after a synchronized pulse of 1 μm 5-HT (B), fixed, stained with anti-5-HT2AR-v5 antibody (visualized with Alexa Fluor 568-conjugated secondary antibody (red)) and anti-c-Cbl antibody (visualized with Alexa Fluor 488-conjugated secondary antibody (green)), and analyzed by confocal microscopy. White asterisk shows position of c-Cbl-depleted cells. Data shown are representative of three independent experiments. Bar, 5 μm.
Differential Association of c-Cbl Mutants with 5-HT2AR
Our microscopy studies presented in Fig. 4B clearly demonstrate that c-Cbl is involved in 5-HT2A receptor trafficking. Because ubiquitin activity of c-Cbl is critical for agonist-induced EGFR degradation (14) and recycling (1), it was essential to investigate whether ubiquitin ligase activity of c-Cbl played any role in the recycling of the 5-HT2A receptor. Accordingly, in the next set of experiments, we compared the effects of wild-type c-Cbl (c-Cbl-WT) with two C-terminal truncation c-Cbl mutants, i.e. c-Cbl-(1–436)-GFP, which maintained ubiquitin ligase activity and c-Cbl-N-GFP without ubiquitin ligase activity (Fig. 5A).
FIGURE 5.

Differential association of c-Cbl mutants with 5-HT2AR. A, schematic representation of full-length c-Cbl (WT) and its two C-terminal truncated mutants (c-Cbl-(1–436) and c-Cbl-N); TKB, tyrosine kinase-binding domain; L, linker region; RF, RING finger domain; P, proline-rich region; UBA, ubiquitin-associated domain. HEK293 cells, which had been either NT or transiently transfected with c-Cbl-WT-GFP, c-Cbl-(1–436)-GFP, or c-Cbl-N-GFP (B) and 5-HT2AR-v5 for 24 h (C and D), were (B) immunoblotted with anti-c-Cbl-GFP antibody, or (C and D) treated with 1 μm 5-HT for (C) 0 or 180 min, or (D) 0 or 5 min, subjected to immunoprecipitation (IP) with an antibody to the 5-HT2AR-v5, followed by immunoblotting (IB) with antibodies to 5-HT2AR-v5 (C) or c-Cbl-GFP (D). Blots in D were then stripped and reprobed for the total amount of 5-HT2AR-v5. Cell lysates in C and D used for immunoprecipitation were also probed for the total amount of c-Cbl-WT-GFP, c-Cbl-(1–436)-GFP, and c-Cbl-N-GFP to normalize for protein expression. Data shown are representative of three independent experiments. Results are mean ± S.E. (n = 3). ** indicates a p of <0.01 versus c-Cbl-(1–436) “0”; ## indicates a p of <0.01 versus c-Cbl-(1–436) “5.”
First, we assessed the expression levels of truncated c-Cbl mutants by Western blotting (Fig. 5B). Briefly, HEK293 cells were transiently transfected with the expression plasmids encoding c-Cbl-WT-GFP and two truncated mutants of c-Cbl also tagged with GFP. Following lysis of the cells, we determined in parallel the expression of all c-Cbl variants by immunoblotting with anti-GFP antibody. As seen in Fig. 5B, the three proteins were uniformly expressed in HEK293, with expected relative mobilities on a polyacrylamide gel. We further established whether c-Cbl mutants affected the expression levels of 5-HT-induced 5-HT2A receptors (Fig. 5C), and whether c-Cbl mutants maintained the previously observed interactions between 5-HT2AR and c-Cbl (Fig. 5D). To test this, we co-transfected 5-HT2AR with c-Cbl-WT, c-Cbl-(1–436), or c-Cbl-N in HEK293 cells and exposed the cells to 1 μm 5-HT for 180 (Fig. 5C) or 5 min (Fig. 5D). Following immunoprecipitation of v5-tagged 5-HT2A receptors, we observed that neither c-Cbl-WT nor c-Cbl mutants significantly altered the overall levels of 5-HT2A receptors upon exposure to 5-HT (Fig. 5C). Most importantly, however, the quantitative analysis of c-Cbl band intensities, normalized to the total 5-HT2AR, revealed that in contrast to c-Cbl-WT (Fig. 5D, second and third lanes), c-Cbl-(1–436) and c-Cbl-N (Fig. 5D, fourth and fifth and sixth and seventh lanes, respectively) associated poorly with the 5-HT2A receptors. Interestingly, the interaction between the two proteins in cells transfected with wild-type c-Cbl remained unchanged despite exposure to 5-HT for 5 min (compare second and third lanes in Fig. 5D) and lasted throughout the 180-min stimulation (data not shown). These results could indicate that 5-HT2A receptors and c-Cbl constitutively interact with each other in the absence of the agonist. This work could also suggest that the C-terminal region of c-Cbl is mandatory for its interaction with 5-HT2A receptors, and furthermore, that the decreased interaction between c-Cbl and 5-HT2AR was independent of c-Cbl ubiquitin ligase activity. However, the situation is much more complex in that the truncation mutants both appear to function as dominant-negative constructs by preventing normal 5-HT2AR recycling by endogenous c-Cbl.
Role of c-Cbl Carboxyl Terminus in 5-HT2AR Recycling
Next, we investigated whether the carboxyl terminus of c-Cbl was involved in 5-HT2AR intracellular trafficking. HEK293 cells overexpressing 5-HT2A receptors and c-Cbl mutants were treated with a synchronized pulse of 1 μm 5-HT and subsequently followed for 3 h. We found that expression of wild-type c-Cbl did not change the 5-HT2AR intracellular processing profile (Fig. 6A, top panels). Indeed, the synchronization studies revealed a pattern of receptor surface expression, which was similar to that obtained with the endogenous wild-type c-Cbl (compare Fig. 1A, bottom panels, 180 min, with Fig. 6, A, top panels, c-Cbl-WT). In contrast, c-Cbl-N and cCbl-(1–436) did not induce changes in 5-HT2AR internalization (data not shown), both mutants altered the degree of receptor recycling. These effects were clearly illustrated in confocal images, where 5-HT2A receptors were found sequestered in tightly packed, centrally located intracellular vesicles (Fig. 6A, middle panels for c-Cbl-N and bottom panels for c-Cbl-(1–436)). Our confocal microscopy analysis was confirmed by quantitative measurements of 5-HT2AR attenuated recycling. As seen in Fig. 6B, cell surface receptor biotinylation with a cleavable biotin revealed that c-Cbl-N and c-Cbl-(1–436) mutants impaired efficient 5-HT2A receptor recycling. This was evidenced by the gain of intracellular receptors following biotin stripping of cells transfected with c-Cbl-N and c-Cbl-(1–436) (Fig. 6B, compare fourth lane, c-Cbl-WT I, with seventh, c-Cbl-N I, and tenth lanes, c-Cbl-1–436 I). It should be noted that, consistent with our c-Cbl mutant results, we demonstrated that c-Cbl knockdown significantly inhibited efficient receptor recycling, as compared with cells transfected with control scrambled siRNA (supplemental Fig. S1, compare fourth lane, SCR I, and seventh lane, c-Cbl I). We next characterized receptor-containing vesicles in cells transfected with c-Cbl-N (Fig. 6C) and c-Cbl-(1–436) (data not shown) using dual-label confocal microscopy. Fig. 6C shows representative single-cell confocal images demonstrating 5-HT2AR localization following a synchronized pulse of 5-HT relative to early endosome, recycling endosome, late endosome, and lysosome markers. Our analysis revealed substantial colocalizations between 5-HT2A receptors with EEA1 and Rab11, and very minimal colocalization with M6PR and LAMP in cells expressing c-Cbl-N or c-Cbl-(1–436).
FIGURE 6.

Role of c-Cbl carboxyl terminus in 5-HT2AR recycling. 180 min following a synchronized pulse of 1 μm 5-HT, serum-deprived HEK293 cells, transiently co-transfected with (A) c-Cbl-WT-GFP, or c-Cbl-(1–436)-GFP, or c-Cbl-N-GFP and 5-HT2AR-v5, or (C) c-Cbl-N-GFP and 5-HT2AR-v5 for 24 h, were fixed, stained with anti-v5 antibody (visualized with Alexa Fluor 568-conjugated secondary antibody (red)) and anti-EEA1, -Rab11, -M6PR, or -LAMP antibodies (visualized with Alexa Fluor 633-conjugated secondary antibody (green)), and analyzed by confocal microscopy. Data shown are representative of three independent experiments. Yellow indicates colocalization. The bar is 5 μm. B, following cell surface biotinylation with a cleavable biotin, serum-deprived HEK293 cells, which had been either NT or transiently transfected with c-Cbl-WT-GFP, c-Cbl-(1–436)-GFP, or c-Cbl-N-GFP and 5-HT2AR-v5 for 24 h, were incubated on ice with 1 μm 5-HT for 60 min, washed free of unbound ligand, and either collected following stripping of cell surface biotin (biotin removed, R), or collected without biotin stripping (cell surface receptors, CS), or collected following exposure to prewarmed ligand-free medium at 37 °C for 180 min and cell surface biotin stripping (intracellular receptors, I). Cell lysates were then subjected to immunoprecipitation (IP) with an antibody to 5-HT2AR-v5, followed by immunoblotting (IB) with an antibody to 5-HT2AR-v5. Cell lysates used for immunoprecipitation were also probed for the total amount of c-Cbl-WT-GFP, c-Cbl-(1–436)-GFP, and c-Cbl-N-GFP. Data shown are representative of three independent experiments. Results are mean ± S.E. (n = 3). D, the colocalizations between the 5-HT2AR-v5 and the respective markers observed in panels C were quantified, as explained in the legend of Fig. 2. ** indicates a p of <0.001 versus M6PR and LAMP.
To quantitatively assess the extent of 5-HT2A receptor colocalization within subplasma membrane intracellular regions, we determined the colocalization coefficients in the same manner as previously described in Fig. 2B (see Fig. 6D). These results demonstrate that expression of c-Cbl mutants lacking the carboxyl terminus lead to increased 5-HT2A receptor localization in EEA1-positive vesicles, when contrasted with results with endogenous wild-type c-Cbl in Fig. 2B, in which 5-HT2AR localized more substantially to Rab11-positive vesicles and less to EEA1-positive endosomes. It should be noted that similar results were obtained with the c-Cbl-(1–436) mutant (data not shown). Together, these findings support an essential role for the carboxyl-terminal domain of c-Cbl in the transition of receptors from early endosomes to Rab11-containing recycling vesicles, and in the efficient recycling of 5-HT2A receptor back to the plasma membrane.
Role of Carboxyl Terminus of c-Cbl in 5-HT2AR Resensitization
Based on our data in Fig. 6 we hypothesized that impaired trafficking of 5-HT2AR in the presence of c-Cbl mutants could affect receptor responsiveness. To evaluate a potential functional significance of the interaction of 5-HT2A receptors with c-Cbl, HEK293 cells transiently co-transfected with 5-HT2AR and c-Cbl constructs were subjected to a synchronized ligand pulse. 5-HT2A receptor responsiveness was subsequently assessed by analysis of Ca2+ mobilization following exposure to either buffer (“untreated”) or 1 μm 5-HT (“post-pulse”) (Fig. 7). As shown in Fig. 7A, 5-HT-induced (“post-pulse”) Ca2+ mobilization in cells expressing c-Cbl-WT and 5-HT2AR was not significantly altered following receptor trafficking in the absence (“veh”) or presence (“5-HT”) of an agonist. This suggests that expression of exogenous c-Cbl does not affect the rates of 5-HT2AR resensitization. In contrast, consistent with our observations that both c-Cbl mutants lacking the carboxyl terminus altered receptor trafficking (Fig. 6A), resensitization of 5-HT2A receptors was significantly inhibited in the presence of c-Cbl-(1–436) or c-Cbl-N (Fig. 7, B and C, respectively). A similar observation was made when 5-HT2AR resensitization was assessed under control and c-Cbl-knockdown conditions in HEK293 cells transiently expressing 5-HT2AR; c-Cbl-knockdown significantly impaired 5-HT2A receptor resensitization (supplemental Fig. S2). These results show a dramatic reduction in the peak amplitude of Ca2+ mobilization between receptors that trafficked in the absence (“veh”) and presence (“5-HT”) of an agonist (see Fig. 7D and supplemental Fig. S2C for quantification). It should be noted that no change was observed in the plateau phases of Ca2+ mobilization. Together, the work in Fig. 7 demonstrates that the carboxyl terminus of c-Cbl is essential for effective 5-HT2AR resensitization.
FIGURE 7.

Role of carboxyl terminus of c-Cbl in 5-HT2AR resensitization. 180 min following a synchronized pulse of 10 μm 5-HT, serum-deprived HEK293 cells transiently co-transfected with 5-HT2AR and c-Cbl-WT (A), c-Cbl-(1–436) (B), or c-Cbl-N (C) for 24 h were loaded with Calcium 3 dye. The responsiveness of 5-HT2AR, which had trafficked in the absence (veh) or presence (5-HT) of an agonist, was assessed with fluorometric imaging plate reader by analysis of Ca2+ mobilization following exposure to either buffer (untreated) or 1 μm 5-HT (post-pulse). Data shown are representative of seven independent experiments. D, the maximum changes in intracellular Ca2+ observed in panels A-C were quantified using GraphPad Prism. Results are mean ± S.E. (n = 7). Asterisks indicate a p of <0.01 versus c-Cbl-WT.
DISCUSSION
It is now generally appreciated that one of the mechanisms of regulation of GPCR function involves receptor movement through endocytic pathway(s). Although much has been learned about the intracellular trafficking of GPCRs (9, 15), many aspects of GPCR sorting machinery and specific proteins involved in determining the fate of GPCRs are incomplete. In this study, we investigated the roles of c-Cbl, the ubiquitin ligase known to regulate sorting and degradation of receptor tyrosine kinases, in 5-HT2AR trafficking and receptor responsiveness. What is new about this work is that we show that c-Cbl is a mediator of recycling of the 5-HT2A receptors to the plasma membrane. To the best of our knowledge, this is the first demonstration that c-Cbl mediates efficient exit of GPCRs from early endosomes into Rab11-positive recycling pathways. Our studies also implicate a role for c-Cbl in controlling GPCR resensitization.
It has been demonstrated that both agonists and antagonists induce endocytosis of 5-HT2AR (16). Although binding of β-arrestin proteins to phosphorylated receptors facilitates internalization of many GPCRs (2), it has been proposed that 5-HT2AR endocytosis is arrestin-independent (17). A previous study also implicated protein kinase C in agonist-induced 5-HT2AR internalization (18). Nevertheless, there are virtually no reports thus far in the literature characterizing post-endocytic intracellular trafficking of 5-HT2A receptors. Following endocytosis, internalized receptors are sorted to either lysosomes to promote receptor proteolytic degradation, or to a recycling pathway to promote receptor return to the plasma membrane. In that regard, our results suggest that serotonin targets 5-HT2AR to a Rab11-positive recycling pathway. It is worth noting, however, that following prolonged exposure to agonists, some recycling GPCRs, e.g. β2-adrenergic receptors, can undergo significant down-regulation (9). Intriguingly, we recently demonstrated that, unlike β2-adrenergic receptors, sustained agonist stimulation induces 5-HT2AR sequestration in the perinuclear region (19), suggesting a more complex mechanism of 5-HT2AR endocytic trafficking.
The most critical observation in the current studies is that knockdown of c-Cbl as well as overexpression of c-Cbl mutants, dysregulates 5-HT2AR recycling. Our studies demonstrate that diminished 5-HT2AR recycling coincides with inefficient interaction between c-Cbl and 5-HT2AR and reduced transition of agonist-activated 5-HT2AR from early endosomes to the Rab11-postive recycling endosomes. As much as these results are somewhat surprising due to known role of c-Cbl in mediating receptor targeting to lysosomal degradation (20), they are consistent with our recent findings implicating c-Cbl as a critical mediator of active sorting of the EGFR to the recycling endosomes (1). Together these findings suggest that c-Cbl may have a novel regulatory role in controlling a common checkpoint during sorting to the recycling pathway. It should be noted that c-Cbl appears to exhibit some specificity toward regulated versus constitutive intracellular endocytic pathways, i.e. knockdowns of c-Cbl had negligible effects on the constitutive trafficking of transferrin receptors (21). Our results do not rule out a role for c-Cbl in the endocytosis of 5-HT2AR, as we did not study the endocytotic process in detail.
One particularly surprising result of the current work is that the carboxyl-terminal domain of c-Cbl, but not necessarily c-Cbl ubiquitin ligase activity, is required for efficient trafficking and recycling of 5-HT2A receptors to the plasma membrane. This was demonstrated by comparing the effects of wild-type c-Cbl with two, previously characterized (22), C-terminal truncation mutants of c-Cbl. The finding that c-Cbl effects on 5-HT2AR recycling are independent of c-Cbl ligase activity is quite intriguing. Indeed, we found that 5-HT2AR was continuously ubiquitinated (data not shown) in the absence or presence of the agonist (potentially caused by a high level of 5-HT2AR constitutive trafficking (23)), yet both mutants, i.e. c-Cbl-(1–436)-GFP, which maintained ubiquitin ligase activity, and c-Cbl-N-GFP without ubiquitin ligase activity, had compromised 5-HT2AR recycling. This warrants further investigation to determine whether c-Cbl is the ubiquitin ligase responsible for 5-HT2AR ubiquitination and to determine the role of receptor ubiquitination in 5-HT2AR trafficking and signaling.
The situation is complicated further by the observation that not only were the truncation mutants unable to support 5-HT2AR recycling, but also showed dominant-negative characteristics in that they were capable of blocking the essential function of endogenous c-Cbl to support recycling. If the truncation mutants merely were unable to support recycling of the 5-HT2AR, one might reasonably expect that native c-Cbl would be able to associate with the receptors leading to unaltered trafficking.
Although our work is consistent with a role for c-Cbl in selectively regulating 5-HT2AR recycling, it is also possible that c-Cbl plays a more general role in vesicular trafficking. Indeed, the fact that two distinct c-Cbl mutants lose appropriate cellular localizations suggests perhaps a general recycling defect. On the other hand, our previous work showed that knockdown of c-Cbl had negligible effects on the constitutive trafficking of transferrin receptors (21). Clearly, more work is needed to determine whether c-Cbl specifically modulates 5-HT2AR recycling, or has more generalized effects on vesicle trafficking.
Nonetheless, our data point to a novel regulatory mechanism whereby an adaptor function for c-Cbl, rather than its ubiqutin ligase activity, seems to modulate 5-HT2AR recycling. Indeed, the multidomain structure of c-Cbl allows it to interact with many partners. During recent years, ∼150 proteins have been identified as targets of c-Cbl (24). The C-terminal region of c-Cbl encompasses the major sites of tyrosine phosphorylation that enable interactions of c-Cbl with SH2 domain-containing proteins; as well as leucine zipper and ubiquitin-associated domains, which mediate dimerization of multiple proteins and bind to ubiquitin residues, respectively (25). Interestingly, c-Cbl has been shown to regulate phosphorylation and the level of hepatocyte growth factor-regulated tyrosine kinase substrate (26), which appears to be required for efficient recycling of some GPCRs (27, 28). It is tempting to speculate that the c-Cbl carboxyl terminus bridges the interaction(s) between 5-HT2AR and protein(s) that control efficient exit of cargo from the early endosomes into the recycling compartments. In addition, the fact that the c-Cbl truncation mutants block the recycling functions of endogenous c-Cbl suggests that the interactions between c-Cbl, the 5-HT2AR, and other partners are highly complex. Future studies are needed to determine how the C terminus of c-Cbl regulates 5-HT2AR trafficking and to identify other possible 5-HT2AR/c-Cbl interacting partners.
It is now evident that receptor recycling pathways promote resensitization of receptor-mediated cellular responsiveness (9). The functional significance of the role of c-Cbl in 5-HT2AR recycling is evident in that c-Cbl is required for resensitization (Fig. 7 and supplemental Fig. S2). This work is significant in that it demonstrates that c-Cbl, which typically mediates receptor degradation, also plays key roles in receptor recycling and resensitization of EGFR (1). Indeed, we recently showed that members of the ESCRT complex, which is intimately involved in protein degradation, also controls recycling of EGF receptors (21). Therefore, the current work adds to an emerging unsuspected paradigm that implicates proteins involved in receptor degradation (c-Cbl, ESCRT proteins) in receptor recycling and resensitization.
In summary, we have discovered a novel role for c-Cbl, the ubiquitin ligase typically associated with receptor degradation, in the recycling and resensitization of the 5-HT2A receptor. This study demonstrates that: 1) 5-HT2A receptors undergo specialized recycling to the plasma membrane through Rab11-positive vesicles, 2) the carboxyl terminus of c-Cbl, but not its ubiquitin ligase activity, is required for efficient exit of 5-HT2AR from early endosomes into Rab11-positive vesicles, and 3) an interaction between 5-HT2AR and carboxyl terminus of c-Cbl is critical for functional receptor recovery and responsiveness. Because dysregulated GPCR trafficking has been associated with a variety of human diseases (15), future insights into the regulation of GPCR endocytic trafficking may provide new approaches for new therapeutic strategies.
Supplementary Material
Acknowledgment
We thank the MUSC Hollings Cancer Center Molecular Imaging Facility for the use of Zeiss confocal microscope.
This work was supported, in whole or in part, by National Institutes of Health Grants DK052448 and GM063909 (to J. R. R.), Department of Veterans Affairs Merit and Research Enhancement Award Program grants (to J. R. R.), a American Heart Association (Mid-Atlantic) fellowship (to A. B.), and laboratory endowments jointly supported by the Medical University of South Carolina Division of Nephrology and Dialysis Clinics, Inc. (to J. R. R.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
- GPCRs
- G protein-coupled receptors
- 5-HT2AR
- 5-hydroxytryptamine 2A receptor
- EEA1
- early endosome antigen 1
- EGFR
- epidermal growth factor receptor
- LAMP
- lysosome-associated membrane protein
- M6PR
- mannose 6-phosphate receptor
- 5-HT
- serotonin
- NT
- not transfected
- SCR
- scrambled.
REFERENCES
- 1. Baldys A., Göoz M., Morinelli T. A., Lee M. H., Raymond J. R., Jr., Luttrell L. M., Raymond J. R., Sr. (2009) Biochemistry 48, 1462–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Drake M. T., Shenoy S. K., Lefkowitz R. J. (2006) Circ. Res. 99, 570–582 [DOI] [PubMed] [Google Scholar]
- 3. Seachrist J. L., Ferguson S. S. (2003) Life Sci. 74, 225–235 [DOI] [PubMed] [Google Scholar]
- 4. Neves S. R., Ram P. T., Iyengar R. (2002) Science 296, 1636–1639 [DOI] [PubMed] [Google Scholar]
- 5. Ferguson S. S. (2001) Pharmacol. Rev. 53, 1–24 [PubMed] [Google Scholar]
- 6. Shenoy S. K. (2007) Circ. Res. 100, 1142–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jacob C., Cottrell G. S., Gehringer D., Schmidlin F., Grady E. F., Bunnett N. W. (2005) J. Biol. Chem. 280, 16076–16087 [DOI] [PubMed] [Google Scholar]
- 8. Acconcia F., Sigismund S., Polo S. (2009) Exp. Cell Res. 315, 1610–1618 [DOI] [PubMed] [Google Scholar]
- 9. Hanyaloglu A. C., von Zastrow M. (2008) Annu. Rev. Pharmacol. Toxicol. 48, 537–568 [DOI] [PubMed] [Google Scholar]
- 10. Lill N. L., Douillard P., Awwad R. A., Ota S., Lupher M. L., Jr., Miyake S., Meissner-Lula N., Hsu V. W., Band H. (2000) J. Biol. Chem. 275, 367–377 [DOI] [PubMed] [Google Scholar]
- 11. Singh A. J., Meyer R. D., Navruzbekov G., Shelke R., Duan L., Band H., Leeman S. E., Rahimi N. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 5413–5418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Berry S. A., Shah M. C., Khan N., Roth B. L. (1996) Mol. Pharmacol. 50, 306–313 [PubMed] [Google Scholar]
- 13. Berg K. A., Harvey J. A., Spampinato U., Clarke W. P. (2005) Trends Pharmacol. Sci. 26, 625–630 [DOI] [PubMed] [Google Scholar]
- 14. Levkowitz G., Waterman H., Zamir E., Kam Z., Oved S., Langdon W. Y., Beguinot L., Geiger B., Yarden Y. (1998) Genes Dev. 12, 3663–3674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Marchese A., Paing M. M., Temple B. R., Trejo J. (2008) Annu. Rev. Pharmacol. Toxicol. 48, 601–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gray J. A., Roth B. L. (2001) Brain Res. Bull. 56, 441–451 [DOI] [PubMed] [Google Scholar]
- 17. Bhatnagar A., Willins D. L., Gray J. A., Woods J., Benovic J. L., Roth B. L. (2001) J. Biol. Chem. 276, 8269–8277 [DOI] [PubMed] [Google Scholar]
- 18. Bhattacharyya S., Puri S., Miledi R., Panicker M. M. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 14470–14475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Idkowiak-Baldys J., Baldys A., Raymond J. R., Hannun Y. A. (2009) J. Biol. Chem. 284, 22322–22331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Duan L., Miura Y., Dimri M., Majumder B., Dodge I. L., Reddi A. L., Ghosh A., Fernandes N., Zhou P., Mullane-Robinson K., Rao N., Donoghue S., Rogers R. A., Bowtell D., Naramura M., Gu H., Band V., Band H. (2003) J. Biol. Chem. 278, 28950–28960 [DOI] [PubMed] [Google Scholar]
- 21. Baldys A., Raymond J. R. (2009) Biochemistry 48, 9321–9323 [DOI] [PubMed] [Google Scholar]
- 22. Bonita D. P., Miyake S., Lupher M. L., Jr., Langdon W. Y., Band H. (1997) Mol. Cell. Biol. 17, 4597–4610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gray J. A., Bhatnagar A., Gurevich V. V., Roth B. L. (2003) Mol. Pharmacol. 63, 961–972 [DOI] [PubMed] [Google Scholar]
- 24. Schmidt M. H., Dikic I. (2005) Nat. Rev. Mol. Cell Biol. 6, 907–918 [DOI] [PubMed] [Google Scholar]
- 25. Swaminathan G., Tsygankov A. Y. (2006) J. Cell. Physiol. 209, 21–43 [DOI] [PubMed] [Google Scholar]
- 26. Stern K. A., Visser Smit G. D., Place T. L., Winistorfer S., Piper R. C., Lill N. L. (2007) Mol. Cell. Biol. 27, 888–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hanyaloglu A. C., McCullagh E., von Zastrow M. (2005) EMBO J. 24, 2265–2283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hanyaloglu A. C., von Zastrow M. (2007) J. Biol. Chem. 282, 3095–3104 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.