

Abstract
In addition to soluble acid hydrolases, many nonlysosomal proteins have been shown to bear mannose 6-phosphate (Man-6-P) residues. Quantification of the extent of mannose phosphorylation and the relevance to physiological function, however, remain poorly defined. In this study, we investigated the mannose phosphorylation status of leukemia inhibitory factor (LIF), a previously identified high affinity ligand for the cation-independent mannose 6-phosphate receptor (CI-MPR), and we analyzed the effects of this modification on its secretion and uptake in cultured cells. When media from LIF-overexpressing cells were fractionated using a CI-MPR affinity column, 35–45% of the total LIF molecules were bound and specifically eluted with free Man-6-P thus confirming LIF as a bona fide Man-6-P-modified protein. Surprisingly, mass spectrometric analysis of LIF glycopeptides enriched on the CI-MPR column revealed that all six N-glycan sites could be Man-6-P-modified. The relative utilization of these sites, however, was not uniform. Analysis of glycan-deleted LIF mutants demonstrated that loss of glycans bearing the majority of Man-6-P residues leads to higher steady-state levels of secreted LIF. Using mouse embryonic stem cells, we showed that the mannose phosphorylation of LIF mediates its internalization thereby reducing extracellular levels and stimulating embryonic stem cell differentiation. Finally, immunofluorescence experiments indicate that LIF is targeted directly to lysosomes following its biosynthesis, providing another mechanism whereby mannose phosphorylation serves to control extracellular levels of LIF. Failure to modify LIF in the context of mucolipidosis II and its subsequent accumulation in the extracellular space may have important implications for disease pathogenesis.
Keywords: Cytokine, Embryonic Stem Cell, Glycoprotein Secretion, Lysosomal Glycoproteins, Lysosomal Storage Disease, Leukemia Inhibitory Factor, Mannose 6-Phosphate, Mucolipidosis II
Introduction
Mannose 6-phosphate (Man-6-P)3-dependent targeting of acid hydrolases to the lysosome remains one of the best studied functions for protein-bound glycans. The biosynthesis of these residues on lysosomal hydrolases and other glycoproteins proceeds via a two-step enzymatic process. The initiating step, catalyzed by the heterohexameric enzyme UDP-N-acetylglucosamine:lysosomal enzyme N-acetylglucosamine-1-phosphotransferase (GlcNAc-1-phosphotransferase), involves the transfer of GlcNAc-1-phosphate to mannose residues on selected high mannose glycans. The second step requires the removal of the GlcNAc residues by the enzyme GlcNAc-1 phosphodiester α-N-acetylglucosaminidase or “uncovering enzyme” to expose Man-6-P monoesters (1, 2). These uncovered Man-6-P monoesters serve as recognition markers for the sorting of newly synthesized lysosomal proteins by the cation-dependent and the cation-independent mannose 6-phosphate receptors (CD-MPR and CI-MPR, respectively) (3, 4). Recently, it has been demonstrated that covered Man-6-P residues can also bind to the CI-MPR via a distinct domain on this receptor (5, 6).
The importance of Man-6-P biosynthesis to human health is highlighted by the fact that mutations in the genes encoding subunits of the GlcNAc-1-phosphotransferase enzyme (GNPTAB and GNPTG) result in the lysosomal disorders, mucolipidosis II (MLII) and mucolipidosis III (ML-III) (4, 7, 8). The clinical manifestations of these diseases are diverse, encompassing skeletal and craniofacial defects, impaired speech, cognitive function, and recurrent lung infections (9). In the absence of GlcNAc-1-phosphotransferase activity, lysosomal hydrolases are not Man-6-P-modified, and most are instead secreted from the cell. The molecular and cellular mechanisms that drive pathology in the various affected tissues of ML-II patients and the specific Man-6-P-modified proteins implicated in individual tissues are incompletely understood. Elucidating such mechanisms will provide a starting point for the development of much needed therapies for these disorders.
In addition to acid hydrolases, several nonlysosomal proteins, including latent transforming growth factor β1 (TGF-β1) (10), leukemia inhibitory factor (LIF) (11, 12), proliferin (13), renin precursor (14), and T-cell activation antigen CD26 (15), have been identified as Man-6-P-modified glycoproteins. However, the manner by which mannose phosphorylation mediates the normal function of these proteins and the extent to which they are modified in a physiological context is not well defined. Previous studies on the mannose phosphorylation of secreted proteins have suggested a wide range of possible functions. Man-6-P residues on latent TGF-β are thought to facilitate its interaction with the CI-MPR at the plasma membrane and subsequent activation via the urokinase plasminogen activator receptor/plasminogen cascade (16–18). In the case of LIF, Man-6-P-mediated internalization by the CI-MPR limits its extracellular levels, in a manner analogous to the down-regulation of IGF-II by this same receptor (11). The biological functions of LIF signaling are diverse, encompassing the coordination of cytokine expression in immune cells, maintenance, and modulation of stem cells (19, 20) and bone homeostasis (21, 22). Moreover, LIF has been implicated in several disease states, including multiple sclerosis (23), cancer (24, 25), and arthritis (26). Because LIF has been shown to play a role in the homeostasis of bone and cartilage (two tissues strongly affected in ML-II), we have hypothesized that loss of Man-6-P modification on this cytokine in the context of ML-II might initiate or propagate the disease process.
To begin to address this hypothesis, we undertook a detailed biochemical analysis of the glycosylation status of human LIF (hLIF) and mouse LIF (mLIF) expressed in multiple cell lines and interrogated the role of mannose phosphorylation on LIF in cultured cells. Our results confirmed the status of LIF as a bona fide Man-6-P-modified glycoprotein and unexpectedly revealed that any of the six N-glycans on this protein can bear Man-6-P residues. Our data further demonstrated that the mannose phosphorylation of LIF serves as a mechanism to control its extracellular levels through direct intracellular degradation of Man-6-P-modified LIF within the lysosome as well as rapid Man-6-P-mediated uptake following secretion. The implications of these novel findings in the pathogenesis of ML-II and relevance of mannose phosphorylation on other nonlysosomal proteins are discussed.
MATERIALS AND METHODS
Cell Lines, Plasmids, and Reagents
CHO and HeLa cells were obtained from the ATCC, and CHO with stable expression of mLIF (CHO-mLIF) was a generous gift from Dr. Steven Dalton (University of Georgia, Athens). All cells were maintained in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum (FBS) supplemented with 100 μg/ml penicillin in addition to streptomycin and maintained in a humidified 5% CO2 atmosphere. All site-directed mutagenesis experiments were performed using the manufacturer's protocol (Agilent/Stratagene, Santa Clara, CA). Cell cultures were metabolically labeled with Tran35S-label (MP Biomedicals, Solon, OH) in methionine- and cysteine-free DMEM (Invitrogen). The CI-MPR lectin affinity column was a generous gift from Dr. Peter Lobel (Robert Wood Johnson Medical School). LIF polyclonal antiserum was obtained from Anne Godard, and the anti-DsRed (mCherry) antibody was purchased from Clontech. Free mannose 6-phosphate and glucose 6-phosphate, alkaline phosphatase (Escherichia coli recombinant and bovine intestine), naphthyl phosphate disodium salt, and Fast Red TR salt were obtained from Sigma. Kifunensine was obtained from Cayman Chemical, and endoglycosidase Hf and N-glycosidase F (PNGase F) were purchased from New England Biolabs (Ipswich, MA). The transfection reagents Lipofectamine PLUS and Opti-MEM were obtained from Invitrogen, and ESGRO® LIF (Millipore, Billerica, MA) was a kind gift from Dr. Lianchun Wang (University of Georgia, Athens).
Cell Transfection and Preparation of Media Samples
Cells were plated in a 35-mm (6-well) dish 4 h before transfection at a cell density of 2.5 × 105 cells/ml. Transfection of cell monolayers with 1 μg of DNA from each construct was performed using the Lipofectamine PLUS system. This transfection medium was replaced the next day with 1 ml of serum-free media. Samples were collected 48 h after the initial transfection (unless otherwise indicated) and concentrated using Centricon 10 tubes to a volume of 250 μl. For the kifunensine experiments, transfection medium was replaced with 1 ml of serum-free media supplemented with 2 μg/ml kifunensine, and cells were incubated overnight at 37 °C prior to analysis.
CI-MPR Affinity Chromatography and Western Blot Analysis
Concentrated media samples (100 μl) and purified LIF (1 μg) were diluted into 1 ml of CI-MPR column buffer (50 mm imidazole, pH 6.0, 150 mm NaCl, 0.05% Triton X-100, and 5 mm EDTA) and then applied to a 0.6-ml CI-MPR affinity column. Fractions (1 ml each) were eluted first with column buffer and then 5 mm glucose 6-phosphate and finally with 5 mm mannose 6-phosphate to elute the Man-6-P-modified LIF. Protein within the column fractions was precipitated using 9 volumes of 100% cold ethanol overnight at 20 °C followed by centrifugation at 1,450 × g for 30 min. Precipitates were washed with 90% ethanol and centrifuged for 10 min at 4 °C and 19,400 × g. Samples were solubilized by boiling in 3% SDS and analyzed by SDS-PAGE on a 12% gel. Transferred proteins were probed with an anti-LIF polyclonal antibody overnight at 4 °C followed by incubation with an anti-rabbit HRP secondary antibody and reactive bands visualized using ECL detection reagents (GE Healthcare). In all cases, Western blot analysis was performed multiple times to verify reproducibility. In some cases, media samples (40 μl) were first treated for 1.5 h with either 50 units of endoglycosidase H or 15 units of PNGase F at 37 °C or no enzyme (control) prior to SDS-PAGE and Western blot analysis. For alkaline phosphatase (AP) experiments, purified human LIF was treated in combination with 5 units of bovine intestine and 1 unit of E. coli recombinant alkaline phosphatase in AP buffer (5 mm Tris, pH 7.5, 1 mm MgCl2, and 1 mm CaCl2) overnight at 37 °C and then analyzed as described above. For densitometry analysis, ImageJ 1.43u (27) was used to quantify the Western blot bands.
Purification of Recombinant hLIF and Isolation of Unmodified and Man-6-P-modified LIF Glycoforms
Recombinant human LIF was purified as described previously (28). For the isolation of the two distinct LIF glycoform pools, 20 μg of purified hLIF was fractionated over the CI-MPR column, and unmodified (unbound) LIF and Man-6-P-modified LIF (5 mm Man-6-P eluted) were collected. Buffer was exchanged twice with PBS and concentrated to 50 μl using Centricon 10 tubes (Millipore, Billerica, MA), followed by BCA protein assay (Pierce) to determined protein concentration.
CI-MPR Chromatography Enrichment of LIF and MS Analysis of Glycopeptides
Enrichment of Man-6-P-modified LIF glycopeptides was performed as described previously (29) with modifications. Briefly, tryptic peptides generated following reduction and alkylation of the protein were transferred to a tube containing a 50-μl bed volume of CI-MPR and rocked for 1 h at 4 °C. The mixture was then placed in a microcolumn containing an additional 50-μl bed volume of CI-MPR and centrifuged for 30 s at 100 × g in a bench-top microcentrifuge to remove flow-through but careful not to completely dry the resin. The flow-through was reloaded 10 times and repeated as described. Then the column was washed twice with 10% EtOH in water and subsequently washed three times with 500 μl of PBS, once with 500 μl of PBS containing 10 mm glucose 6-phosphate, and finally with 500 μl of 20 mm sodium phosphate buffer, pH 6.9, and then centrifuged again to remove excess buffer. The beads were resuspended in 100 μl of 20 mm sodium phosphate buffer, pH 6.9, containing 10 mm Man-6-P and incubated on ice for 5 min. The column was then centrifuged briefly, and the first fraction was collected. Another 100 μl of Man-6-P-containing buffer was added, and the column was then centrifuged to remove all water and reapplied. Finally, the column was washed with 50 μl of water to remove any remaining glycopeptides and centrifuged again. Man-6-P fractions were dried and resuspended to a final concentration of 20 mm sodium phosphate, pH 6.9, in 18O-H2O. Likewise, flow-through or peptide samples before column enrichment were similarly resuspended. All peptide samples were digested with PNGase F overnight at 37 °C as described previously (30). Resulting deglycosylated peptides were desalted and then analyzed on a hybrid linear ion trap-Orbitrap mass spectrometer (LTQ-Orbitrap-XL; ThermoFisher) via nLC-MS/MS essentially as described previously (30). All full MS spectra (300–2000 m/z) were captured in the Orbitrap at 60,000 resolution, and the top five peaks were analyzed in the linear ion trap following CID (dynamic exclusion of 2 for 30 s). MS/MS data were analyzed using Bioworks (Sequest algorithm) against the Swiss-Prot human/mouse/rat data base allowing for dynamic modifications +16 Da for methionine (oxidation), +57 Da for cysteine (carboxyamidomethylation), and +3 Da for Asn (glycosylated Asn to Asp (18O) conversion) with a parent mass tolerance of 20 ppm and stringently filtered for 2+/3+ ions having Xcorr greater than 3.0/4.0, respectively. This filtering resulted in a false discovery rate of less than 1% for all modified peptides. Furthermore, the position of a 3-dalton addition was manually confirmed to be at the site of the formerly glycosylated Asn in the MS/MS spectra.
Metabolic 35S Labeling
Metabolic labeling was carried out as described previously (33).
Transfection of Triple Glycan-deleted LIF Mutants and Secretion Assay
N-Sequon sites were mutated using the QuickChange XL site-directed mutagenesis kit (Agilent Technology, Santa Clara, CA). Triple glycan-deleted mutants (TDMs) were transfected into CHO cells (as described above), and media aliquots were either subjected to CI-MPR lectin chromatography to assess levels of mannose phosphorylation or secretion assays were performed. For secretion assays, transiently transfected TDM LIF mutants were incubated for various culture times in serum-free media, collected, concentrated, and subjected to Western blot analysis or Coomassie staining (to determine equivalent amounts of secreted protein). In addition, media samples were normalized to total secreted protein using micro BCA protein assay.
Self-renewal Assay Using Embryonic Stem Cells (ESCs)
The self-renewal assay was performed as described previously (31) with modifications. Briefly, ESCs were seeded at clonal density and cultured for 5 days in ES medium (without LIF) supplemented with 10% FBS in equal concentrations of ESGRO® (nonglycosylated) LIF (control), unmodified LIF, or isolated Man-6-P-modified LIF. Equivalent cultures were treated with 5 mm Man-6-P. The amount of LIF used was based on the ESGRO® LIF concentration needed to maintain the cells in self-renewal. For the alkaline phosphatase experiments, cells were washed and fixed in 4% paraformaldehyde, and AP staining was determined by incubation with staining buffer containing 8 mm MgCl2, 25 mm Tris-HCl, pH 9.0, 0.4 mg/ml naphthyl phosphate, and 1 mg/ml Fast Red TR. In each independent experiment, 150 colonies in each well were scored, and the percentage of AP-positive colonies was calculated. AP-positive stained colonies with tightly packed dome-shaped morphology were scored as undifferentiated, and colonies with a flattened, nonuniform morphology and variable staining were scored as differentiated.
LIF, LIF-mCherry, sp-mCherry, and Plasmid Preparation
Human LIF (full sequence, including signal peptide) I.M.A.G.E. clone (clone I.D. 7939578) was obtained from Open Biosystems (Thermo Scientific, Waltham, MA) and was subcloned, using PCR, with end primers, including flanking BamHI and XbaI restriction sites. The PCR product was ligated into the pcDNA 3.1 expression vector. For LIF-mCherry, the mCherry plasmid was obtained from Clontech and PCR subcloned with flanking EcoRI and XbaI restriction sites concurrently with LIF, adding BamHI and EcoRI restriction sites to the ends of its sequence. Both PCR products were subjected to triple ligation with pcDNA 3.1 to create the LIF-mCherry fusion. Generation of mCherry plasmid with a signal peptide (sp-mCherry) was synthesized by GeneArt (Invitrogen) with flanking restriction sites (BamHI and EcoRI) and subsequently subcloned into pcDNA 3.1.
Immunofluorescence and Confocal Microscopy
For immunofluorescence experiments, cells were seeded on coverslips and cultured overnight prior to analyses. Cells were washed with phosphate-buffered saline, fixed in 3.7% formaldehyde in PBS, and stained as described previously (32). The standard dilution buffer for both primary and secondary antibody incubations contained PBS, pH 7.4, 0.1% Triton X-100, and 1 mg/ml bovine serum albumin. Confocal images were acquired using an Olympus FV1000 laser scanning microscope equipped a ×60 oil immersion (numerical aperture 1.4) objective. Stacks of 0.25-μm (based on calculated optimums) optical sections were collected in the z-dimension and subsequently collapsed into a single image (maximum intensity or z-projection) unless otherwise noted.
RESULTS
Overexpression in CHO and HeLa Cells Results in the Secretion of LIF That Is Highly Man-6-P-modified
In an effort to quantify the relative amount of secreted LIF protein that contains mannose-phosphorylated glycans, we subjected purified human LIF (hLIF) and culture media from CHO cells stably expressing mouse LIF (CHO-mLIF) to affinity chromatography using an immobilized CI-MPR column. Protein from each column fraction was precipitated and resolved by SDS-PAGE, and LIF levels were analyzed by Western blot. As shown in Fig. 1A, a substantial fraction of purified hLIF and CHO-mLIF bound to the CI-MPR column, indicating that one or more of its glycans contained Man-6-P residues. We noted in all cases that the bound LIF had a consistently slightly faster electrophoretic mobility, likely due to differences in glycan processing upon modification with Man-6-P residues. Similar results were obtained when hLIF was expressed in HeLa and CHO cells by transient transfection (Fig. 1B). Interaction with the CI-MPR column was Man-6-P-mediated because treatment of CHO-mLIF with alkaline phosphatase prior to analysis effectively abolished column binding (Fig. 1C). Quantitative densitometry of the Western blots showed that 35–45% of the total LIF molecules were decorated with Man-6-P-containing glycans. In general, these values were higher than earlier estimates of the mannose phosphorylation on endogenous LIF (5%) or transfected CHO cells (15%) (11). These discrepancies may reflect the different techniques utilized to gauge the level of mannose phosphorylation in the respective studies. Nevertheless, the extent of mannose phosphorylation of secreted LIF in CHO and HeLa cells is comparable with the level seen on overexpressed cathepsin D in these cell lines indicating that LIF is a good substrate for the GlcNAc-1-phosphotransferase enzyme (33).
FIGURE 1.
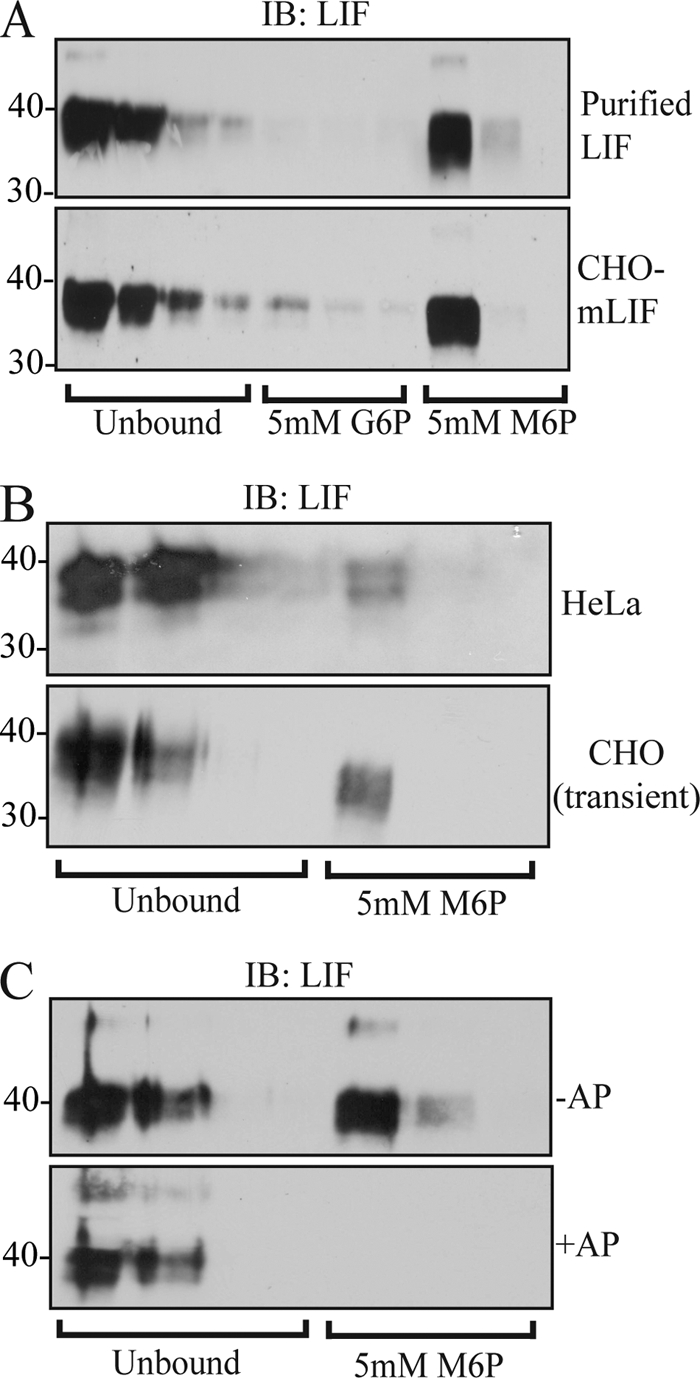
Secreted mouse and human LIF are modified with Man-6-P residues. A, immunoblot (IB) analysis of purified hLIF (top panel) and secreted mLIF from stably overexpressing CHO cells (CHO-mLIF; bottom panel) fractionated on a CI-MPR affinity column. M6P, Man-6-P; G6P, Glc-6-P. B, immunoblot analysis of precipitated column fractions following transient expression of hLIF in HeLa and CHO. C, treatment of CHO-LIF media with AP attenuates binding of LIF to the CI-MPR column, demonstrating that the interaction is specifically mediated by Man-6-P-modified glycans.
Changes in N-Glycan Processing Do Not Result in Substantial Differences in the Extent of LIF Mannose Phosphorylation
We treated CHO cells that were transiently transfected with hLIF with the Golgi mannosidase I inhibitor kifunensine to determine whether changes in N-glycan processing would alter the overall level of mannose phosphorylation. Inhibition of Golgi mannosidase I action is known to prevent the processing of Man8GlcNAc2 to Man5GlcNAc2 structures, resulting in failure of these glycans to be converted into a complex type (34). As shown in Fig. 2A, kifunensine treatment caused all of the N-glycans on LIF to remain as high mannose type. Despite the lack of further glycan processing, no significant difference in the amount of LIF that bound to the CI-MPR column was noted (Fig. 2B). These data indicate that the mannose phosphorylation of LIF does not appear to be influenced by changes in the processing of its N-glycans. Furthermore, it is also consistent with the observations that GlcNAc-1-phosphotransferase is localized earlier within the Golgi than Golgi mannosidase I (35).
FIGURE 2.

Increasing the high mannose glycoforms on LIF does not potentiate further mannose phosphorylation. A, immunoblots (IB) of secreted hLIF following endoglycosidase Hf (eHf) and PNGase F (PNG) treatment of transfected CHO cells. B, immunoblot analysis of hLIF from transfected CHO cells following kifunensine (Kfn) treatment (2 μg/ml) and CI-MPR column chromatography. Note that although kifunensine treatment completely inhibits glycan maturation, mannose phosphorylation of LIF does not increase.
Site Mapping of CI-MPR-enriched Glycopeptides on Purified hLIF Reveals That All Six Glycans Can Bear Man-6-P Residues
Whereas the experiments above clearly demonstrated that LIF expressed in multiple cell culture systems is mannose-phosphorylated, they do not provide any information regarding which N-glycans bear Man-6-P residues. To address this question, we undertook a mass spectrometric analysis of glycopeptides generated from purified human LIF. Our initial analysis demonstrated complete occupancy of the six potential N-glycan sites on human LIF (data not shown). The purified hLIF was next subjected to proteolysis with trypsin, and the resulting glycopeptides were enriched using a CI-MPR affinity microcolumn. The expected profile of LIF glycopeptides following tryptic digestion is shown in Fig. 3. Following collection of unbound peptides and the elution of Man-6-P-bearing glycopeptides with free Man-6-P, these two pools were deglycosylated via PNGase F in O18-H2O and then analyzed by tandem mass spectrometry. Surprisingly, we found that all five glycopeptides could be enriched on the CI-MPR column, likely indicating that any of the six N-glycans on LIF can be modified with Man-6-P residues (Table 1). It should be noted that one of the glycopeptides contains two occupied Asn residues, and thus it is possible that only one of these N-linked sites contains a Man-6-P-modified glycan. Importantly, the nonglycosylated peptides were only recovered in the unbound pool demonstrating the specificity of the column (data not shown).
FIGURE 3.

Amino acid sequence, tryptic peptides, and N-glycan sites of human LIF. hLIF is a 181-amino acid protein containing six potential N-glycan sites (shown in boldface). These N-linked sites are conserved in the mouse sequence. Digestion of hLIF with trypsin yields five glycopeptides (underlined) with one peptide bearing two glycans. The LIF sequence is shown without the signal peptide.
TABLE 1.
Tandem mass spectrometry identification of modified peptides following CI-MPR chromatography enrichment
N* represents a +3-dalton modification (conversion of glycosylated Asn to Asp by PNGase F in 18O-H2O).
| LIF-glycosylated peptides | Charge state (z) | Data assignment |
|
|---|---|---|---|
| Xcorr | Mass accuracy | ||
| ppm | |||
| (−)SPLPITPVN*ATCAIR(H) | 2 | 4.37 | −2.4 |
| 3 | 4.12 | −1.7 | |
| (R)SQLAQLN*GSANALFILYYTAQGEPFPNNLDK(L) | 3 | 5.64 | −4.6 |
| (K)LCGPN*VTDFPPFHAN*GTEK(A) | 2 | 3.93 | 0.3 |
| 3 | 4.27 | −3.2 | |
| (R)IVVYLGTSLGN*ITR(D) | 2 | 4.34 | −2.9 |
| (K)LN*ATADILR(G) | 2 | 3.49 | −2.2 |
Decreased Levels of LIF Mannose Phosphorylation Correlate with Greater Extracellular Accumulation
We next generated several glycan-deleted LIF mutants to investigate the relative utilization of the various N-glycan sites for Man-6-P addition and to determine the impact of reduced mannose phosphorylation of extracellular LIF levels. These mutants were expressed in CHO cells, and secreted LIF was subjected to analysis by CI-MPR affinity chromatography and Western blot. Interestingly, we found that all of the single glycan deletion mutants exhibited the same overall level of column binding compared with wild type LIF, suggesting that loss of no single glycan substantially altered the mannose phosphorylation of the protein (supplemental Fig. 1). In contrast to the single deletion mutants, the TDMs that were generated displayed a wide variability in the level of mannose phosphorylation, depending on which sites were mutated (Fig. 4A). One such mutant, TDM31, failed to bind the column, suggesting a complete lack of Man-6-P-modified glycans on secreted LIF, whereas another mutant TDM95 showed substantially increased CI-MPR column binding compared with fully glycosylated LIF. The column binding of a third mutant TDM56 was similar to wild type LIF. Thus, despite the fact that all glycopeptides could be identified as containing Man-6-P residues, their relative utilization was not uniform.
FIGURE 4.

Loss of mannose phosphorylation results in the extracellular accumulation of LIF. A, representative immunoblots (IB) of triple glycan-deleted LIF mutants (TDMs) fractionated on the CI-MPR affinity column as follows: TDM31, N31:56:85Q; TDM56, N56:85:95Q; and TDM95, N95:118:138Q. M6P, Man-6-P. B, CHO cells were transfected with TDM31, -56, or -95 constructs, and secreted LIF levels were assayed by Western blot (n = 3) over the various culture times, with and without PNGase F (PNG). As a loading control, secreted protein was normalized using micro BCA protein assay in addition to Coomassie Blue stains (B, bottom panel). This analysis revealed that similar amounts of protein were synthesized for the various glycan-deleted mutants, suggesting that removal of N-glycan sites does not generally affect the synthesis or glycosylation of these mutants. C, autoradiograph of immunoprecipitated TDM31 and TDM56 LIF mutants from CHO cells pulse-labeled for 2 h with [35S]methionine. D, quantification of densitometry analysis comparing the fold change of TDM31 and -56 to TDM95 at 96 h post-transfection.
To determine the relationship between mannose phosphorylation and extracellular levels, CHO cells were transfected with the various TDM constructs, and culture media were collected over several days. Aliquots of media, normalized to total secreted protein, were subjected to SDS-PAGE and Western blot analysis to gauge the extracellular levels of LIF (Fig. 4B). Parallel experiments revealed no major differences in the amount of secreted proteins in the media samples as detected by Coomassie staining. Furthermore, metabolic labeling and analysis of newly synthesized cellular LIF from transfected cultures showed that equal amounts of two of these mutants, TDM31 and TDM56, were being made (Fig. 4C). Although levels of LIF were comparable for the three constructs tested at 48 h post-transfection, clear differences in the amount of extracellular LIF can be noted at 96 h post-transfection. Analysis of the Western blots with the PNGase F-treated samples by densitometry (Fig. 4D) revealed that, on average, TDM31 and -56 were present at higher levels compared with TDM95 (2.25 ± 0.12- and 1.44 ± 0.61-fold, respectively). Although the mechanism is unclear, we consistently observed a PNGase F-resistant form of TDM95 in these experiments. The reduced levels of TDM95 and TDM56 compared with TDM31 correlated well with the increased mannose phosphorylation on these mutants (Fig. 4A).
Extracellular Levels of Man-6-P-modified LIF and Its Self-renewal Capacity Can Be Controlled through Internalization by the Plasma Membrane-associated CI-MPR
Increased extracellular levels of some LIF mutants at steady state could result from either decreased intracellular sorting to lysosomes or reduced uptake and degradation in lysosomes. To address a role of mannose phosphorylation in the control of extracellular LIF levels via its uptake by cell surface CI-MPR, we performed a self-renewal assay in ESCs. It is well documented that the presence of LIF is sufficient to maintain mouse ESCs in self-renewal (36). Removal of LIF from the culture media (or decreases in its concentration) causes mouse ESCs to spontaneously differentiate, a process that is accompanied by a decrease in alkaline phosphatase activity. Mouse ES cells were treated with various concentrations of unmodified LIF, Man-6-P-modified LIF, and nonglycosylated LIF for 5 days. The extent of self-renewal in these cultures was then determined by quantifying the number of colonies that were positive for alkaline phosphatase staining. As shown in Fig. 5, there was no significant difference in the ability of unmodified LIF and nonglycosylated LIF to maintain ESCs in self-renewal at all concentrations tested. In contrast, we observed a clear difference in the self-renewal capacity of Man-6-P-modified LIF and unmodified LIF. These results suggested that Man-6-P-dependent uptake of modified LIF reduced its concentration in the media, resulting in less self-renewal. To directly test this possibility, free Man-6-P was added to ES cultures that were treated with Man-6-P-modified LIF. As shown in Fig. 5, ES cell self-renewal was significantly increased upon addition of free Man-6-P. Addition of Man-6-P to cultures treated with unmodified LIF had no effect on the percent of AP-positive colonies, indicating that free Man-6-P alone does not influence the extent of self-renewal (data not shown). Because the presence of free Man-6-P has been shown to block the uptake of Man-6-P-modified proteins by cell surface CI-MPR, we believe that the recovery of ESC self-renewal in these cultures (to levels comparable with unmodified LIF) strongly supports the fact that mannose-phosphorylated LIF is rapidly internalized, ultimately leading to reduced extracellular LIF activity and spontaneous ESC differentiation.
FIGURE 5.

Activity of Man-6-P-modified LIF in ESC self-renewal is reduced due to internalization by cell surface Man-6-P receptor. Nonglycosylated ESGRO® LIF (black), unmodified LIF (white), Man-6-P (M6P)-modified LIF (gray), and Man-6-P-modified LIF + 5 mm Man-6-P (dark gray) were added to separate ESC cultures plated at clonal density. After 5 days, AP staining was performed, and the number of AP-positive colonies (undifferentiated) compared with the number of unstained colonies (differentiated) was scored based on overall AP stain intensity and cell morphology. Assays were performed as triplicates of the same experiment. Error bars represent the S.E. of independent triplicate experiments. p values were given based on a t test determined from the independent triplicate experiments (*, p < 0.05; **, p < 0.001).
Man-6-P-modified LIF Is Subject to Intracellular Lysosomal Targeting and Degradation
We next performed experiments to explore whether LIF was being directly targeted to the lysosome following its biosynthesis and mannose phosphorylation in the Golgi. Intracellular LIF was undetectable by Western blotting with an anti-LIF polyclonal antibody (Fig. 6A). However, it is possible that LIF (or the epitope recognized by the antibody) is subject to rapid degradation upon arrival in the lysosome. To determine whether LIF is in fact targeted to lysosomes, we generated a chimera with WT LIF fused to the fluorophore mCherry at the C-terminal end. Unlike GFP, mCherry has increased stability in the lysosomal compartment (37) and can therefore serve as a marker for the Man-6-P-mediated transport of LIF to the lysosome. HeLa cells were transfected with this chimeric construct, and intracellular localization was monitored by immunofluorescence. As shown in Fig. 6B, mCherry staining was clearly detected in the lysosomal compartment as assessed by co-localization with LAMP-1. As a control, we next expressed mCherry with a signal peptide attached (Fig. 6C). Although some intracellular Cherry staining was noted, there was no co-localization with LAMP-1, indicating that the presence of mCherry staining in the lysosomes in the earlier experiments was a result of the Man-6-P-modified LIF portion of the chimeric protein and not the ability of mCherry itself to traffic to this compartment. Addition of Man-6-P to the culture media of transfected cells did not appear to alter overall mCherry staining, suggesting that the lysosomal staining we observed is most likely due to direct targeting of the LIF chimera to the lysosome and not its secretion and uptake via the CI-MPR (Fig. 6D). Some intracellular staining might also be attributed to uptake of LIF by gp130 + LIF receptor. We also generated and tested a TDM31 LIF-mCherry construct (Fig. 6E). Although Man-6-P-modified forms of this LIF mutant cannot be detected in the media, we clearly observed lysosomal staining. Therefore, it is likely that any Man-6-P-modified forms of this LIF mutant are not secreted but instead are being targeted to the lysosome. In support of its lysosomal degradation, we further showed that intracellular LIF could be readily detected in the lysosomal compartment using the anti-LIF polyclonal antibody in the CHO-LIF cells in the presence (but not absence) of protease inhibitors (supplemental Fig. 2).
FIGURE 6.

Man-6-P-modified LIF is subject to intracellular lysosomal targeting and degradation. A, HeLa cells were transfected with various LIF-mCherry or mCherry (with signal peptide) constructs and grown in the presence or absence of 5 mm Man-6-P. Cell and media (M) pools were then analyzed by Western blot using either anti-LIF or anti-mCherry antisera. IB, immunoblot. B–E, confocal analysis of HeLa cells transfected with LIF-mCherry or mCherry constructs (red) and co-stained with the lysosomal marker LAMP-1 (green). B, LIF-mCherry. Co-localization indicates lysosomal targeting of LIF (see arrowheads). C, signal peptide-containing mCherry. D, LIF-mCherry-transfected cells grown in the presence of 5 mm Man-6-P. E, TDM31 LIF-mCherry.
Culture media from transfected HeLa cells and cellular lysates were subjected to Western blot with both anti-LIF and anti-mCherry antibodies to assess the status of the chimeric protein in these cultures (see Fig. 6A). LIF was primarily detected as two distinct molecular weight bands, likely representing forms of the molecule with variable glycosylation because PNGase F treatment reduces these forms to a single band (data not shown). Importantly, we only observed the 29-kDa mCherry protein by Western blot in the WT LIF-mCherry transfected cells, again suggesting that LIF itself is rapidly degraded upon arrival in the lysosome. Fully intact chimeric protein could be readily detected in the culture media of WT LIF-mCherry transfectants using either antibody. Moreover, we noted only the mCherry band by Western blot in lysates of transfected cultures treated with free Man-6-P. These results suggest that direct intracellular targeting may indeed be the primary means by which the LIF-mCherry chimera reaches the lysosome. Collectively, these data indicate that LIF is normally targeted to lysosomes, providing a mechanism to limit its secretion via Man-6-P targeting.
DISCUSSION
The observation that some nonlysosomal and secreted proteins have been demonstrated to bear Man-6-P residues on their N-glycans has raised questions regarding the biological relevance of this modification. Despite the fact that Man-6-P residues can be detected on several nonlysosomal proteins when overexpressed, it is unclear whether these proteins are modified in a physiological context or whether the residues mediate some aspect of their biological activity. Because current enrichment and mass spectrometric methodologies are capable of detecting very small amounts of Man-6-P-modified glycopeptides, these analyses may identify glycoproteins in which the Man-6-P modification represents a low frequency or rare event. Additional experimentation is necessary, however, to determine whether the degree of mannose phosphorylation is comparable with that found on lysosomal hydrolases and whether the interaction of these proteins with the GlcNAc-1-phosphotransferase enzyme has been a process driven by their molecular evolution (29, 38, 39). Earlier studies on the cytokine LIF identified this protein as a high affinity ligand for the CI-MPR and revealed that exogenous LIF is subject to Man-6-P-mediated internalization and degradation (11, 12). In this study, we extend these findings by demonstrating extensive Man-6-P modification on secreted LIF and an unexpectedly broad distribution of the Man-6-P tag on its glycans. Furthermore, our data show that the mannose phosphorylation of LIF is important for controlling its extracellular levels by multiple mechanisms (Fig. 7). Most significantly, we show that LIF is targeted to the lysosome and degraded, limiting the amount that can be secreted from the cell. This mechanism represents yet another example whereby glycosylation controls the half-life and activity of proteins and bears similarities to the regulation of hormone clearance by sulfated and sialic acid-containing glycans (40–42) as well as the control of cell surface half-life of the glucose transporter Glut-2 by the N-acetylglucosaminyltransferase GlcNAcTIVa (43).
FIGURE 7.

Mannose phosphorylation controls LIF extracellular levels by multiple mechanisms. In WT cells, mannose phosphorylation and subsequent lysosomal trafficking of newly synthesized LIF can limit its secretion, and uptake of secreted LIF via cell surface CI-MPR can also help to reduce its extracellular levels. In ML-II cells, loss of GlcNAc-1-phosphotransferase activity and impaired mannose phosphorylation of proteins will “unhinge” these control mechanisms leading to accumulation of extracellular LIF and increased LIF signaling and activity.
In comparison with other IL-6 subfamily members, mammalian LIF contains an unusually high number of N-glycans, with six consensus sites in both the human and mouse sequences. The demonstration that these glycans do not play a role in the ability of LIF to bind its receptor suggests that their presence serves another purpose (11). Enrichment of LIF glycopeptides on a CI-MPR affinity column in conjunction with MS analysis revealed that any of the six N-glycans on LIF are capable of bearing Man-6-P residues, indicating a high degree of flexibility in the manner by which GlcNAc-1-phosphotransferase can access LIF. It is important to note that despite the fact that each enriched glycopeptide is detectable, the relative utilization at each site in the purified protein (% Man-6-P-modified peptide versus total peptide) cannot be accurately determined with this method. Our results with the triple deletion mutants, however, clearly showed that some sites harbor glycans with a higher relative utilization for mannose phosphorylation than others. Nonetheless, the detection of multiple Man-6-P-bearing glycopeptides on LIF is consistent with the analysis of lysosomal hydrolases by several groups, suggesting that LIF has evolved to be a highly mannose-phosphorylated protein despite the fact that it does not have any known function within lysosomes. Interestingly, we found that deletion of any of the six N-glycan sites on LIF failed to significantly alter the overall level of mannose phosphorylation, even though obvious differences in the ability of the triple deletion mutants to bind the CI-MPR affinity column were demonstrated. One interpretation for this finding is that loss of certain glycan sites reduces the competition for the active site of the GlcNAc-1-phosphotransferase enzyme, allowing glycans with less favorable orientations to be modified. Investigation into the protein determinants that mediate binding of LIF to GlcNAc-1-phosphotransferase is currently underway and should shed light on whether the presence of specific lysine residues aid in its mannose phosphorylation as noted with multiple lysosomal hydrolases (33, 44–48) and secreted proteins such as DNase I (49, 50).
We believe the inability to control extracellular levels of LIF via mannose phosphorylation of its glycans may have important implications for the initiation and progression of disease symptoms in ML-II/ML-III. Based on this mechanism, failure to modify endogenous LIF due to a reduction or loss of GlcNAc-1-phosphotransferase activity would be expected to increase its extracellular levels in tissues where LIF is expressed, including the lung (51–53), growth plate chondrocytes (54), and osteoblasts (19, 21). Increased extracellular LIF could subsequently lead to inappropriate signaling and altered tissue homeostasis, in particular in bone and cartilage, where LIF plays important roles in coordinating the function of several types. Indeed, increased levels of LIF in the synovial fluid have been associated with rheumatoid arthritis, a condition that is clinically similar to ML-II/ML-III in its characteristic loss of articular cartilage (55–57). LIF has also been shown to negatively regulate osteoclast apoptosis (22) and can induce osteoclast differentiation (58). Therefore, excess LIF signaling may result in overactive osteoclast activity, another hallmark of these disorders. It is worthwhile to note that the novel “gain-of-function” mechanism described here is clearly distinct from the consequences expected from a complete lack of LIF, as evidenced by the fact that LIF knock-out mice do not exhibit any phenotypes consistent with ML-II.
Our experiments using the glycan-deleted LIF mutants demonstrated that an increase in the level of mannose phosphorylation correlated with decreased accumulation in the culture media upon transfection in CHO cells. The fact that low levels of mannose phosphorylation on LIF are detected in certain tissues may indicate that most of the LIF that is made is actually targeted to the lysosome for degradation. Such a mechanism would provide an additional measure of control, beyond regulation of LIF expression at the transcriptional level, by creating a critical threshold of protein expression that would need to be exceeded to obtain LIF secretion in quantities necessary for a sustained signaling response. Under pathological or certain physiological scenarios where LIF expression is greatly up-regulated (i.e. inflammation, placental formation, or tumorigenesis), the loss of post-translational Man-6-P-dependent regulation may compound the increase in extracellular levels and exacerbate disease pathology. In addition, certain cell types may down-regulate the GlcNAc-1-phosphotransferase expression as a means of stimulating LIF secretion. In support of this, Braulke and co-workers (59) recently demonstrated the GlcNAc-1-phosphotransferase enzyme itself might be subjected to regulation through proteolysis of its γ-subunit in macrophages.
In summary, our data have revealed that mannose phosphorylation can serve as a novel post-translational mechanism to control the secretion and extracellular levels of certain nonlysosomal proteins, including LIF. It will be of interest to determine whether other nonlysosomal proteins are regulated in this manner. As demonstrated by earlier studies, other cytokines such as macrophage colony-stimulating factor also bear Man-6-P residues on their N-glycans (11). Thus, the mechanism we propose for LIF may help explain the relationship between glycosylation and activity that was observed by other investigators (60, 61). Although the extent of mannose phosphorylation on secreted LIF was significant (30–40%), these residues can be detected on proteins at much lower frequencies than what was observed for LIF. Indeed, our current evidence suggests that other cytokines previously reported to be Man-6-P-modified such as latent TGF-β exhibited much lower levels of these residues.4 Thus, a determination of this extent remains an important consideration for assessing the potential function of mannose phosphorylation on this class of proteins.
Supplementary Material
Acknowledgments
We acknowledge Dr. Peter Lobel for the gift of CI-MPR affinity matrix and Dr. Steve Dalton for providing key reagents. We thank Dr. Daniel Kraushaar and Dr. Lianchun Wang for their help with ESC self-renewal assays. In addition, we acknowledge Dr. Heather Flanagan-Steet for assistance with the confocal imaging.
This work was supported, in whole or in part, by National Institutes of Health Grant 1R01GM086524-01 from NIGMS (to R. S.) and Grant 5P41RR018502 from NCRR (to L. W.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1 and 2.
J. Barnes and R. Steet, unpublished results.
- Man-6-P
- mannose 6-phosphate
- LIF
- leukemia inhibitory factor
- CI-MPR
- cation-independent mannose 6-phosphate receptor
- ESC
- embryonic stem cell
- TDM
- triple deletion mutant
- PNGase F
- N-glycosidase F
- AP
- alkaline phosphatase
- hLIF
- human LIF
- mLIF
- mouse LIF.
REFERENCES
- 1. Do H., Lee W. S., Ghosh P., Hollowell T., Canfield W., Kornfeld S. (2002) J. Biol. Chem. 277, 29737–29744 [DOI] [PubMed] [Google Scholar]
- 2. Rohrer J., Kornfeld R. (2001) Mol. Biol. Cell 12, 1623–1631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dahms N. M., Olson L. J., Kim J. J. (2008) Glycobiology 18, 664–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kornfeld S. (1987) FASEB J. 1, 462–468 [DOI] [PubMed] [Google Scholar]
- 5. Chavez C. A., Bohnsack R. N., Kudo M., Gotschall R. R., Canfield W. M., Dahms N. M. (2007) Biochemistry 46, 12604–12617 [DOI] [PubMed] [Google Scholar]
- 6. Olson L. J., Peterson F. C., Castonguay A., Bohnsack R. N., Kudo M., Gotschall R. R., Canfield W. M., Volkman B. F., Dahms N. M. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 12493–12498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Raas-Rothschild A., Cormier-Daire V., Bao M., Genin E., Salomon R., Brewer K., Zeigler M., Mandel H., Toth S., Roe B., Munnich A., Canfield W. M. (2000) J. Clin. Invest. 105, 673–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Reitman M. L., Kornfeld S. (1981) J. Biol. Chem. 256, 11977–11980 [PubMed] [Google Scholar]
- 9. Cathey S. S., Leroy J. G., Wood T., Eaves K., Simensen R. J., Kudo M., Stevenson R. E., Friez M. J. (2010) J. Med. Genet. 47, 38–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Purchio A. F., Cooper J. A., Brunner A. M., Lioubin M. N., Gentry L. E., Kovacina K. S., Roth R. A., Marquardt H. (1988) J. Biol. Chem. 263, 14211–14215 [PubMed] [Google Scholar]
- 11. Blanchard F., Duplomb L., Raher S., Vusio P., Hoflack B., Jacques Y., Godard A. (1999) J. Biol. Chem. 274, 24685–24693 [DOI] [PubMed] [Google Scholar]
- 12. Blanchard F., Raher S., Duplomb L., Vusio P., Pitard V., Taupin J. L., Moreau J. F., Hoflack B., Minvielle S., Jacques Y., Godard A. (1998) J. Biol. Chem. 273, 20886–20893 [DOI] [PubMed] [Google Scholar]
- 13. Lee S. J., Nathans D. (1988) J. Biol. Chem. 263, 3521–3527 [PubMed] [Google Scholar]
- 14. Faust P. L., Chirgwin J. M., Kornfeld S. (1987) J. Cell Biol. 105, 1947–1955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ikushima H., Munakata Y., Ishii T., Iwata S., Terashima M., Tanaka H., Schlossman S. F., Morimoto C. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 8439–8444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Godár S., Horejsi V., Weidle U. H., Binder B. R., Hansmann C., Stockinger H. (1999) Eur. J. Immunol. 29, 1004–1013 [DOI] [PubMed] [Google Scholar]
- 17. Dennis P. A., Rifkin D. B. (1991) Proc. Natl. Acad. Sci. U.S.A. 88, 580–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang L., Tredget E. E., Ghahary A. (2000) Wound Repair Regen. 8, 538–546 [DOI] [PubMed] [Google Scholar]
- 19. Metcalf D. (2003) Stem Cells 21, 5–14 [DOI] [PubMed] [Google Scholar]
- 20. Gonzalez-Perez O., Jauregui-Huerta F., Galvez-Contreras A. Y. (2010) Curr. Immunol. Rev. 6, 167–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sims N. A., Walsh N. C. (2010) BMB Rep. 43, 513–523 [DOI] [PubMed] [Google Scholar]
- 22. Ruan M., Pederson L., Bradley E. W., Bamberger A. M., Oursler M. J. (2010) Endocrinology 151, 1713–1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Slaets H., Hendriks J. J., Stinissen P., Kilpatrick T. J., Hellings N. (2010) Trends Mol. Med. 16, 493–500 [DOI] [PubMed] [Google Scholar]
- 24. Shin J. E., Park S. H., Jang Y. K. (2011) Mol. Cells 31, 181–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Maruta S., Takiguchi S., Ueyama M., Kataoka Y., Oda Y., Tsuneyoshi M., Iguchi H. (2009) Clin. Exp. Metastasis 26, 133–141 [DOI] [PubMed] [Google Scholar]
- 26. Kapoor M., Martel-Pelletier J., Lajeunesse D., Pelletier J. P., Fahmi H. (2011) Nat. Rev. Rheumatol. 7, 33–42 [DOI] [PubMed] [Google Scholar]
- 27. Abramoff M. D., Magelhaes P. J., Ram S. J. (2004) Biophotonics Int. 11, 36–42 [Google Scholar]
- 28. Godard A., Heymann D., Raher S., Anegon I., Peyrat M. A., Le Mauff B., Mouray E., Gregoire M., Virdee K., Soulillou J. P., et al. (1992) J. Biol. Chem. 267, 3214–3222 [PubMed] [Google Scholar]
- 29. Sleat D. E., Zheng H., Qian M., Lobel P. (2006) Mol. Cell. Proteomics 5, 686–701 [DOI] [PubMed] [Google Scholar]
- 30. Lim J. M., Aoki K., Angel P., Garrison D., King D., Tiemeyer M., Bergmann C., Wells L. (2009) J. Proteome Res. 8, 673–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kraushaar D. C., Yamaguchi Y., Wang L. (2010) J. Biol. Chem. 285, 5907–5916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Steet R., Kornfeld S. (2006) Mol. Biol. Cell 17, 2312–2321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Steet R., Lee W. S., Kornfeld S. (2005) J. Biol. Chem. 280, 33318–33323 [DOI] [PubMed] [Google Scholar]
- 34. Hering K. W., Karaveg K., Moremen K. W., Pearson W. H. (2005) J. Org. Chem. 70, 9892–9904 [DOI] [PubMed] [Google Scholar]
- 35. Lazzarino D. A., Gabel C. A. (1989) J. Biol. Chem. 264, 5015–5023 [PubMed] [Google Scholar]
- 36. Niwa H., Ogawa K., Shimosato D., Adachi K. (2009) Nature 460, 118–122 [DOI] [PubMed] [Google Scholar]
- 37. Katayama H., Yamamoto A., Mizushima N., Yoshimori T., Miyawaki A. (2008) Cell Struct. Funct. 33, 1–12 [DOI] [PubMed] [Google Scholar]
- 38. Sleat D. E., Wang Y., Sohar I., Lackland H., Li Y., Li H., Zheng H., Lobel P. (2006) Mol. Cell. Proteomics 5, 1942–1956 [DOI] [PubMed] [Google Scholar]
- 39. Sleat D. E., Della Valle M. C., Zheng H., Moore D. F., Lobel P. (2008) J. Proteome Res. 7, 3010–3021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mi Y., Shapiro S. D., Baenziger J. U. (2002) J. Clin. Invest. 109, 269–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Steirer L. M., Park E. I., Townsend R. R., Baenziger J. U. (2009) J. Biol. Chem. 284, 3777–3783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Park E. I., Mi Y., Unverzagt C., Gabius H. J., Baenziger J. U. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 17125–17129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ohtsubo K., Takamatsu S., Minowa M. T., Yoshida A., Takeuchi M., Marth J. D. (2005) Cell 123, 1307–1321 [DOI] [PubMed] [Google Scholar]
- 44. Dustin M. L., Baranski T. J., Sampath D., Kornfeld S. (1995) J. Biol. Chem. 270, 170–179 [DOI] [PubMed] [Google Scholar]
- 45. Cuozzo J. W., Tao K., Cygler M., Mort J. S., Sahagian G. G. (1998) J. Biol. Chem. 273, 21067–21076 [DOI] [PubMed] [Google Scholar]
- 46. Warner J. B., Thalhauser C., Tao K., Sahagian G. G. (2002) J. Biol. Chem. 277, 41897–41905 [DOI] [PubMed] [Google Scholar]
- 47. Baranski T. J., Cantor A. B., Kornfeld S. (1992) J. Biol. Chem. 267, 23342–23348 [PubMed] [Google Scholar]
- 48. Cantor A. B., Baranski T. J., Kornfeld S. (1992) J. Biol. Chem. 267, 23349–23356 [PubMed] [Google Scholar]
- 49. Nishikawa A., Gregory W., Frenz J., Cacia J., Kornfeld S. (1997) J. Biol. Chem. 272, 19408–19412 [DOI] [PubMed] [Google Scholar]
- 50. Nishikawa A., Nanda A., Gregory W., Frenz J., Kornfeld S. (1999) J. Biol. Chem. 274, 19309–19315 [DOI] [PubMed] [Google Scholar]
- 51. Knight D. A., Lydell C. P., Zhou D., Weir T. D., Robert Schellenberg R., Bai T. R. (1999) Am. J. Respir. Cell Mol. Biol. 20, 834–841 [DOI] [PubMed] [Google Scholar]
- 52. Elias J. A., Zheng T., Whiting N. L., Marcovici A., Trow T. K. (1994) Am. J. Physiol. 266, L426–L435 [DOI] [PubMed] [Google Scholar]
- 53. Wang J., Chen Q., Corne J., Zhu Z., Lee C. G., Bhandari V., Homer R. J., Elias J. A. (2003) J. Biol. Chem. 278, 31226–31232 [DOI] [PubMed] [Google Scholar]
- 54. Grimaud E., Blanchard F., Charrier C., Gouin F., Redini F., Heymann D. (2002) Cytokine 20, 224–230 [DOI] [PubMed] [Google Scholar]
- 55. Lotz M., Moats T., Villiger P. M. (1992) J. Clin. Invest. 90, 888–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Villiger P. M., Geng Y., Lotz M. (1993) J. Clin. Invest. 91, 1575–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Upadhyay A., Sharma G., Kivivuori S., Raye W. S., Zabihi E., Carroll G. J., Jazayeri J. A. (2009) Cytokine 46, 332–338 [DOI] [PubMed] [Google Scholar]
- 58. Richards C. D., Langdon C., Deschamps P., Pennica D., Shaughnessy S. G. (2000) Cytokine 12, 613–621 [DOI] [PubMed] [Google Scholar]
- 59. Pohl S., Tiede S., Marschner K., Encarnação M., Castrichini M., Kollmann K., Muschol N., Ullrich K., Müller-Loennies S., Braulke T. (2010) J. Biol. Chem. 285, 23936–23944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Altmann S. W., Prystowsky M. B. (1992) Arch. Biochem. Biophys. 293, 349–355 [DOI] [PubMed] [Google Scholar]
- 61. Okamoto M., Nakai M., Nakayama C., Yanagi H., Matsui H., Noguchi H., Namiki M., Sakai J., Kadota K., Fukui M., et al. (1991) Arch. Biochem. Biophys. 286, 562–568 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.