

Abstract
Lysoplasmalogenase (EC 3.3.2.2 and EC 3.3.2.5) is an enzyme that catalyzes hydrolytic cleavage of the vinyl ether bond of lysoplasmalogen, forming fatty aldehyde and glycerophosphoethanolamine or glycerophosphocholine and is specific for the sn-2-deacylated form of plasmalogen. Here we report the purification, characterization, identification, and cloning of lysoplasmalogenase. Rat liver microsomal lysoplasmalogenase was solubilized with octyl glucoside and purified 500-fold to near homogeneity using four chromatography steps. The purified enzyme has apparent Km values of ∼50 μm for both lysoplasmenylcholine and lysoplasmenylethanolamine and apparent Vm values of 24.5 and 17.5 μmol/min/mg protein for the two substrates, respectively. The pH optimum was 7.0. Lysoplasmalogenase was competitively inhibited by lysophosphatidic acid (Ki ∼20 μm). The predominant band on a gel at ∼19 kDa was subjected to trypsinolysis, and the peptides were identified by mass spectrometry as Tmem86b, a protein of unknown function. Transient transfection of human embryonic kidney (HEK) 293T cells showed that TMEM86b cDNA yielded lysoplasmalogenase activity, and Western blot analyses confirmed the synthesis of TMEM86b protein. The protein was localized in the membrane fractions. The TMEM86b gene was also transformed into Escherichia coli, and its expression was verified by Western blot and activity analyses. Tmem86b is a hydrophobic transmembrane protein of the YhhN family. Northern blot analyses demonstrated that liver expressed the highest level of Tmem86b, which agreed with tissue distribution of activity. Overexpression of TMEM86b in HEK 293T cells resulted in decreased levels of plasmalogens, suggesting that the enzyme may be important in regulating plasmalogen levels in animal cells.
Keywords: Enzyme Purification, Intestinal Metabolism, Liver Metabolism, Membrane Lipids, Membrane Proteins, Phospholipid Metabolism, Plasmalogen, Gene Identification, Lysoplasmalogen, Lysoplasmalogenase
Introduction
Plasmalogens (1-alk-1′-enyl- 2-acyl-sn-glycero-3-phosphoethanolamine or 1-alk-1′-enyl-2-acyl-sn-glycero-3-phosphocholine) are abundant membrane glycerophospholipids found in anaerobic bacteria and throughout the invertebrate and vertebrate animal kingdom (1, 2). They are different from other membrane glycerophospholipids by having an alk-1′-enyl ether-linked chain at the glycerol sn-1 carbon. In mammals, plasmalogen levels are cell- and tissue-specific and range between 4 and 32% of total phospholipid mass (1). The highest amounts are in the brain, the heart, and circulating immune cells, and the lowest amounts are in the liver and small intestinal mucosa (3). The unique structure and chemistry of the plasmalogens due to the presence of the vinyl ether bond affect the physical and chemical properties of membranes (for reviews and references therein, see Refs. 2, 4, and 5). Plasmalogens are critical for normal cell function and development. Many diseases are associated with altered plasmalogen levels. Plasmalogen levels are decreased in the peroxisomal disorders (5), Alzheimer disease (6), and Down syndrome (7), and they are elevated in tumors and in cell lines derived from tumors (8). Moreover, the amount of ether lipid content corresponds to the degree of malignancy within a tissue (9). Thus, the metabolism of plasmalogen and its regulation are vital areas of study.
Lysoplasmalogens (1-alk-1′-enyl-2-hydroxy-sn-glycero-3-phosphoethanolamine or 1-alk-1′-enyl-2-hydroxy-sn-glycero-3-phosphocholine) occupy an important metabolic junction in plasmalogen metabolism. They are formed from plasmalogen in a hydrolysis reaction that releases polyunsaturated fatty acids from glycerol sn-2 carbon and forms lysoplasmalogen. This reaction is catalyzed by a plasmalogen-selective iPLA2 (plsPLA2),2 a ubiquitous enzyme that has been purified from several tissues (4). This plsPLA2 catalyzed reaction is important in plasmalogen turnover and membrane remodeling and also in the release of bioactive molecules. pls-PLA2 is activated in physiological and pathophysiological conditions (4).
The released lysoplasmalogen is a bioactive metabolite. Lysoplasmalogens are amphiphilic molecules, and at levels near their critical micelle concentration, they produce membrane-perturbing effects and cell lysis (10). Lysoplasmalogens increase membrane fluidity and are involved in cell fusion (11) and cause circulating inflammatory cells to migrate to the endothelium (12). Lysoplasmenylcholine activates cAMP-dependent protein kinase A, suggesting involvement in signal transduction (13). Lysoplasmalogen is important in the synthesis of platelet activating factor (14). Higher levels of lysoplasmalogen have been linked to electrophysiological disturbances in myocytes (15) and to inhibition of Na+-K+-ATPase in renal cells (16).
Lysoplasmalogens are normally maintained at very low levels in cells (17). The lysoplasmalogen that is formed in a plsPLA2 reaction may be converted back to plasmalogen in a transacylation reaction, thus reforming plasmalogen (remodeling pathway) (18). Alternatively, the lysoplasmalogen may be degraded enzymatically by hydrolytic enzymes that include a phospholipase C (19), a phospholipase D (20), and lysoplasmalogenase.3 Lysoplasmalogenase (EC 3.3.2.2; EC 3.3.2.5), also known as alkenyl ether hydrolase, catalyzes the hydrolysis of the vinyl ether bond of lysoplasmenylcholine or lysoplasmenylethanolamine (21–23) and of 1-alkenyl-glycerol (24) in reactions that form a long chain aldehyde and glycerophosphocholine, glycerophosphoethanolamine, or glycerol, depending on the substrate (22, 23). Lysoplasmalogenase is a microsomal enzyme highly specific for the sn-2-deacylated form of plasmalogen and has no activity with diradyl plasmalogen (21–26). The enzyme has been identified and studied in microsomes from the liver (21–23) and small intestinal mucosa (26), where its specific activities are high. The enzyme has also been found in brain microsomes, where its activity is low, only of liver activity (24, 25). Thus, in these tissues, brain, liver, and small intestinal mucosa, there exist reciprocal relationships between content of plasmalogens and the activity of lysoplasmalogenase.
As the enzyme that cleaves the vinyl ether bond, lysoplasmalogenase is probably an important enzyme in maintaining the balance between plasmalogen and lysoplasmalogen and thereby preserving membrane stability and function. If levels of lysoplasmalogen rise, they may disrupt and lyse cell membranes. Conversely, if lysoplasmalogen levels are too low, the transacylation reaction cannot occur, and membrane plasmalogen levels will decrease, thereby disturbing membrane structure and function by that route.
In our earlier work, we solubilized, partially purified, and characterized lysoplasmalogenase from rat liver microsomes (23, 27) and identified and characterized the enzyme in digestive tract mucosa (26). In the present work, we aimed to further purify lysoplasmalogenase and to identify the protein and its gene. The putative gene was transfected into human embryonic kidney (HEK) 293T cells and into Escherichia coli. Characterization of the expressed proteins provides evidence that this is the lysoplasmalogenase. We tested the hypothesis that overexpression of the gene in HEK cells would result in decreased levels of plasmalogens.
EXPERIMENTAL PROCEDURES
Chemicals and Materials
Bovine heart lysoplasmenylcholine (A-354), bovine brain plasmenylethanolamine, 1-palmitoyl-2-linoleoyl-phosphatidylcholine, and bovine brain diradyl glycerophosphoethanolamine (A-356) were from Doosan-Serdary Research Laboratories (Toronto, Canada). Cardiolipin, phosphatidylethanolamine, phosphatidylserine, phosphatidylinositol, sphingomyelin, and lysophosphatidic acid were from Avanti Polar Lipids, Inc. (Alabaster, AL). 1-O-n-Octyl-β-d-glucopyranoside (octyl glucoside) was from Inalco Biochemicals (San Luis Obispo, CA). Diethylaminoethyl cellulose (DE52) was obtained from Whatman Chemical Separation Ltd. The preformed 4–20% acrylamide gels, the Bradford protein assay, and the hydroxyapatite CHT5 columns were from Bio-Rad. The gel electrophoresis column (Superdex 200; 3-ml volume), the Mono Q (5/50 GL; 1-ml volume) column, and the FPLC system were from GE Healthcare. The Amicon Ultra centrifugal filter units (4 ml) (10,000 molecular weight cut-off) were from Millipore Corp. (Temecula, CA). TRIS base, KCl, NaCl, HEPES, mannitol, and sucrose were of reagent grade and were from Sigma-Aldrich. Yeast alcohol dehydrogenase (LS001070) was from Worthington Biochemical Corporation (Lakewood, NJ). The antibodies against MEK1/2 and cytochrome c oxidase were from Cell Signaling (catalogue no. 9126), and antibodies against DDK were from OriGene Technologies, Inc. (Rockville, MD).
Lipid Substrates
The ethanolamine lysoplasmalogen substrate was prepared from bovine brain ethanolamine glycerophospholipids by mild alkaline hydrolysis, followed by chromatography to separate the lysoplasmalogen from fatty acids and diradyl compounds, and quantified as described (19, 23).
Aqueous suspensions of lipid substrates were prepared about every 3 days from stock solutions stored in chloroform/methanol (2:1, v/v) at −20 °C. Aliquots were dried under N2, and appropriate amounts of 70 mm glycylglycine-NaOH buffer (pH 7.1) were added, and suspensions were vortexed, sonicated, and maintained at 2 °C (23).
For a portion of the experiments shown in Fig. 3, the ethanolamine and choline lysoplasmalogen substrates were prepared in liposomes. Chloroform/methanolic solutions of lysoplasmalogen and bovine brain glycerophosphoethanolamine and 1-oleoyl-2-linoleoyl-sn-glycero-3-phosphocholine were mixed in molar ratios of 6, 47, and 47%, respectively, and dried as a thin film in a glass conical tube under N2. 70 mm glycylglycine-NaOH (pH 7.1) was added to make aqueous suspensions of 1.0 mm with respect to the lysoplasmalogen substrates and 8.3 mm for each diradyl glycerophospholipid. The mixture was vortexed and sonicated for ∼15 s four times. The liposomes were turbid and were maintained at 2 °C.
FIGURE 3.
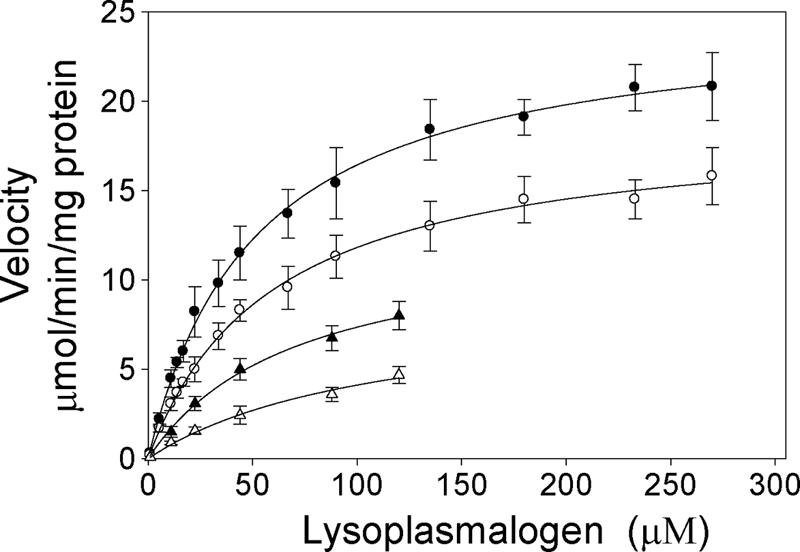
Michaelis-Menten kinetics graph showing dependence of liver lysoplasmalogenase velocity on substrate concentration. Velocities represent initial velocities. Lysoplasmalogenase catalyzed hydrolysis of both choline (black filled symbols) and ethanolamine (white filled symbols) lysoplasmalogen substrates. Top two curves, substrates were prepared in 70 mm glycylglycine (circles). Final concentrations of substrates included 5.5, 11, 22, 44, 88, 135, 180, and 270 μm lysoplasmalogen. Bottom two curves, lysoplasmalogen was prepared at 6 mol % in liposomes of 94% diradyglycerophospholipids (47 mol % GPE and 47 mol % GPC) (triangles). Activity was measured using the coupled enzyme assay. The substrate was added to the reaction mixture before initiating the reaction by the addition of lysoplasmalogenase (0.21 μg of Mono Q sample). Reaction volumes were 0.52 ml. Velocity is given as μmol of aldehyde converted to alcohol/min/mg of protein. Values represent the means with S.D. (error bars) (n = 3).
Assays for Lysoplasmalogenase
Assay a, Coupled Enzyme Assay Using Yeast Alcohol Dehydrogenase
This assay was carried out as described (23, 29). In this coupled enzyme assay, the aldehyde that is produced by hydrolysis of lysoplasmalogen is reduced to an alcohol in a second enzymic reaction, catalyzed by exogenously added yeast alcohol dehydrogenase. In the second reaction, NADH is oxidized in a 1:1 stoichiometry between the mol of aldehyde produced and mol of NADH oxidized. The reaction is monitored at 340 nm, and the absorbance change is converted into nmol of NADH oxidized using the extinction coefficient for NADH (6220 m−1 cm−1). The standard reaction mixture contained 70 mm glycylglycine-NaOH, pH 7.1, 1 mm DTT, 0.20 mm NADH, 0.35 mg of fatty acid-free bovine serum albumin per ml, 1 mg (250 IU) of yeast alcohol dehydrogenase per ml, in a total volume of 0.52 ml. Efficacy of the coupling enzyme was evaluated under different conditions by the addition of 10–20 nmol of tetradecanaldehyde (aqueous suspension) to the reaction assay cuvette. The reaction rates are initial velocities usually obtained within the first few min of the reaction.
For determining the pH optimum, the buffers in the coupled enzyme assay included the following: for pH 5.5–6.7, 80 mm 3-(N-morpholino)ethanesulfonic acid (NaOH); for pH 6.5–7.7, 80 mm glycylglycine (NaOH); and for pH 7.5–8.6, 80 mm Tris-HCl. The amount of alcohol dehydrogenase in each assay was adjusted appropriately to keep the aldehyde to alcohol reaction non-rate-limiting.
Assay b, a Two-dimensional TLC Procedure for Determination of Lysoplasmalogenase Activity
This procedure was used in the stoichiometric studies in which changes in the amounts of lysoplasmalogen, water-soluble glycerphosphoethanolamine, and aldehyde were compared, using purified lysoplasmalogenase. It was also used in the inhibitor study with p-chloromercuribenzoate because this compound inhibited the coupling enzyme, alcohol dehydrogenase. Finally, assay b was used to study possible hydrolytic activity of the purified lysoplasmalogenase with monoacyl glycerophospholipids and with 1-acyl-2-lyso-glycero-3-phosphatidic acid (LPA) as substrates. These lipids were present at 125 and 250 μm. This procedure was performed as described in earlier work (23) with the following modifications. The reaction times were 0, 3, 8, and 16 min. The reactions were stopped by the addition of 12 ml of chloroform/methanol (2:1, v/v); the lower phase (lipids) were concentrated under N2 and applied to a 10 × 10-cm TLC plate. The solvent system for first and second dimensions was chloroform, methanol, ammonia (65:35:4). Following the first dimension, the plate was dried and inverted over a dish of 40 ml of concentrated HCl for 5 min, dried, and run in the second dimension. The lipid spots were visualized using I2 vapors, relevant areas were scraped into glass tubes, and phosphorus was analyzed (28). The water-soluble components were identified and quantified as described (23).
Isolation and Solubilization of Rat Liver Microsomal Proteins
The harvesting of livers from seven male Sprague-Dawley rats and isolation of microsomes were carried out at 2–4 °C as described earlier (23, 27) with a few modifications. The minced livers were homogenized in 9 volumes (w/v) of homogenizing buffer consisting of 20 mm Tris-HCl buffer, pH 7.8, 120 mm mannitol, 70 mm sucrose, 0.25 mm EGTA, and BSA (fatty acid-free) 0.5 mg/ml. The homogenate was centrifuged at 600 × g for 10 min, and the resultant supernatant was further centrifuged at 14,000 × g for 20 min. The next resultant supernatant was centrifuged at 105,000 × g for 60 min to obtain the light microsomes. The microsomes were suspended in 420 ml of washing buffer containing 20 mm Tris-HCl (pH 7.4), 100 mm KCl, 70 mm mannitol, 30 mm sucrose, 1 mm DTT, 0.05 mm leupeptin and centrifuged at 105,000 × g for 65 min. The washed microsomal pellets were suspended by homogenization in 100 ml of solubilizing buffer consisting of 20 mm Tris-HCl (pH 7.8), 120 mm mannitol, 70 mm sucrose, 1 mm DTT, 0.05 mm leupeptin, 0.1 mm PMSF. Protein concentration was 9 mg/ml. Solubilization was achieved by adding 11 ml of 20% octyl glucoside (in H2O) dropwise with stirring. The detergent/protein ratio was ∼2:1 (w/w), and the detergent concentration was ∼2% (∼68 mm). After 20 min, the suspension was centrifuged at 105,000 × g for 65 min.
Purification of Lysoplasmalogenase Using Column Chromatography
DEAE Anion Exchange Chromatography
The clear amber-colored 100,000 × g supernatant was applied to a 60-ml DEAE-cellulose column pre-equilibrated in 25 mm Tris-HCl, pH 7.9, per the manufacturer's instructions, followed by equilibrating in DEAE buffer A containing 25 mm Tris-HCl, pH 7.9, 0.75 mm DTT, 0.05 mm leupeptin, and 0.75% octyl glucoside. The column was washed with 40 ml of DEAE buffer A, and then a 600-ml KCl gradient (0–0.85 m KCl in DEAE buffer A) was opened, and the proteins were eluted (flow rates were 0.6 ml/min). Enriched fractions were pooled, assayed for activity and protein, and divided into two aliquots.
Hydroxyapatite Chromatography (FPLC), Dialysis, and Concentration
The 5.0-ml hydroxyapatite (HA) CHT5 column was prepared and equilibrated per the manufacturer's directions, using HA buffer A (10 mm potassium phosphate (KPi) buffer, pH 7.18, 1.0 mm DTT, 0.2 mm CaCl2, 0.05 mm leupeptin, and 0.75% octyl glucoside) and HA buffer B (1.0 m KPi buffer, pH 7.18, 1.0 mm DTT, 0.01 mm CaCl2, 0.75% octyl glucoside, and 0.05 mm leupeptin). One-half of the pooled enriched DEAE fractions were loaded onto an HA column at 0.6 ml/min. The column was washed with 15 ml of HA buffer A, followed by a 65-ml gradient from 17% buffer B (83% buffer A) to 70% buffer B (30% buffer A) at a flow rate of 0.6 ml/min. The application of enzyme to column and elution were repeated for the second aliquot of enriched DEAE fractions. The enriched fractions were pooled and dialyzed (10,000 molecular weight cut-off) against 100 volumes of DEAE buffer A overnight at 2–4 °C. The dialyzed HA sample (18.5 ml) was divided into three parts, and each was concentrated to ∼50 μl by centrifugation in 4-ml Millipore filtration units that had been precentrifuged with DEAE buffer A.
Gel Filtration Chromatography (FPLC)
The 3-ml volume Superdex 200 gel filtration column was equilibrated per the manufacturer's directions using an FPLC system. The GF buffer contained 150 mm NaCl, 50 mm Tris-HCl (pH 7.9), 1.0% octyl glucoside, 1.0 mm DTT, 0.05 mm leupeptin. 50 μl of the HA fraction were applied to the GF column at 0.01 ml/min, followed by elution at 0.10 ml/min, collecting 0.11 ml/tube. This application of enzyme to column and elution were repeated for the second and third aliquots (50 μl each) of the HA fraction.
The fractions were assessed for activity and protein, and the enriched fractions were pooled (2.4 ml) and concentrated to about 75 μl in the 4-ml equilibrated filtration units. The concentrated protein was diluted with 75 μl of DEAE buffer A in order to dilute the salt in the sample.
Mono Q Chromatography (FPLC)
A Mono Q 5/50 GL 1-ml volume column was equilibrated via the manufacturer's directions using DEAE buffers A and B. The GF fraction (135 μl) was loaded onto the column at 0.01 ml/min. The column was washed with 3 ml of DEAE buffer A, and the proteins were eluted using a 25-ml gradient (0–0.8 m KCl in DEAE buffer A) at 0.15 ml/min. After analysis of protein and activity, the Mono Q fractions 27 and 28 were combined, as were fractions 29 and 30. These fractions and also fraction 31 were concentrated about 5-fold using Millipore 4-ml filtration units.
SDS-PAGE Analysis of the Purified Proteins
Sufficient 5× loading buffer was added to each purified sample to obtain final concentrations (1×) of 40 mm Tris-HCl, pH 6.8, 10% glycerol, 2.4% SDS, 50 mm dithiothreitol, and 0.004% bromphenol blue. Samples were maintained at 25 °C for ∼30 min before loading into the well. The fractions were analyzed using 4–20% gradient gels using Tris-glycine buffer and the Laemmli procedure (31). Proteins were made visible using silver stain or using Coomassie stain for gels in which bands were used for amino acid sequencing.
Mass Spectrometry Analyses of Peptide Fragments Followed by Protein Identification
The predominant band at ∼19 kDa from a Coomassie-stained gel was excised and sent to the W. M. Keck Foundation Biotechnology Research Laboratory at Yale University School of Medicine, where it was digested and analyzed for amino acid sequence, followed by protein identification. The gel slices were first washed with 50% acetonitrile, 50% water, followed by 50 mm ammonium bicarbonate, 50% acetonitrile, 50% water. It was then dried, rehydrated with trypsin in 10 mm ammonium bicarbonate, and digested at 37 °C for 16 h. The resultant peptides were separated using a Waters Symmetry C 18 trap column and a nanoAcquity UPLC separating column. Elution was performed with a 52-min linear gradient of 5–50% acetonitrile in 0.05% formic acid. The amino acid sequences were obtained using mass spectrometry (Orbitrap instrument). The sequence data were searched for analogy to known peptides/protein using the Mascot algorithm (version 2.20) for uninterpreted MS/MS spectra and searching the NCBInr data base. The significance score relied on multiple matches to more than one peptide from the same protein.
Expression of Human TMEM86b in HEK 293T Cells
HEK 293T cells were grown in DMEM supplemented with 10% fetal bovine serum, penicillin (100 units/ml), and streptomycin (100 μg/ml) at 37 °C with 5% CO2. A mammalian expression construct that contains the 226-amino acid open reading frame of human transmembrane protein 86b (TMEM86b; accession number NP_776165) upstream of the Myc-DDK tag in pCMV6 Entry vector was purchased (OriGene Technologies, Inc.). The construct or empty vector control (5 μg each) was transfected into HEK 293T cells by Lipofectamine (Invitrogen) according to the manufacturer's instructions. The transfected cells were also plated onto slides for immunohistochemistry analyses.
Subcellular Fractionation of Transfected 293T Cells into Membranes/Membrane Compartments and Cytosol and Assessment of Protein Expression by Western Blot and by Coupled Enzyme Assay
All procedures were carried out at 2–4 °C. After 72 h of transfection, empty vector- and TMEM86b-transfected cells (∼20 million of each) were rinsed with 50 ml of PBS and suspended in 1.5-ml Eppendorf tubes with 750 μl of Isolation Buffer, pH 7.4: 0.3 m mannitol, 0.1% BSA, 0.2 mm EDTA, 20 mm HEPES-NaOH, and 0.01 mm leupeptin hydrochloride. The plasma membranes of cells were disrupted by means of nitrogen cavitation using a cooled Parr cell disruption vessel (Parr Instrument Co., Moline, Illinois). The cell suspension was loaded into the bottom of the disruption chamber for 20 min at a pressure of 500 p.s.i. (32). The pressure was released, disrupting the plasma membrane and releasing the organelles (33). The suspension was centrifuged at 125,000 × g for 45 min. The pellet included the nuclei, mitochondria, plasma membrane, and microsomes, and the supernatant was the cytosol fraction. The pellet was suspended in 120 mm mannitol, 70 mm sucrose, 10 mm Tris-HCl (MST) at pH 7.4 by homogenization. Proteins were assessed using the Bradford assay, and lysoplasmalogenase activity was analyzed using the coupled enzyme assay. Expression of TMEM86b in the fractions was determined by Western blot analyses using anti-DDK monoclonal antibody (Origene TA50011) and performed as described (34). The antibody against cytochrome c oxidase was used to detect mitochondrial protein, and MEK1/2 was used to detect the cytosol.
Lipid Extraction and Plasmalogen Determinations in Transfected HEK Cells
About 5 million empty vector- and 5 million TMEM86b-transfected cells were divided into three portions and placed in 20-ml glass vials in a volume of 2 ml. Lipids were extracted using a modified Bligh and Dyer method (35). For every 1 ml of sample, 3.75 ml of chloroform/methanol (1:2, v/v) were added and vortexed for 10 min. Another 1.25 ml of chloroform was added and mixed for 1 min, followed by 1.25 ml of 1 m NaCl and mixing. The samples were centrifuged for 15 min at 2,000 × g. The upper phase and interphase were treated with acetone to precipitate the protein; proteins were solubilized with 1 n NaOH and measured by the Lowry method (36). The lower phase containing the lipids was analyzed for phospholipids and for plasmalogens. The iodometric assay (37, 38) was used to detect total plasmalogen content. Duplicate aliquots of the lipid extract were evaporated to dryness under nitrogen gas and dissolved in 75 μl of methanol. To one of the samples, 75 μl of 3% aqueous KI was added as a blank, and to the other sample, 75 μl of iodine solution (6 × 10−4 n I2 in 3% KI) was added. These were mixed vigorously and then incubated at room temperature for 20 min. Last, 600 μl of 95% ethanol were added, and the absorbance at 355 nm was measured. A control tube was included in which the lipid sample was omitted. The extinction coefficient is 27,500 M−1 cm−1.
Phospholipid classes were analyzed using TLC 20 × 20-cm Silica Gel H plates (Analtech, Newark, NJ) scored into lanes. Seven lanes were used for standards, and samples were run in duplicate. The plates were developed up to 5 cm below the top of the plate with chloroform/methanol/acetic acid/acetone/water (35:25:4:14:2, v/v/v/v/v), dried, developed up to 3 cm below the top of the plate with chloroform/methanol/acetic acid (190:10:1, v/v/v), dried, and then developed to 1 cm below the top of the plate with hexane/diethyl ether/acetic acid (80:20:2) and dried a final time. Lipids were visualized by spraying the plates with a solution of 3% cupric acetate in 8% phosphoric acid, charring them in an oven for 40 min at 160 °C, and scanning (Hewlett Packard) the plates. The quantity of lipids in each band was determined through photodensitometry using the UNIX version of IMAL (T. J. Nelson). Standards included phosphatidylcholine and phosphatidylethanolamine, cardiolipin, phosphatidylserine, phosphatidylinositol, and sphingomyelin, which were run in serial dilution from a concentration of ∼2.5 mg/ml to ∼0.16 mg/ml.
Expression of TMEM86b in E. coli
A bacterial expression vector, pEX-N-His-GST-TMEM86b, was purchased (Origene Technologies, Inc.) and transformed into competent E. coli strains (overexpressing C41(DE3), C43(DE3), C41(DE3)pLysS, and C43(DE3)pLysS) (Lucigen Corp., Middleton, WI). The production of fusion proteins was performed as described previously (39). Briefly, isopropyl β-d-1-thiogalactopyranoside was added to log phase bacterial cultures to a final concentration of 1 mm, and cultures were harvested at various time points over a 24-h period. The pellets were suspended in 20 mm Tris-HCl buffer, pH 7.4, 120 mm mannitol, 70 mm sucrose buffer by very brief sonication. Expression of TMEM86b protein was assessed by Western blot analyses using anti-His and anti-GST antibodies. Activity of lysoplasmalogenase was measured using the coupled enzyme assay.
Northern Blot Analysis
A multiple-tissue RNA blot was purchased (Ambion, FirstChoice Mouse Blot 1). A 32P-labeled probe was prepared from a DNA fragment generated by PCR using the TMEM86b expression vector as a template and the forward primer 5′-GCCCTTGTGTGCTCGGCTGT-3′ and reverse primer 5′-ATAGGCTGCCACCGGCAGGA-3′. The Northern blot analysis was performed as described previously (34).
Immunocytochemistry
Immunohistochemistry was performed with 293T cells transiently transfected with the TMEM86b construct and grown on 8-chamber slides. The cells were fixed with 10% buffered formalin, washed with PBS plus 2% donkey serum (Chemicon, Temecula, CA), 1% BSA, 0.3% Triton X-100, 0.1% sodium azide). They were incubated with primary anti-DDK antibodies overnight, followed by washing and incubation with secondary antibodies. Cells were incubated with 4,6-diamidino-2-phenylindole (DAPI) for nuclear staining. Cells were viewed with a Nikon Axioscope at 63-fold magnification.
RESULTS
Solubilization and Purification of Lysoplasmalogenase from Seven Rat Livers (Table 1 and Fig. 1)
TABLE 1.
Summary of the purification of microsomal lysoplasmalogenase from seven rat livers
Enzymes were measured by a coupled enzyme assay, and proteins were measured by a Bradford assay.
| Purification step | Protein | Units | Specific activity | Purification | Recovery |
|---|---|---|---|---|---|
| mg/fraction | nmol/min/fraction | nmol/min/mg | -fold | % | |
| 1. Microsomes | 925.8 | 43,820 | 47.3 | 1.0 | 100 |
| 2. 100,000 × g supernate ∼100 ml | 694.0 | 49,594 | 71.4 | 1.5 | 113 |
| 3. DEAE-cellulose (fractions 54–69) | 80.6 | 40,300 | 500 | 10.6 | 92 |
| 4. Hydroxyapatite (fractions 42–48) from two columns | 3.0 | 12,360 | 4,120 | 87.0 | 28 |
| 5. Gel filtration (fractions 17–24) from three columns | 0.491 | 4,023 | 8,194 | 173.2 | 9.2 |
| 6. Mono Q (fractions 27 and 28) | 0.035 | 660 | 18,800 | 397 | 1.5 |
| Mono Q (fraction 29) | 0.040 | 689 | 17,225 | 364 | 1.6 |
| Mono Q (fraction 30) | 0.040 | 979 | 24,500 | 518 | 2.2 |
| Mono Q (fraction 31) | 0.023 | 362 | 15,760 | 333.0 | 0.83 |
FIGURE 1.

Purification of lysoplasmalogenase; elution profiles from four columns. This figure is from one purification experiment in which microsomes from seven rat livers were solubilized and purified using these four column chromatography steps. A, anion exchange chromatography on DEAE-cellulose open column. B, hydroxyapatite chromatography CHT5 column, FPLC. C, gel filtration chromatography on Superdex S-200, FPLC. D, anion exchange chromatography on Mono Q 5/50 GL column, FPLC. Fractions were tested for lysoplasmalogenase activity (black filled circles) using the coupled enzyme assay and were expressed in nmol/min/fraction. The protein concentrations (white filled circles) were assessed by Bradford assay and given in mg/fraction (A) or μg/fraction (B–D).
The following purification results are from one experiment that was similar to at least six experiments. Following homogenization of livers in isotonic buffer, the light microsomal fraction was isolated by differential centrifugation and comprised 926 mg of protein and 44,000 units of activity. The washed microsomes were solubilized with 2% octyl glucoside, and 75% of protein (694 mg) and 113% of the units (49,594 nmol/min) were recovered in the 100,000 × g supernatant (a 1.5-fold purification). Purification on four sequential chromatography columns followed: DEAE-cellulose open column, hydroxyapatite (FPLC), gel filtration 200 (FPLC), and last a Mono Q 5/50 GL column (FPLC).
The 100 K supernatant (105 ml) was applied directly to an open DEAE column, and the enriched fractions 54–69 eluted between 0.4 and 0.5 m KCl in the KCl gradient. These fractions were purified 10.6-fold with 92% recovery (Table 1 and Fig. 1A). The enriched fractions were pooled (88 ml) and loaded onto two HA columns. The enriched fractions were eluted as a sharp peak late in the KPi gradient (Fig. 1B). Combining the enriched fractions from the two HA columns, the enzyme was purified ∼87-fold with 28% recovery, and volume was 14 ml. The HA-purified enzyme lost ∼35% of its activity when it was concentrated in the HA elution buffer (high KPi and pH 7.18) (data not shown). Thus, the HA-purified fractions were dialyzed overnight against DEAE buffer concentrated from 14 ml to ∼150 μl before applying to the gel filtration column in three different applications of ∼50 μl each. The gel filtration elution pattern seen in Fig. 1C was typical of the other two elutions. The enriched fractions from the three GF columns were pooled and were 173-fold purified with 9.2% recovery, and volume was 2.4 ml. These fractions were concentrated to 75 μl and diluted 1:1 with DEAE buffer A in order to lower the salt concentration of sample to about 75 mm NaCl and 25 mm Tris-HCl, a necessary step for retention of lysoplasmalogenase on the Mono Q column. The column was developed with 0–0.8 m KCl gradient, and active fractions eluted as a single peak between 0.33 and 0.48 m KCl. Fractions 27–31 were highly purified. Combined fractions 27 and 28 were purified 483-fold with 1.5% recovery. Fraction 29 was purified 364-fold with 1.6% recovery; fraction 30 was the most highly purified fraction (518-fold) and contained 1.6% of the units. After pooling fraction 27 with 28 and fraction 29 with 30, these fractions and also fraction 31 were concentrated in preparation for PAGE and for kinetic analyses.
Purity of the Enriched Fractions from Purification Steps Was Assessed Using SDS-PAGE (Fig. 2)
FIGURE 2.

SDS-PAGE analysis of purity of enriched fractions isolated during purification of lysoplasmalogenase. The gel was a 4–20% polyacrylamide, and the Laemmli procedure was used (31). Samples were prepared as described under “Experimental Procedures.” Approximately 10 μg of protein were added per well except in lanes 7 and 9, which received less. The approximate units of lysoplasmalogenase loaded into each well are listed in parentheses. Lanes 1 and 10, standards; lane 2, microsomal fraction (0.5 nmol/min); lane 3, 100 K supernatant (0.75 nmol/min); lane 4, DEAE-cellulose (5 nmol/min); lane 5, hydroxyapatite (41 nmol/min); lane 6, gel filtration (82 nmol/min); lane 7, Mono Q fractions 27 and 28 (20 nmol/min), lane 8, Mono Q fractions 29 and 30 (200 nmol/min); lane 9, Mono Q fraction 31 (30).
The enriched fractions were examined by SDS-PAGE using a 4–20% polyacrylamide gel and silver staining. In preparing the samples with loading buffer, it was important that the samples were not heated. Heating above ∼45 °C caused aggregation of the lysoplasmalogenase protein, and it remained in the well of the gel or near top of the gel (data not shown). In the gel shown in Fig. 2, ∼10 μg of each sample were loaded into the wells, except for lanes 7 and 9, which received less. In the absence of heating, a distinct band appeared at about ∼19 kDa that correlated with the activity added to the well.
Kinetic Properties and Lipid Substrate Specificity of the Purified Liver Lysoplasmalogenase
Mono Q-purified enzyme samples with specific activities between 17 and 20 μmol/min/mg protein were the enzyme sources for characterization studies. The reaction velocity was dependent on time for >20 min and was also dependent on concentration of lysoplasmalogenase. Enzyme exposed to 95 °C for 3 min had no activity. We compared the stoichiometry between the mol of ethanolamine lysoplasmalogen that disappeared and the mol of products, glycerophosphoethanolamine and free aldehyde, formed. The results of the experiment showed that there was a 1:1:1 correlation. Enzyme assay b was used to assess the amount of lysoplasmalogen and amount of glycerophosphoethanolamine after 10 min of reaction (data not shown).
The enzyme catalyzed the hydrolysis of both choline and ethanolamine lysoplasmalogen substrates. The effect of increasing choline or ethanolamine lysoplasmalogen concentration on initial velocity is shown in Fig. 3. The lysoplasmalogen substrates were prepared as micelles in aqueous buffer in the upper two curves (circles) and as components of liposomes in lower two curves (triangles).
The velocities followed rectangular hyperbolas characteristic of the binding isotherm. A fit of the Michaelis-Menten equation to the data of the upper two curves gave apparent Km values of ∼50 μm for both substrates and Vmax values of 24.5 μmol/min/mg protein and 17.5 μmol/min/mg protein for lysoplasmenylcholine and lysoplasmenylethanolamine, respectively. By use of the molecular weight derived from the gene sequence for the rat Tmem86b protein (25,140 Da), the turnover numbers for choline and ethanolamine substrates are 616 and 438 min−1, respectively.
We examined the question of whether lysoplasmalogenase was able to catalyze the hydrolysis of lysoplasmalogen when the substrate was presented as a component of a phospholipid bilayer, thus mimicking the more physiological conditions in a cell. Lysoplasmalogen substrates were prepared in liposomes at concentrations that might be found in cell membranes (6 mol %). The lower two curves of Fig. 3 show that the purified enzyme catalyzed the hydrolysis of both substrates, albeit at lower rates compared with reactions in which the substrates were presented as aqueous suspensions (upper two curves).
The substrate selectivity of lysoplasmalogenase was further investigated with 1-alkenyl-2-acyl-sn-glycero-3-phosphocholine or 1-alkenyl-2-acyl-sn-glycero-3-phosphoethanolamine, 1-acyl-sn-glycero-3-phosphocholine or 1-acyl-sn-glycero-3-phosphoethanolamine, and LPA. Enzyme assay B was used as well as 125 and 250 μm levels of potential substrates. The purified liver enzyme had no activity with any of these lipids.
Lysoplasmalogenase has a pH optimum of 7.0 with both ethanolamine and choline substrates. 50% of maximal velocity occurred at pH 5.5 and 8; thus, the decline in activity with change in pH was a little steeper at higher pH values (data not shown).
The purified lysoplasmalogenase was unaffected by low millimolar concentrations (<5 mm) of Ca2+, Mg2+, Mn2+, and EDTA. NaF or PMSF did not inhibit the enzyme. Phosphatidic acid, sphingosine 1-phosphate, monoacylglycerophosphocholine, and monoacylglycerophosphoethanolamine did not inhibit the alkenyl hydrolase activity of purified lysoplasmalogenase. The concentrations of the compounds tested were between 100 and 500 μm (data not shown). However, the metabolite lysophosphatidic acid was found to be a strong competitive inhibitor of lysoplasmalogenase, with a Ki value of ∼20 μm (Fig. 4).
FIGURE 4.
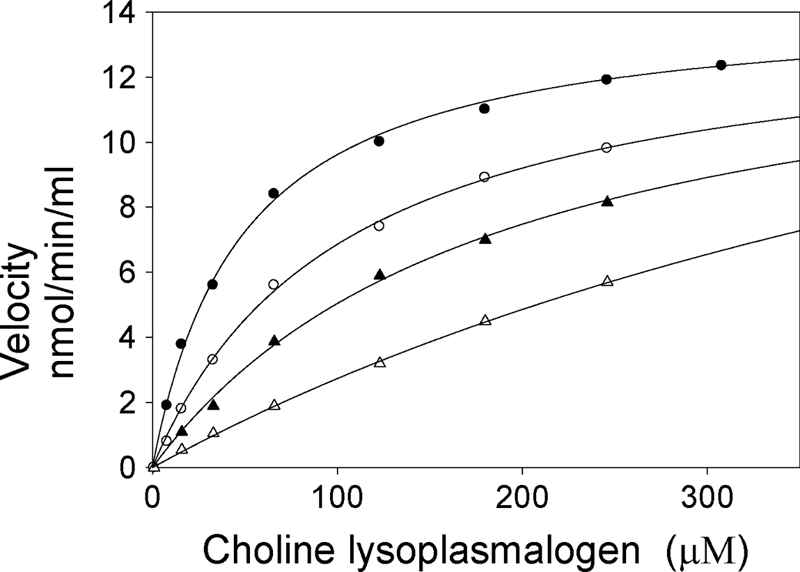
Inhibitory effects of lysophosphatidic acid on lysoplasmalogenase activity. The curve with the black filled circles had no inhibitor. The velocities of the reactions are shown at three concentrations of lysophosphatidic acid: 22 μm (white filled circles), 44 μm (black filled triangles), and 111 μm (white filled triangles). The choline lysoplasmalogen concentrations were 16, 33, 66, 123, 180, and 246 μm. The velocities were measured using the coupled enzyme assay and represent initial velocities. The assays were started with enzyme addition (0.35 μg of a Mono Q sample). Each data point was the average of 2–3 assays, and S.D. values were less than 10%. The experiment was representative of three independent experiments.
Identification of the ∼19 kDa Band as Tmem86b Protein
A similar gel to that shown in Fig. 3 was stained with Coomassie Blue stain, and the prominent band at 19 kDa was excised and subjected to protein digestion followed by mass spectrometric analyses (Keck Research Laboratories at Yale University, New Haven, CT). Seven peptides obtained from this 19 kDa band matched exactly to a 20-residue region near the N terminus or a 51-residue region near the C terminus of the rat transmembrane protein 86B (Tmem86b; GenBankTM accession number NP_001103074) (Fig. 5). Together, the two regions covered 31% of the Tmem86 molecule.
FIGURE 5.

Partial digestion of the purified protein band followed by sequencing using mass spectrometry identified the protein as Tmem86b. Partial digestion of the purified protein band was followed by sequencing using mass spectrometry. A, the seven peptides identified with a score greater than identity score obtained from MALDI-MS and peptide mass data base searching. B, the locations of the peptides in the Tmem86b protein are shown by underlining. The number of occurrences of the peptides in the protein is reflected by the thickness of the lines (thin lines, 1 occurrence; medium line, 2 occurrences; thickest line, 3 occurrences).
The entire purification from rat liver microsomes and the analysis of the band at ∼19 kDa were repeated with the same identification of Tmem86b as the predominant protein. Moreover, Tmem86b had also been found as a minor species in the 19-kDa region from mass spectrometric analyses of heat-treated samples from earlier preparations.
Tmem86b is an uncharacterized protein that was cloned from rat brain in a National Institutes of Health full-length cDNA project (40). A sequence analysis shows that Tmem86b has 42% identity to another protein, Tmem86a (accession number NP_001128488) (Fig. 6), but has no significant homologies to other rat sequences in the GenBankTM database. Both Tmem86a and Tmem86b belong to the YhhN family, a family involved in lipid metabolism. Tmem86b has only been identified in vertebrates, including humans, mice, rats, cows, dogs, and zebrafish. A multiple-sequence alignment and a dendrogram showing the phylogenetic relationships of the Tmem86b proteins in vertebrates are shown in Fig. 7.
FIGURE 6.

Sequence alignment of rat Tmem86a and Tmem86b. The region homologous to the YhhN family of proteins is highlighted in gray.
FIGURE 7.

Phylogeny of Tmem86b proteins in vertebrates. A, multiple-sequence alignment. B, dendrogram.
Isochorismatase enzymes are the only other family of proteins that catalyze the hydrolysis of a vinyl ether bond (41). A sequence alignment of human isochorismatase domain-containing protein 2 (accession number NP_078986) and TMEM86b (accession number NP_776165) revealed 35% identity between the two proteins in a region of 26 amino acids that is within the YhhN homologous region (Table 2).
TABLE 2.
Sequence alignment of human isochorismatase (ISOC2) and human transmembrane protein 86B (TMEM86b)
Shown is sequence alignment of human isochorismatase domain-containing protein 2, mitochondrial isoform 2 (ISOC2, 221 amino acids, accession number NP_078986), and transmembrane protein 86B (TMEM86b, 226 amino acids, accession number NP_776165).
![]()
Molecular Cloning of TMEM86b in Mammalian Cells
To determine whether tmem86b encoded the lysoplasmalogenase, we first expressed TMEM86b in HEK 293T cells. A mammalian expression construct encoding a complete human TMEM86b-Myc-DDK fusion protein or the empty vector were transiently transfected into HEK 293T cells.
72 h after transfection, high levels of lysoplasmalogenase activity were obtained from cells transfected with the TMEM86b construct (1951 units; 197 nmol/min/mg protein), and minimal activities were found in the control cells (16 units; 1.6 nmol/min/mg protein). The activity was predominantly in the membranes/membrane compartments, with only ∼1% in cytosol. The specific activity in the membrane fractions was 327 nmol/min/mg protein (Table 3).
TABLE 3.
293T cells transfected with TMEM86b gene express lysoplasmalogenase activity in membrane/membrane compartment fractions
The expression of TMEM86b into HEK cells and the isolation of total lysates, membrane/membrane compartment fractions (plasma membranes, nuclei, mitochondria, and microsomes), and cytosol are described under “Experimental Procedures.” Lysoplasmalogenase activity was measured using the coupled enzyme assay with choline lysoplasmalogen (200 μm). 15 μg of protein were added to the cuvette (0.52 ml total volume). The protein concentrations were determined using the Bradford procedure. Values are means from triplicate assays, and S.D. values are <12% of the mean.
| 293T cells | Empty vector |
TMEM86b |
||||
|---|---|---|---|---|---|---|
| Total cell lysate | Membrane fraction | Cytosol | Total cell lysate | Membrane fraction | Cytosol | |
| Protein (μg/fraction) | 10,340 | 6,590 | 3,475 | 9,917 | 6,333 | 3,384 |
| Lysoplasmalogenase activity (nmol/min/fraction) | 16 | 14 | 0 | 1,951 | 2,068 | 23 |
| Specific activity (nmol/min/mg protein) | 1.6 | 2.1 | 0 | 197 | 327 | 7 |
Lysoplasmalogenase in the transfected 293T cells catalyzed the hydrolysis of both the choline and ethanolamine substrates (Fig. 8). With the choline substrate, a typical saturation kinetics curve was obtained. With the ethanolamine lysoplasmalogen, higher levels of the substrate appear to be inhibiting. The reasons for this are not known. The Km values for both substrates are estimated to be ∼70 μm.
FIGURE 8.
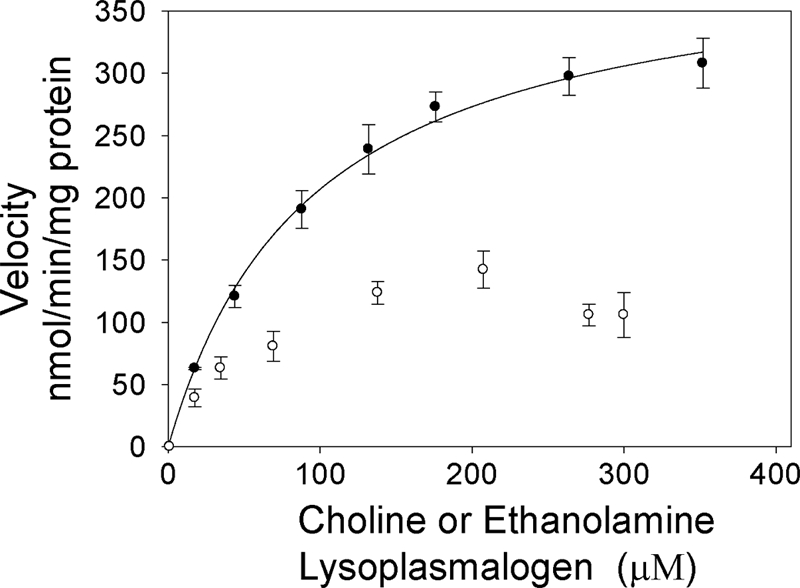
Lysoplasmalogenase activity in human 293T cells transfected with TMEM86b. The initial velocities of lysoplasmalogenase activity versus substrate concentration with ethanolamine or choline lysoplasmalogen were plotted in a Michaelis-Menten kinetics plot. Lysoplasmalogenase activity was assayed using the coupled enzyme assay under standard assay conditions. 15 μg of total membrane protein were added to 0.52 ml of the reaction assay. Error bars, S.D.
In Western blot analysis, anti-DDK antibody recognized two protein species of ∼27 and ∼54 kDa in total lysates (T) prepared from cells transfected with the TMEM86b construct but not from cells transfected with the empty vector (Fig. 9A, lanes 4 and 1). The size of the 27-kDa species corresponds to the recombinant TMEM86b protein (226 amino acid residues from TMEM86b and 25 residues from the Myc-DDK tags), whereas the 54-kDa species could possibly be a dimer.
FIGURE 9.

TMEM86b encodes a membrane bound protein that conferred lysoplasmalogenase activity in mammalian cells (activity is shown in Table 2). A, Western blot analysis of HEK 293T cells transiently transfected with TMEM86b construct or empty vector (EV). At the top is shown the structure of the TMEM86b/Myc/DDK fusion protein. At the bottom is a Western blot analysis of total cell lysates (T), membrane fractions (M), and cytosolic fractions (C) with the indicated antibodies. A 4–20% acrylamide Laemmli gel was run with 72-h total lysates, total membranes, and cytosol from the TMEM86b-transfected cells (lanes 4–6) and the empty vector-transfected cell lysates (lanes 1–3). The protein was transferred on to a PDVF membrane and reacted with anti-DDK antibody, with anti-MEK1/2 antibody that was specific for cytosol, and with anti-cytochrome c oxidase antibody, specific for mitochondrial membrane. B, immunofluorescence of TMEM86b-transfected cells provides evidence that lysoplasmalogenase is located in the cytoplasm. Top photograph, anti-DDK antibody (green). Middle photograph, DAPI (blue) stain for DNA/nuclei. The arrow points to an apparently untransfected cell. The bottom photograph shows a merged image of anti-DDK antibodies and DAPI stain. C, the lysoplasmalogenase topology was predicted using the transmembrane hidden Markov model (42). The x axis shows the numbers of the residues in the amino acid chain from the N terminus at 0 to the C terminus at residue 226. The y axis shows the probability score and indicates the probability that a particular residue of the amino acid chain will be located in a certain region of the membrane. The red lines indicate the probability that these residues have a transmembrane location, and the six transmembrane domains of Tmem86b are shown. The blue line indicates the probability that these residues are located on the cytoplasmic side (inside) of the membrane, and the violet line indicates the probability that these residues are located on the noncytoplasmic side (outside) of the membrane (42).
Subcellular fractionation further showed that TMEM86b was associated with the cell membrane fractions and was not present in the cytosol (Fig. 9, lanes 5 and 6). Anti-cytochrome c oxidase antibody confirmed similar loading of membranes and total cell lysate in the empty vector- and the TMEM86b-transfected cells. Anti-MEK1/2 (a soluble cytosolic protein) antibody confirmed that the fractionation procedure separated the cytosol from the membrane fractions. This transfection experiment was repeated two times with different cultures of 293T cells on different days, and the same results were obtained.
Immunohistochemistry was performed with the 293T cells transfected with the TMEM86b construct using anti-DDK antibody (Fig. 9B). The cells were also incubated with DAPI stain for nuclei (blue). The immunofluorescence provided evidence that TMEM86b is located throughout the cytoplasm (green).
Evidence from many experiments suggest that the native liver lysoplasmalogenase is a membrane-associated protein. Analyses of the molecular structure of Tmem86b reveal a protein with many hydrophobic side chains. The Tmem86b protein topology was predicted using the transmembrane hidden Markov model (42), and this analysis suggests that the protein has six transmembrane domains (Fig. 9C).
Expression of TMEM86b in E. coli
The data suggested that TMEM86b either corresponded to lysoplasmalogenase or encoded a gene product that up-regulates lysoplasmalogenase in HEK 293T cells. To differentiate between these two possibilities, we next expressed TMEM86b in E. coli, which do not have the tmem86b gene, plasmalogens (2), or lysoplasmalogenase activity.4 A prokaryotic expression vector for the production of His-GST-TMEM86b recombinant protein was constructed and transformed into E. coli (Fig. 10A). The production of recombinant proteins was induced with isopropyl β-d-1-thiogalactopyranoside and monitored over a 24-h period. Total lysates were analyzed by SDS-PAGE followed by protein staining and Western blot analysis (Fig. 10, B and C). Western blot analysis showed that the expression of the 53-kDa fusion protein, which reacted with anti-His antibodies, was induced 2 h after isopropyl 1-thio-β-d-galactopyranoside induction, peaked at 4 h, and then declined afterward (Fig. 10C). Activity of lysoplasmalogenase was measured using the coupled enzyme assay. Significantly, lysoplasmalogenase activities were proportional to the amounts of fusion protein produced (Fig. 10D). The specific activities, apparent Km values, and pH optimum of the E. coli enzyme are very close to those of the purified liver enzyme (Table 4). Because lysoplasmalogenase activity was obtained when TMEM86b was expressed in E. coli, we concluded that TMEM86b encoded lysoplasmalogenase.
FIGURE 10.

Expression of TMEM86b in E. coli. A, structure of the His-GST-TMEM86b fusion protein. The N-terminal His-tagged region is represented by an unfilled box. B, LB + ampicillin (50 mg/liter) and chloramphenicol (34 mg/liter) were inoculated overnight with a 1:100 (v/v) bacterial culture and grown to an OD of 0.8. Subsequently, fusion protein production was induced with isopropyl 1-thio-β-d-galactopyranoside (1 mm final concentration). Lysates were prepared from bacteria harvested at time points shown at the top of each lane, subjected to SDS-PAGE, and analyzed by Coomassie Brilliant Blue staining (B) and Western blotting with anti-His antibody (C). D, activity of lysoplasmalogenase in E. coli cells.
TABLE 4.
E. coli cells transfected with TMEM86b gene express lysoplasmalogenase activity
The enzyme activity was assayed by a coupled enzyme assay. The proteins were estimated by the Bradford procedure. For this experiment, n = 3 assays, and S.D. is shown in parentheses. The results shown here are similar to those obtained from other E. coli strains expressing the TMEM86b gene.
| Substrate lysoplasmalogen | pH optimum | Activity | Apparent Km |
|---|---|---|---|
| nmol/min/mg protein | μm | ||
| Choline | 7.05 | 580 (58) | 45 |
| Ethanolamine | 6.95 | 505 (60) | 53 |
Tissue Distribution of Lysoplasmalogenase
The tissue distribution of lysoplamalogenase was determined by Northern blot analysis. A 32P-labeled DNA fragment from the human TMEM86b cDNA yielded a 2-kb transcript in the liver (Fig. 11, lane 3). Weaker signals were also observed in the brain, testis, and embryo. A smaller transcript of 1.3 kb was observed in the heart (lane 1). The origin of the smaller transcript remains to be established and possibly was generated by alternative splicing. A search of the GEO Profiles database revealed specific expression of Tmem86b in the mouse liver and intestinal tract (GEO accession number GDS522) (43), including duodenum, jejunum, ileum, and colon (Fig. 11B). The distribution of mRNA expression of Tmem86b showed the highest levels in the jejunum, where also the highest activity of lysoplasmalogenase was found (26). The jejunum is an important site of lipid absorption, metabolism, and distribution. Collectively, the high levels of Tmem86b transcripts in the liver and small intestines correlated with empirical findings of high contents of lysoplasmalogenase activity in those tissues (23, 25, 26).
FIGURE 11.

Expression of TMEM86b transcripts in mouse tissues. A, Northern blot analysis. Poly(A)+ RNA was hybridized to 32P-labeled TMEM86b probes (top) or cyclophilin probes as control (bottom). B, specific expression of TMEM86b along the mouse intestinal tract. Data were retrieved from GEO accession code GDS522 (43). Error bars, S.D.
Plasmalogen Levels Are Decreased in HEK 293T Cells with tmem86b Overexpression. (Table 5)
TABLE 5.
Plasmalogen levels are decreased in HEK 293T cells with TMEM86b overexpression
293T cells were transfected with empty vector or TMEM86b expression vector. The lipids were extracted (Bligh and Dyer procedure) from three aliquots of cells (1.5 × 106 cells/aliquot) transfected with TMEM86b (n = 3 aliquots of cells); the empty vector cells were similarly extracted (n = 3 aliquots of cells). The lipid extracts were used to determine total phospholipids diradyl glycerophosphoethanolamines, and diradyl glycerophosphocholines by TLC methods. Plasmalogens were measured using the iodometric assay. The proteins were assessed by Lowry. The numbers represent the means (n = 3), and the S.E. values are shown in parentheses. EV, empty vector; PL, phospholipids; PE, diradyl glycerophosphoethanolamines; PC, diradyl glycerophosphocholines.
| 293T cells | Protein | Total PL | Total PE + PC | Plasmalogen | Plasmalogen levels |
||
|---|---|---|---|---|---|---|---|
| nmol/mg protein | Percentage of PL | Percentage of PC + PE | |||||
| μg | nmol | nmol | nmol | nmol/mg | % | % | |
| EV ∼1.5 × 106 | 467 (12) | 301 (22) | 235 (20) | 26.2 (1.5) | 56.1 | 8.8 | 11.1 |
| Tmem86b ∼1.5 × 106 | 488 (18) | 320 (15) | 259 (8) | 21.5 (0.4) | 44.1 | 6.7 | 8.3 |
| Percentage decrease in plasmalogen | |||||||
| % | % | % | |||||
| 21.4 | 23.0 | 25.2 | |||||
The hypothesis that lysoplasmalogenase might be important in controlling the levels of plasmalogens in cells was tested. The 293T cells that were transfected with TMEM86b for 72 h were harvested, and the lipids were extracted by the method of Bligh and Dyer (35). The lipid extract was used to determine total phospholipids, diradyl glycerophosphoethanolamine, and diradyl glycerophosphocholine by the TLC method and the total plasmalogen levels by iodometric analyses. The results showed a decrease in plasmalogen levels in the cells overexpressing lysoplasmalogenase when compared with the empty vector cells by the following percentages: 18% in nmol of plasmalogen/million cells; 21% in nmol of plasmalogen/mg of protein; 23% in nmol of plasmalogen as a percentage of total phospholipid; and 24% in nmol of plasmalogen as a percentage of (phosphatidylcholine + phosphatidylethanolamine) (Table 5). These results are similar to another independent transfection experiment in which plasmalogen levels were decreased in the TMEM86b-overexpressing cells (data not shown).
DISCUSSION
This is the first report to purify lysoplasmalogenase to near homogeneity and to identify the protein and DNA sequence. The identity of the gene, tmem86b, was confirmed by molecular cloning and protein expression in human embryonic kidney cells and in E. coli cells. The function of this gene was previously unknown.
Purification of Lysoplasmalogenase and Characterization of the Purified Enzyme
In our previous work (23), we had solubilized and purified the liver microsomal enzyme 160-fold using two columns, the DEAE column and the hydroxyapatite FPLC column. In the present work, the enzyme was further purified to 500-fold, and the protein band on an SDS-polyacrylamide gel was identified. Changes in procedures between the earlier and the present work included the addition of a gel filtration column and a Mono Q column, increasing the amount of octyl glucoside in all buffers from 0.5 to 0.75% and to 1.0% in the GF buffer, and equilibrating the Millipore filtration units with appropriate buffer before concentrating the protein solutions. In preparing the samples for PAGE, the concentration of bromphenol blue dye was lowered to 0.004% to prevent protein aggregation. We also eliminated the heating step because heating of the protein sample caused aggregation, and lysoplasmalogenase remained in the stacking gel or migrated as a smear near the top of a gradient gel. With these improvements, the enzyme was purified to near homogeneity, and the predominant protein band at about 19 kDa was excised, partially digested, and sequenced.
Properties of the Mono Q-purified Enzyme
This enzyme was highly selective for the deacylated form of ethanolamine or choline plasmalogen and had no activity with diradyl plasmalogens. The apparent Km values were 50 μm with both substrates, and the apparent Vmax values were 24.5 and 17.5 μmol/min/mg protein with lysoplasmenylcholine and lysoplasmenylethanolamine, respectively. These relatively low Km values as well as the high Vmax and Kcat values are compatible with a role for lysoplasmalogenase in maintaining levels of lysoplasmalogen below the levels of monoradylglycerophospholipids known to perturb membrane structure and function (17). The values are also similar to those reported for macrophage-like cell line P388D1 lysophospholipases, which act on monoacyl glycerophospholipids (44).
Moreover, the Km values are high enough that lysoplasmalogen would be available to enter into the transacylase reaction in which an acyl group is transferred to lysoplasmalogen from 1-alkyl-2-acyl-sn-glycero-3-phosphocholine, thus reforming diradyl plasmalogen (18). This CoA-independent transacylase has an apparent Km for lysoplasmalogen in the 5–30 μm range (14, 18) and is present in many tissues (45).
The properties of the purified liver lysoplasmalogenase, including substrate specificity, pH optimum, integral microsomal membrane protein, effects of divalent cations, and inhibition by p-chloromercuribenzoate and Triton X-100, are very similar to the properties of the native liver microsomes (21, 22, 46) and to the partially purified liver enzyme (23). The enzyme properties are also similar to those of rat brain microsomes with the exception that activity levels are much lower in the brain (24, 25). A highly sensitive assay for detecting hydrolysis of the vinyl ether bond was developed in the laboratory of Debuch (24, 25), in which the plasmenylethanolamine and lysoplasmenylethanolamine substrates were radiolabeled with 14C at the C-1 of the alkenyl group (19). These workers found no evidence for an enzyme that catalyzed hydrolysis of the vinyl ether group of the intact plasmalogen molecule in any subcellular fraction of rat brain (25). A lysoplasmalogenase activity was also identified in oligodendroglial cell enriched fractions from bovine brain that had properties similar to those of purified liver lysoplasmalogenase in its neutral pH optimum and lack of requirement for Mg2+ (47). A lysoplasmalogenase activity was reported in guinea pig heart microsomes and mitochondria, using a coupled enzyme assay with aldehyde dehydrogenase as coupling enzyme (48). This heart lysoplasmalogenase differed from the purified liver and brain lysoplasmalogenase in the following ways. The enzyme strictly required deoxycholate for activity, Mg2+ activated enzyme, and the pH optimum was 8.5. We were unable to detect a vinyl ether-hydrolyzing enzyme in any subcellular fractions of rat or guinea pig heart using a coupled enzyme assay or iodometric assay.4
Identification, Cloning, and Properties of the Expressed TMEM86b Proteins
After identification of the 19 kDa band as Tmem86b, the human cDNA for this protein was transfected into HEK 293T cells. The transfected cells produced lysoplasmalogenase enzyme activity, and Western blot analyses confirmed the expression of TMEM86b. The expressed protein had properties similar to those of the purified liver protein. It catalyzed hydrolysis of the vinyl ether bond of both ethanolamine and choline lysoplasmalogen with Km values of about 70 μm. The recombinant protein had a pH optimum of 7.05, was inhibited by LPA (50% inhibition at 50 μm LPA), was associated with the membrane fraction, and was absent from the cytosol.
To eliminate the possibility that the TMEM86b gene could have coded for a transcription factor that in turn up-regulated lysoplasmalogenase, we expressed TMEM86b into E. coli, a bacteria that has no gene for Tmem86b, no plasmalogens (2), and no lysoplasmalogenase activity. The E. coli expressed high levels of lysoplasmalogenase activity, and Western blot analyses confirmed the production of a recombinant protein of the size of the His-GST-TMEM86b construct. The expressed protein had similar enzymic properties to the purified liver enzyme in substrate specificity, Km, and pH optimum (Table 4). We concluded that tmem86b was the gene for lysoplasmalogenase.
Tmem86b Protein
The National Institutes of Health database revealed that lysoplasmalogenase is highly conserved in evolution in rat, pig, mouse, chimpanzee, human, dog, cow, and zebrafish. The human protein TMEM86b contains 226 amino acids and has a molecular mass of ∼24,500 Da. TMEM86b is a transmembrane protein of previously unknown function and is a member of the YhhN family.
Tmem86b is composed of many hydrophobic amino acids and is predicted to have six transmembrane domains (Fig. 9C). This hydrophobicity of the molecule is consistent with our empirical findings that the protein is tightly integrated with the membrane and requires 2% octyl glucoside to “solubilize” the molecules as well as the continued presence of detergent or phospholipids for maintenance of activity at 2–4 °C. It requires either 50% glycerol or 1–4 mm phospholipids for maintenance of activity at −20 °C. Our empirical finding that, in the presence of SDS, gentle heating followed by cooling induced the molecule to aggregate is probably due to its hydrophobic nature (49).
The isochorismatases catalyze hydrolysis of vinyl ether bonds of isochorismate-related compounds in the enterobactin (41) and in the phenazine (50) biosynthetic pathways of microorganisms. Isochorismatases are the only other known enzyme that catalyzes hydrolysis of a vinyl ether bond. The structure of the TMEM86b protein is different from isochorismatase except for a 26-amino acid region in which there is low (35%) identity. This area does not include the aspartate 38 amino acid that is critical for catalysis in isochorismatase (50). It will be interesting to learn the catalytic mechanism for lysoplasmalogenase.
Abundance of Lysoplasmalogenase and Its Possible Role in Liver and Small Intestinal Mucosa
Lysoplasmalogenase is a very abundant protein in liver microsomes; about 0.2% of the protein mass is lysoplasmalogenase, and the Kcat (400–600 min−1) is very high. The results from the Northern blot showed the highest expression of mRNA in the liver, and GEO data found high mRNA expression for Tmem86b in small intestinal mucosa that coincided with our activity studies in that tissue (26). The biological function for this high activity in the liver and small intestines is not known. Liver and small intestinal mucosa contain very low levels of plasmalogens (1, 3) yet high activities of plasmalogen-synthesizing enzymes (51). Vance (52) shed light on this apparent paradox when she discovered that the liver cells synthesized plasmalogens, rich in docosahexaenoic acid and arachidonic acid, and incorporated them into the nascent HDL and LDL, which were then secreted into the blood stream. It is only during the de novo synthesis of plasmalogens that docosahexaenoic acid and arachidonic acid become linked to the sn-2 carbon of the intermediate metabolite (1-alkyl-2-hydroxy-sn-glycero-3-phosphate) that will become plasmalogen (53). When the lipoprotein remnants return from the peripheral tissue to the liver, they are catabolized and recycled. Thus, we hypothesize that lysoplasmalogenase may be especially important in liver and in small intestinal mucosa for hydrolyzing the vinyl ether bond of lysoplasmalogen in this recycling process. Developmental studies of the specific activities of lysoplasmalogenase in liver and small intestinal mucosa of rat pups support a role for lysoplasmalogenase in nutrient metabolism and/or distribution (26).
Possible Relationship between Levels of Lysoplasmalogenase and Levels of Plasmalogen in Cells
We and others (22, 23) observed an inverse relationship between plasmalogen levels and lysoplasmalogenase activity in tissues. Plasmalogen levels are high in the brain and heart, where lysoplasmalogenase activities are low (except see Ref. 48). On the contrary, plasmalogen levels are low in liver and small intestinal mucosa, where lysoplasmalogenase activities are high. It has long been known that tumors and cell cultures derived from them contain high levels of ether lipids, both 1-O-alkyl and 1-O-alkenyl (plasmalogen) glycerophospholipids and neutral lipids (8). Also, the levels of the ether lipids are proportional to the degree of malignancy (9).
Moreover, tumor cells contain low or nil levels of 1-alkyl-glycerophosphocholine or 1-alkyl-glycerophosphoethanolamine monooxygenase, the enzyme that cleaves the ether bond of the 1-O-alkyl-glycerophospholipids or 1-O-alkyl-glycerol (54). This enzyme is similar to lysoplasmalogenase in that it is a lysophospholipase and is present in high concentration in the liver and the small intestinal mucosa. It is distinct from lysoplasmalogenase in the structure of the ether bond it cleaves and in the type of reaction catalyzed. The reaction is an oxidative cleavage and requires molecular oxygen and tetrahydrobiopterin (54). The amino acid sequence of this monooxygenase enzyme is not known.
There are no reports in the literature regarding lysoplasmalogenase activity in tumors or tumor cell lines. However, we found no detectable activity (<0.1 nmol/min/mg) of lysoplasmalogenase in the liver hepatoma cell lines Hep G2 and Hep 3B using the coupled enzyme assay,4 and in searching the National Institutes of Health Web site we found there was no mRNA expression for tmem86b in any tumors examined (n = 3030).
In the present work, we tested the hypothesis that HEK 293T cells that had been transfected with TMEM86b may have lower levels of plasmalogens. The results showed that 72 h after transfection, the plasmalogen levels were decreased by 18–25% in the cells with TMEM86b overexpression when compared with the control cells. These cells were examined only 72 h after transfection, and it is possible that the plasmalogen levels may be even lower in stably transfected cells. We are continuing these studies and extending the studies to include Hep G2, Hep 3B, leukemia, and breast cancer cell lines. Also we are examining the effects of the lowered plasmalogen levels on cell viability.
In the present work, we found that lysophosphatidic acid is a competitive inhibitor of lysoplasmalogenase activity (Fig. 4). LPA is a signaling molecule and promotes cell growth, proliferation, migration, and survival (55) and under certain conditions may lead to cancer initiation, progression, and/or metastasis (56). New cell growth and proliferation require synthesis of lipids, including plasmalogens, for the new membranes. LPA inhibition of lysoplasmalogenase may function to conserve the vinyl ether bond and allow more lysoplasmalogen to enter into the transacylation reaction and form plasmalogen by the remodeling pathway.
In summary, we purified lysoplasmalogenase to near homogeneity. Mass spectrometry was used to identify the protein and to identify the putative gene, tmem86b, for which there was no previously known function. The gene was transfected into human 293T cells as well as into E. coli cells. Expression of the protein was confirmed by Western blot analyses and enzyme assays. Overexpression of TMEM86b resulted in decreased levels of plasmalogen in HEK cells, providing evidence that lysoplasmalogenase may be important in determining plasmalogen levels in animal cells.
Acknowledgments
We are thankful for the generosity of Drs. Daniel Schoenberg and Mark Parthun in sharing their FPLC and HPLC apparatuses and for many helpful discussions. We also thank Warren Erdahl, Ron Louters, and Clifford Chapman for valuable assistance. We are grateful to the MS and Proteomics Resource Laboratory of the W. M. Keck Foundation Biotechnology Resource Laboratory at Yale University School of Medicine for running the LC-MS/MS analyses on many gel slices. We thank Drs. Kalpana Ghoshal and Jharna Datta for help with cell culture.
This work was supported by Ohio State University Development fund Grant 306500 (from Daniel S. and Amy E. F. Jurkowitz) (to M. S. J.) and a grant from the Columbus Medical Research Foundation (to L.-C. W.). The work was also supported by National Institutes of Health (NCI) ROI Grant CA149623 (to S. L.).
Names for lysoplasmalogenase include lysoplasmalogenase 1 (to allow for the possibility of more enzymes to be discovered), alkenyl ether hydrolase, alkenylglycerophosphoethanolamine hydrolase, alkenylglycerophosphocholine hydrolase, and alkenylglycerohydrolase. Lysoplasmalogens include lysoplasmenylethanolamine, 1-alk-1′-enyl-sn-glycero-3-phosphoethanolamine; lysoplasmenylcholine, and 1-alk-1′-enyl-sn-glycero-3-phosphocholine. Plasmalogens include plasmenylethanolamine (1-alk-1′-enyl-2-acyl-sn-glycero-3-phosphoethanolamine) and plasmenylcholine (1-alk-1′-enyl-2-acyl-sn-glycero-3-phosphocholine).
M. S. Jurkowitz, unpublished results.
- plsPLA2
- plasmenyl-selective phospholipase A2
- DDK
- DYKDDDDK epitope
- GF
- gel filtration
- KPi
- potassium phosphate buffer
- LPA
- lysophosphatidic acid (1-acyl-sn-glycerol-3-phosphate).
REFERENCES
- 1. Horrocks L. A., Sharma M. (1982) in New Comprehensive Biochemistry (Neuberger A., van Deenan L. L. M. eds) pp. 51–95, Elsevier Biomedical Press, Amsterdam [Google Scholar]
- 2. Goldfine H. (2010) Prog. Lipid Res. 49, 493–498 [DOI] [PubMed] [Google Scholar]
- 3. Broderson S. H. (1967) Histochemical and Chemical Studies of Plasmalogens in the Rat, Ph.D. dissertation, State University of New York at Buffalo, Buffalo, NY [Google Scholar]
- 4. Brites P., Waterham H. R., Wanders R. J. A. (2004) Biochim. Biophys. Acta 1636, 219–231 [DOI] [PubMed] [Google Scholar]
- 5. Nagan N., Zoeller R. A. (2001) Prog. Lipid Res. 40, 199–229 [DOI] [PubMed] [Google Scholar]
- 6. Ginsberg L., Rafique S., Xuereb J. H., Rapoport S. I., Gershfeld N. L. (1995) Brain Res. 698, 223–226 [DOI] [PubMed] [Google Scholar]
- 7. Murphy E. J., Schapiro M. B., Rapoport S. I., Shetty H. U. (2000) Brain Res. 867, 9–18 [DOI] [PubMed] [Google Scholar]
- 8. Snyder F. (1972) in Ether Lipids: Chemistry and Biology (Snyder F. ed) pp. 273–295, Academic Press, Inc., New York [Google Scholar]
- 9. Howard B. V., Morris H. P., Bailey J. M. (1972) Cancer Res. 32, 1533–1538 [PubMed] [Google Scholar]
- 10. Jaffé E. R., Gottfried E. L. (1968) J. Clin. Invest. 47, 1375–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Han X. L., Gross R. W. (1991) Biochim. Biophys. Acta 1069, 37–45 [DOI] [PubMed] [Google Scholar]
- 12. White M. C., Rastogi P., McHowat J. (2007) Am. J. Physiol. Cell Physiol. 293, C1467–C1471 [DOI] [PubMed] [Google Scholar]
- 13. Williams S. D., Ford D. A. (1997) FEBS Lett. 420, 33–38 [DOI] [PubMed] [Google Scholar]
- 14. Nieto M. L., Venable M. E., Bauldry S. A., Greene D. G., Kennedy M., Bass D. A., Wykle R. L. (1991) J. Biol. Chem. 266, 18699–18706 [PubMed] [Google Scholar]
- 15. Corr P. B., Snyder D. W., Cain M. E., Crafford W. A., Jr., Gross R. W., Sobel B. E. (1981) Circ. Res. 49, 354–363 [DOI] [PubMed] [Google Scholar]
- 16. Schonefeld M., Noble S., Bertorello A. M., Mandel L. J., Creer M. H., Portilla D. (1996) Kidney Int. 49, 1289–1296 [DOI] [PubMed] [Google Scholar]
- 17. Corr P. B., Gross R. W., Sobel B. E. (1984) Circ. Res. 55, 135–154 [DOI] [PubMed] [Google Scholar]
- 18. Kramer R. M., Deykin D. (1983) J. Biol. Chem. 258, 13806–13811 [PubMed] [Google Scholar]
- 19. Gunawan J., Vierbuchen M, Debuch H. (1979) Hoppe Seylers Z. Physiol. Chem. 360, 971–978 [DOI] [PubMed] [Google Scholar]
- 20. Wykle R. L., Schremmer J. M. (1974) J. Biol. Chem. 249, 1742–1746 [PubMed] [Google Scholar]
- 21. Warner H. R., Lands W. E. (1961) J. Biol. Chem. 236, 2404–2409 [PubMed] [Google Scholar]
- 22. Gunawan J., Debuch H. (1981) Hoppe Seylers Z. Physiol. Chem. 362, 445–452 [DOI] [PubMed] [Google Scholar]
- 23. Jurkowitz-Alexander M., Ebata H., Mills J. S., Murphy E. J., Horrocks L. A. (1989) Biochim. Biophys. Acta 1002, 203–212 [DOI] [PubMed] [Google Scholar]
- 24. Gunawan J., Debuch H. (1985) J. Neurochem. 44, 370–375 [DOI] [PubMed] [Google Scholar]
- 25. Gunawan J., Debuch H. (1982) J. Neurochem. 39, 693–699 [DOI] [PubMed] [Google Scholar]
- 26. Jurkowitz M. S., Horrocks L. A., Litsky M. L. (1999) Biochim. Biophys. Acta. 1437, 142–156 [DOI] [PubMed] [Google Scholar]
- 27. Jurkowitz-Alexander M. S., Horrocks L. A. (1991) Methods Enzymol. 197, 483–490 [DOI] [PubMed] [Google Scholar]
- 28. Rouser G., Fleischer S., Yamamoto A. (1970) Lipids 5, 494–496 [DOI] [PubMed] [Google Scholar]
- 29. Jurkowitz-Alexander M. S., Hirashima Y., Horrocks L. A. (1991) Methods Enzymol. 197, 79–89 [DOI] [PubMed] [Google Scholar]
- 30. Bradford M. M. (1976) Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 31. Laemmli U. K. (1970) Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 32. Graham J. M. (1993) Methods Mol. Biol. 19, 97–108 [DOI] [PubMed] [Google Scholar]
- 33. Hunter M. J., Commerford S. L. (1961) Biochim. Biophys. Acta. 47, 580–586 [DOI] [PubMed] [Google Scholar]
- 34. Allen C. E., Muthusamy N., Weisbrode S. E., Hong J. W., Wu L. C. (2002) Genes Chromosomes Cancer 35, 287–298 [DOI] [PubMed] [Google Scholar]
- 35. Bligh E. G., Dyer W. J. (1959) Can. J. Biochem. Physiol. 37, 911–917 [DOI] [PubMed] [Google Scholar]
- 36. Lowry O. H., Rosebrough N. J., Farr A. L., Randall R. J. (1951) J. Biol. Chem. 193, 265–275 [PubMed] [Google Scholar]
- 37. Gottfried E. L., Rapport M. M. (1962) J. Biol. Chem. 237, 329–333 [PubMed] [Google Scholar]
- 38. Huque T., Brand J. G., Rabinowitz J., Gavarron F. (1987) Comp. Biochem. Phys. B 86, 135–139 [DOI] [PubMed] [Google Scholar]
- 39. Wu L. C., Mak C. H., Dear N., Boehm T., Foroni L., Rabbitts T. H. (1993) Nucleic Acids Res. 21, 5067–5073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Strausberg R. L., Feingold E. A., Grouse L. H., Derge J. G., Klausner R. D., Collins F. S., Wagner L., Shenmen C. M., Schuler G. D., Altschul S. F., Zeeberg B., Buetow K. H., Schaefer C. F., Bhat N. K., Hopkins R. F., Jordan H., Moore T., Max S. I., Wang J., Hsieh F., Diatchenko L., Marusina K., Farmer A. A., Rubin G. M., Hong L., Stapleton M., Soares M. B., Bonaldo M. F., Casavant T. L., Scheetz T. E., Brownstein M. J., Usdin T. B., Toshiyuki S., Carninci P., Prange C., Raha S. S., Loquellano N. A., Peters G. J., Abramson R. D., Mullahy S. J., Bosak S. A., McEwan P. J., McKernan K. J., Malek J. A., Gunaratne P. H., Richards S., Worley K. C., Hale S., Garcia A. M., Gay L. J., Hulyk S. W., Villalon D. K., Muzny D. M., Sodergren E. J., Lu X., Gibbs R. A., Fahey J., Helton E., Ketteman M., Madan A., Rodrigues S., Sanchez A., Whiting M., Madan A., Young A. C., Shevchenko Y., Bouffard G. G., Blakesley R. W., Touchman J. W., Green E. D., Dickson M. C., Rodriguez A. C., Grimwood J., Schmutz J., Myers R. M., Butterfield Y. S., Krzywinski M. I., Skalska U., Smailus D. E., Schnerch A., Schein J. E., Jones S. J., Marra M. A. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 16899–16903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rusnak F., Liu J., Quinn N., Berchtold G. A., Walsh C. T. (1990) Biochemistry 29, 1425–1435 [DOI] [PubMed] [Google Scholar]
- 42. Krogh A., Larsson B., von Heijne G., Sonnhammer E. L. (2001) J. Mol. Biol. 305, 567–580 [DOI] [PubMed] [Google Scholar]
- 43. Mutch D. M., Anderle P., Fiaux M., Mansourian R., Vidal K., Wahli W., Williamson G., Roberts M. A. (2004) Physiol. Genomics 17, 11–20 [DOI] [PubMed] [Google Scholar]
- 44. Zhang Y. Y., Dennis E. A. (1988) J. Biol. Chem. 263, 9965–9972 [PubMed] [Google Scholar]
- 45. Blank M. L., Smith Z. L., Fitzgerald V., Snyder F. (1995) Biochim. Biophys. Acta. 1254, 295–301 [DOI] [PubMed] [Google Scholar]
- 46. Ellingson J. S., Lands W. E. M. (1968) Lipids 3, 111–120 [DOI] [PubMed] [Google Scholar]
- 47. Freeman N. M., Carey E. M., Carruthers A. (1984) Neurochem. Int. 6, 273–281 [DOI] [PubMed] [Google Scholar]
- 48. Arthur G., Page L., Mock T., Choy P. C. (1986) Biochem. J. 236, 475–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Von Jagow G., Schagger H. (1994) in A Practical Guide to Membrane Protein Purification (Von Jagow G., Schagger H. eds) pp. 68–69, Academic Press, Inc., San Diego, CA [Google Scholar]
- 50. Parsons J. F., Calabrese K., Eisenstein E., Ladner J. E. (2003) Biochemistry 42, 5684–5693 [DOI] [PubMed] [Google Scholar]
- 51. Snyder F. (1972) in Ether Lipids: Chemistry and Biology (Snyder F. ed) pp. 121–156, Academic Press, Inc., New York [Google Scholar]
- 52. Vance J. E. (1990) Biochim. Biophys. Acta 1045, 128–134 [DOI] [PubMed] [Google Scholar]
- 53. Gaposchkin D. P., Zoeller R. A. (1999) J. Lipid Res. 40, 495–503 [PubMed] [Google Scholar]
- 54. Soodsma J. F., Piantadosi C., Snyder F. (1970) Cancer Res. 30, 309–311 [PubMed] [Google Scholar]
- 55. Moolenaar W. H. (1995) Curr. Opin. Cell Biol. 7, 203–210 [DOI] [PubMed] [Google Scholar]
- 56. Mills G. B., Moolenaar W. H. (2003) Nat. Rev. Cancer. 3, 582–591 [DOI] [PubMed] [Google Scholar]